
Abstract
Accurate prediction of risk of progression from smoldering multiple myeloma (SMM) to active multiple myeloma (MM) is paramount to individualized early therapeutic strategies with minimum risk of overtreatment. Current risk stratification models do not account for evolving biomarker trajectories. We assembled a cohort of 2,344 patients with SMM from seven international centers with longitudinal clinical and biological data to train and validate the Precursor Asymptomatic Neoplasms by Group Effort Analysis (PANGEA)-SMM risk models. Four evolving biomarkers were significantly associated with shorter time to progression: M-protein increase ≥0.2 g dl−1, involved/uninvolved serum free light chain ratio increase ≥20, creatinine increase >25% and hemoglobin decrease ≥1.5 g dl−1. PANGEA-SMM outperforms established models, including the 20/2/20 and IMWG models, by more accurately predicting progression (C-statistic = 0.79), even without biomarker history (C-statistic = 0.78) or recent bone marrow biopsy (C-statistic = 0.78). We present PANGEA-SMM to the community as an easy-to-use, open-access tool for risk stratification in SMM. Validation tools are available to compare PANGEA-SMM to established models.
Similar content being viewed by others


Main
Smoldering multiple myeloma (SMM) is a biologically heterogeneous precursor to symptomatic multiple myeloma (MM), characterized by highly varying time to progression1. It is currently defined by the presence of a non-IgM M-protein ≥3 g dl−1 or urinary M-protein ≥500 mg per 24 hours and/or 10–60% clonal bone marrow plasma cells (BMPCs) without evidence of any myeloma-defining events1. Current models to stratify patients by the risk of progression from SMM to MM rely on static biomarkers, without using data on the preceding biomarker trajectory. The commonly used 20/2/20 model includes serum M-protein concentration (>2 g dl−1), involved over uninvolved (involved/uninvolved) serum free light chain (sFLC) ratio (>20) and BMPC infiltration (>20%), which are routinely measured in patients with SMM. The International Myeloma Working Group (IMWG) model additionally includes cytogenetic abnormalities such as t(4;14), t(14;16), del(13/13q) and gain(1q)2,3.
These current risk prediction models are well established and provide simple, reliable clinical scores. However, they have several limitations. Most notably, they fail to account for evolving biomarkers that reflect dynamic disease progression4,5,6,7,8,9,10,11,12, and they require bone marrow (BM) biopsies, which are often unavailable during follow-up and may have inconsistent results due to the patchy nature of plasma cell infiltration13. As a result, some high-risk patients go undetected and develop end-organ damage such as bone lesions, despite close monitoring14,15. Conversely, other patients may be mistakenly labeled as high risk due to the failure to account for stable biomarker trajectories that imply stable tumor mass over time16. Therefore, there is an urgent need for more accurate models that use longitudinal, dynamically evolving biomarkers to identify patients with SMM at the highest risk of progression to overt MM while minimizing the risk of overtreatment or underestimation of progression17,18,19,20,21. This is especially pressing in light of recent data showing the clinical benefit of early treatment with daratumumab in high-risk SMM.
To address these limitations, we analyzed a large cohort of 2,344 patients with untreated SMM to identify new predictive features from evolving biomarkers (including M-protein, sFLC ratio, creatinine and hemoglobin) and assess their contribution to longitudinal risk assessment of progression from SMM to MM. The training cohort consisted of 1,031 patients at the Dana-Farber Cancer Institute (DFCI), and the validation cohort comprised 1,313 patients from six international institutions. Our aim was to develop the Precursor Asymptomatic Neoplasms by Group Effort Analysis (PANGEA)-SMM model for repeated risk monitoring with significantly improved prediction accuracy compared to current models.
Results
Cohort description
We first aimed to develop a dynamic risk monitoring tool using clinical and biological variables that are available in patients with SMM—specifically, those with ≥10% plasma cells on BM biopsy at SMM diagnosis—and serial measurements of monoclonal protein, light chains and other routinely measured parameters. The training cohort included 1,031 patients with SMM (with 231 progressing to MM). The five validation cohorts included a total of 1,313 patients with SMM (with 421 progressing to MM). The median number of timepoints (visits with laboratory measurements) was four in the training cohort (interquartile range (IQR): 2–8), over a median follow-up time of 3.7 years (IQR: 1.8–7.1). In validation cohorts, the median number of timepoints was between one and seven, over a median follow-up time of 1.6−6.4 years. The distribution of disease and demographic characteristics and follow-up data are detailed in Table 1, with the distribution of biomarkers at baseline available in Supplementary Table 1. Unstratified time to progression was similar across cohorts, with 2-year progression rates of 12.1% in the training cohort and 12.0% in validation cohort 1 and broadly similar results in the other cohorts (Extended Data Fig. 1). In the training cohort, unstratified progression rates were 1.3% within 3 months, 3.8% within 6 months and 7.6% within 1 year of baseline (Extended Data Fig. 1). In the large cohorts (training cohort and validation cohorts 1 and 2), unstratified yearly progression rates were approximately 5% per year for the first 5 years and approximately 3% per year afterwards (Extended Data Fig. 1).
Defining dynamic biomarkers
From a baseline multivariate Cox model trained to predict progression to MM using established biomarkers (latest values of M-protein, involved/uninvolved sFLC ratio, creatinine and BMPC) in the training cohort, we used cross-validation to estimate the value of adding time-varying evolving dynamic biomarkers to the model. These dynamic biomarkers were used to provide information beyond merely the current static biomarker values. We identified four dynamic variables—evolving M-protein, creatinine, sFLC ratio and hemoglobin—that significantly improved risk predictions compared to using only rolling static biomarkers. The dynamic variables with associated hazard ratios are defined in Table 2. As outlined in the Methods section, the identification of dynamic biomarker trends in PANGEA-SMM was a two-step process: (1) we first defined classes of candidate trend definitions with varying thresholds for each biomarker, and (2) we then determined the optimal definition and threshold for each biomarker using k-fold cross-validation—that is, the definition and threshold that produced the greatest improvement in predictive performance (cross-validated C-statistic) compared to using no trend information. For example, for the sFLC ratio, larger increases over long time windows were found to be more informative for distinguishing high-risk patients than smaller increases over short time intervals. By contrast, for the M-protein concentration, smaller increases over short time intervals were found to be more informative. This distinction may arise because the sFLC ratio is affected by variability in both the involved and uninvolved chain measurements, whereas M-protein measurements are more stable.
Dynamic changes were detected in 2.9−21.2% of the total visits (and in 9.3−46.1% of patients) in the training cohort, depending on the dynamic biomarker considered (for example, dynamic hemoglobin versus dynamic M-protein; Supplementary Table 2). Evolving biomarkers were detected most frequently for M-protein and least frequently for sFLC ratio. Optimal dynamic definitions were very similar when removing BMPC from the Cox models (in terms of optimal dynamic variables and cross-validated C-statistics), and, therefore, we used the same dynamic definitions in our updated PANGEA-SMM no-BM model to avoid conflicting dynamic definitions and keep the detection of dynamic changes consistent across models.
Training and validation of PANGEA-SMM
Based on those definitions, we trained the PANGEA-SMM BM and no-BM models, which predict MM progression risk using both current biomarkers and dynamic biomarkers. All of the dynamic biomarkers were significant (at a 5% false discovery rate using the Benjamini−Hochberg procedure), indicating additional levels of progression risk beyond current biomarker levels (Extended Data Table 1).
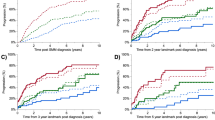
The PANGEA-SMM dynamic risk predictions—when stratified into high-risk, intermediate-risk and low-risk groups—led to different risk classification compared to 20/2/20. Patients were classified as high risk if their predicted 2-year risk of progression to MM was greater than 40%, intermediate risk if their predicted 2-year risk was between 10% and 40% and low risk if their predicted 2-year risk was less than 10%. Notably, the actual 2-year progression rates of the so-called high-risk patients were higher for the PANGEA-SMM classification than for the 20/2/20 classification (Fig. 1, Table 3 and Extended Data Fig. 2). For example, in validation cohort 1, the PANGEA-SMM BM high-risk group had a 2-year progression rate of 49.0% (95% confidence interval (CI): 17.7–68.4%), whereas the 20/2/20 high-risk group had a rate of only 33.2% (95% CI: 20.8–43.7%). Similar differences were observed in the other validation cohorts (Table 3). We note that although the cohorts have somewhat heterogeneous point estimates of 2-year progression rates for the intermediate-risk and high-risk groups, their CIs have substantial overlap (Table 3). These differences are not statistically significant and could be caused by sampling variability. This heterogeneity could also be due to differences across cohorts in the biomarker distributions within the intermediate-risk and high-risk groups.

Results from validation cohorts 1−5 were combined via meta-analysis as described in the Methods section). Plots show Kaplan−Meier estimates non-progression probability stratified by risk category (PANGEA or 20/2/20), with 95% pointwise CIs.
The longitudinal PANGEA-SMM BM high-risk status also had higher estimates of positive predictive value (PPV) for progression within 2 years than 20/2/20 high-risk status (for example, 73.9% versus 44.5% at 2 years in validation cohort 1), and both had similar negative predictive value (NPV) estimates (for example, 87.6% versus 88.6% at 2 years in validation cohort 1). Notably, when results were pooled across cohorts and timepoints, PANGEA-SMM BM had an overall PPV 12.7% greater (95% CI: 1.5−23.9%) than rolling 20/2/20 (Extended Data Table 2). Due to the relatively small number of patients classified as high risk in each cohort, CIs for the cohort-specific and time-specific PPV are wide; full results on the predictive value of PANGEA-SMM and 20/2/20 high-risk status are described in Extended Data Figs. 3 and 4, and results for alternative thresholds to define the PANGEA-SMM high-risk status are described in Supplementary Table 3.
For comparisons, we examined both the rolling 20/2/20 and IMWG models in our cohorts. Time to progression stratified by baseline 20/2/20 risk category was similar across cohorts, with 2-year progression rates similar to the original Mayo results3 of 6.2% for the low-risk group, 17.9% for the intermediate-risk group and 44.2% for the high-risk group (Fig. 1). Time to progression stratified by the IMWG four-factor risk category including cytogenetics was also consistent with previously published results3 (Extended Data Fig. 5).
When looking at individualized predictions, the PANGEA-SMM dynamic models improved the ranking accuracy of risk predictions over the rolling 20/2/20 model (that is, 20/2/20 computed using the patient’s latest biomarker measurements22) as measured by increases in time-specific and overall C-statistics (Fig. 2 and Supplementary Tables 3 and 5). The C-statistic measures the ability of prediction models to correctly rank patients by how quickly they indeed progress (that is, the degree to which patients with higher risk scores have shorter times to progression). For example, in validation cohort 1, the PANGEA dynamic BM model had an overall C-statistic relative increase of 10% over 20/2/20 (from 0.76 (95% CI: 0.72–0.80) to 0.84 (95% CI: 0.79–0.88)) and a 5-year C-statistic relative increase of 37% (from 0.63 (95% CI: 0.50–0.76) to 0.86 (95% CI: 0.79–0.94)). Similar results were seen for the other validation cohorts (Supplementary Table 4). Based on meta-analysis of all five validation cohorts, the PANGEA-SMM BM model demonstrated a significantly higher overall C-statistic compared to the 20/2/20 model, with a difference of 0.07 (9.6% increase) (95% CI: 0.05−0.09; Supplementary Table 5). Even when complete patient history was unavailable, the PANGEA-SMM models achieved higher C-statistics than the rolling 20/2/20 model (Supplementary Table 4; see ‘No history’ columns). Additionally, PANGEA-SMM outperforms the 20/2/20 model in scenarios with reduced visit frequency. For example, see ‘Validation results with less frequent observations’ in the Methods section, in which we present results from a modified version of validation cohort 1 in which the data were restricted to include only one patient visit per year. Finally, to demonstrate the absence of bias due to the presence of patients censored for early intervention in our training cohort, we performed a sensitivity analysis by retraining the PANGEA-SMM BM model after including the 233 patients who received early intervention. The C-statistic moved from 0.8403 (95% CI: 0.8111–0.8695) in the original BM model to 0.8402 (95% CI: 0.8103–0.8701) after exclusion. Estimated coefficients remained similar (<0.045 difference) between both models, as did the 2-year risk predictions (0.4% median difference, correlation 0.999).

The C-statistic at each timepoint (x axis) was computed using only the most recent observation for each remaining patient who is still being followed-up that many years after diagnosis. Results for ‘Validation cohorts 1−5’ are based on meta-analysis (across cohorts) as described in the Methods section. Overall C-statistic difference is based on meta-analysis (across cohorts and timepoints) as described in the Methods section and in Supplementary Table 5).
To illustrate the new model, we depicted the biomarker trajectories, PANGEA-SMM 2-year risk prediction and 20/2/20 2-year risk prediction for an example patient (Fig. 3a). This patient never had an M-protein concentration above 2 g dl−1 and was never considered high risk by the 20/2/20 model. However, the increasing trends in creatinine and sFLC ratio after 2.5 years led the PANGEA-SMM dynamic models to detect a very high risk of progression. The patient progressed to MM 8 months after the detection of an increasing M-protein trend by the PANGEA-SMM model (predicted risk increasing from 7.1% to 18.4%), 6 months earlier than 20/2/20 detected a similar level of risk. Figure 3b,c illustrates the dynamic follow-up of two additional patients, depicting cases where PANGEA-SMM predicted a higher risk than the 20/2/20 criteria, based on dynamic increases in biomarker levels rather than high absolute values, prior to progression. The PANGEA-SMM and 20/2/20 risk scores are similar when all biomarker values are low; otherwise, the risk scores can differ more substantially because PANGEA-SMM offers continuous risk scores, whereas 20/2/20 offers only three discrete levels of predicted risk. Extended Data Fig. 6 shows the predicted risk scores for (first row) three progressor patients with higher PANGEA-SMM risk and lower 20/2/20 risk and for (second row) three non-progressor patients with lower PANGEA-SMM risk and higher 20/2/20 risk. We emphasize that these cases were selected as an example to illustrate that there can be substantial differences between the risk predictions of PANGEA-SMM and 20/2/20. We developed an open-access, online calculator that allows users to enter patient data and view the resulting risk predictions for the PANGEA-SMM BM or no-BM model (https://pangeamodels.org).

a–c, For each of the three patients (a, b and c, respectively), the line color for M-protein, FLC ratio, creatinine and hemoglobin indicates whether an evolving dynamic biomarker is detected at each timepoint. Rolling 20/2/20 risk increases at the latest timepoint, where the patient already shows signs of progression to myeloma (anemia and acute kidney failure). By contrast, a skewed M-protein concentration is observed at the penultimate laboratory report, which translates through PANGEA-SMM as an increased predicted risk of progression.
PANGEA-SMM calibration
To assess the calibration of the PANGEA-SMM dynamic model, we compared its average prediction of the 2-year progression risk to the actual progression rates. In validation cohort 1, the PANGEA-SMM BM model consistently gave risk predictions within 3% of the actual rate of progression for subgroups defined by 20/2/20 category, M-protein concentration, sFLC ratio, BMPC, creatinine and age (Supplementary Table 6). The no-BM model showed similar calibration except when stratified by BM or high sFLC ratio. This level of accuracy is similar to or better than the rolling 20/2/20 model, and similar results were observed in the other cohorts (Supplementary Tables 7−10), except for validation cohort 3, where PANGEA-SMM no-BM showed better calibration results than PANGEA-SMM BM, and for validation cohort 5, where PANGEA evidently overestimated the 2-year progression risk in most subgroups (perhaps due to the slower ascertainment of transitions to MM and the less frequent evaluations in this cohort, despite a slightly higher overall fraction of progressors; compare to Table 1).
Cytogenetic markers
Fluorescence in situ hybridization (FISH) cytogenetic markers improve risk predictions—for example, in the IMWG model3. In our training cohort, cytogenetic markers, including t(4;14), 17p deletion and 1q gain, had significant marginal associations with MM progression in univariate analysis while controlling for no additional covariates (Extended Data Table 3). We observed evidence that translocations t(14;16) and t(14;20) were significant when added to the PANGEA-SMM BM model, but the other translocations (including t(4;14), t(11;14) and MYC rearrangements) and hyperdiploidy alone were not (Extended Data Table 3). We then tested the combined variable integrating multiple cytogenetic abnormalities defined by the International Myeloma Society (IMS) in 2025 (ref. 23): as any of t(4;14), t(14;16) or t(14;20) combined with either 1q gain or 1p deletion. We found that it was associated with a significantly increased risk of progression when added to the PANGEA-SMM model, further improving on the PANGEA-SMM model with the new IMS/IMWG cytogenetic risk stratification. Interestingly, the combination of markers used in the IMWG model of SMM3 (any of hyperdiploidy, 13q deletion, 1q gain or translocation t(4; 14)) was not significant when added to the PANGEA-SMM model but did have a significant independent association with MM progression. In the training cohort, cytogenetic abnormalities (of any kind) were observed in 47.2% of PANGEA-SMM BM high-risk cases, in 41.2% of PANGEA-SMM BM intermediate-risk cases and in 33.1% of PANGEA-SMM BM low-risk cases. Similarly, mixed results were seen in validation cohort 2 (Supplementary Table 11).
Validation and transparency
Finally, to facilitate scientific discussion and transparency, we created an open-access validation application (https://pangeamodels.org). In the application, users can specify a dataset (DFCI or Greek/UK) and explore the performance (ranking accuracy, calibration and predictive value) of PANGEA-SMM and 20/2/20 in flexible patient subpopulations defined by demographics and biomarkers (for example, age, sex and M-protein). This enables the investigation of many more subpopulations than can be discussed in standard tables and figures and can help identify areas in which current risk prediction models have greater room for improvement.
Discussion
In this study, we assembled a cohort of 2,344 patients with SMM with longitudinal clinical and biological follow-up data. We used time-varying multivariate Cox regression models to leverage the information brought by the fine variation of biomarkers for detecting early progression patterns and improve on the current criteria already included in the 20/2/20 and IMWG models. Through global collaborations across seven international centers, we were able to define clinically relevant evolving criteria of the PANGEA-SMM dynamic risk monitoring in patients with SMM.
The diagnosis of SMM defines a subgroup of patients with precursor conditions at a higher, yet very heterogenous, risk of progression to MM. Current static criteria and risk models are limited for guiding clinical management and early intervention. The plurality of biomarkers and risk stratification models with significant value for predicting progression from SMM to MM may lead to confusion and limit the interpretation and applicability in clinical trials. Multiple clinical trials use different criteria, models and risk groups that developed over recent years24,25,26. A homogenous and simple approach is critical for consistency in clinical routine. The 20/2/20 model, developed by Lakshman et al.2 in 2018, which used BMPC, sFLC ratio, and serum M-protein levels, is simple to stratify patients27, but dynamic biomarker trends are not integrated into individualized risk predictions. Additionally, the IMWG score by Mateos et al.3 in 2020 requires cytogenetic data, which may not be available in a substantial number of patients with SMM due to the inability to enrich enough BMPCs for FISH analyses. A unified, straightforward and precise risk stratification model incorporating dynamic biomarkers is essential to facilitate the implementation of therapeutic strategies and improve patient outcomes in SMM.
In recent years, multiple studies have demonstrated an interest in analyzing evolving trajectories of biomarkers and their inclusion in risk stratification models4,5,6,7,8,9,10,11,12. Definitions to capture dynamics were proposed for hemoglobin (increasing anemia) in the initial PANGEA model28, followed by evolving M-protein and/or total immunoglobulin increase and evolving sFLC, and were associated in multiple studies with a significantly shorter time to progression to MM. However, most early approaches were limited to comparing follow-up values to the baseline value, limiting the interception of rapidly evolving patterns late after diagnosis or reassessment of evolving patterns after this period. Furthermore, even more recent attempts were made on limited numbers of patients, leading to varying definitions and thresholds for considering each biomarker as evolving6,10.
In the present study, we identified four dynamic biomarker trends that are significantly associated with shorter time to progression and that can be used in the clinic with data routinely collected for patients followed for MM precursor conditions: an M-protein increase ≥0.2 g dl−1 within 18 months; an sFLC ratio increase ≥20 within 24 months; a creatinine increase ≥25% within 12 months; and a hemoglobin decrease ≥1.5 g dl−1 within 12 months. Those four definitions can be reassessed at any time during follow-up, regardless of baseline values or time of follow-up of the patient. Cox models demonstrated that each of those four evolving definitions independently improves risk stratification. We incorporate these dynamic trends into two dynamic risk monitoring models, enabling predictions that use peripheral blood biomarkers and, optionally, BM results, if available. Due to its dynamic nature and integration of evolving biomarkers, PANGEA-SMM is particularly suitable for dynamic risk monitoring to support clinical decisions.
The PANGEA-SMM model is still applicable in the absence of biomarker histories, recent BM biopsy or FISH results, making it an easy model to use for patients worldwide. PANGEA-SMM performs accurately at baseline or when no biomarker history is available. Notably, PANGEA-SMM consistently had better or similar performance to 20/2/20 in terms of ranking accuracy, calibration and predictive value across our five validation cohorts. This accuracy across heterogeneous datasets supports the applicability of PANGEA-SMM across clinical contexts.
PANGEA-SMM can also classify patients into groups of low, intermediate or high risk of progression to MM. This information could be used at baseline or any time during follow-up to guide inclusion in clinical trials. Furthermore, our results indicate that patients classified as high risk by PANGEA-SMM have a significantly higher PPV and a similar NPV at 2 years compared to patients classified as high risk by 20/2/20. This demonstrates that PANGEA-SMM accurately identifies patients who are truly high-risk SMM and potentially benefit from early treatment. The individual risks of patients classified as high risk by either PANGEA-SMM or 20/2/20 are heterogeneous. Notably, the ability of PANGEA-SMM to predict personalized, continuous progression risks allows a more nuanced and informative assessment of risk compared to only classifying patients into risk groups.
Interestingly, adding FISH results to the PANGEA-SMM BM model improved stratification. In our results, t(14;20) alone and the combined variable of the new IMS/IMWG criteria defined by one cytogenetic abnormality among t(4;14), t(14;16) and t(14;20) combined with either 1q gain or 1p deletion were associated with a significantly increased risk of 2-year progression compared to the prediction of the PANGEA-SMM BM model. By integrating FISH with the ability of PANGEA-SMM to model disease trajectories, risk stratification in patients with SMM is notably improved.
Further refinement of the model could be achieved by leveraging next-generation sequencing from blood and BM samples as well as the addition of circulating tumor cells and the use of highly sensitive technologies for detecting M-protein levels, such as mass spectrometry, which can also be included in future studies16,29,30. It should also be noted that despite the number of centers included in our study, racial and ethnic diversity was limited or not documented in our validation cohorts, given that most of those validation studies were conducted in Europe where race is not captured. Further studies will be needed to allow for the generalization of risk progression assessment in diverse populations and define an optimal observation frequency to improve risk prediction. Finally, although the training cohort includes a non-negligible proportion of patients censored due to early intervention, this proportion is much lower in the independent validation cohorts, which nonetheless demonstrate similar performance. This consistency supports the conclusion that censoring due to early intervention did not introduce meaningful bias into the model’s predictions. Also, censoring is only informative when it depends on factors that (1) affect the time to event and (2) are not already controlled for in the model31. In our case, early intervention typically occurred based on clinical markers that are included in the PANGEA-SMM models (for example, worsening trends or biomarker thresholds), reducing the potential that censoring due to early intervention was informative and a source of bias. Finally, retraining the BM model after excluding patients censored due to early intervention showed no meaningful impact on the results, supporting our approach.
There is a broad consensus in the literature on risk prediction modeling that validating models across multiple, diverse and heterogeneous cohorts is essential for assessing model performance and avoiding overly optimistic estimates of accuracy32,33. In our study, both the training and validation cohorts represent heterogeneous populations and standards of care, spanning institutions in the United States and several European countries. Although this diversity strengthens the generalizability of our findings, it also introduces complexity when comparing performance metrics across cohorts. To enhance the transparency and interpretability of our results, we developed an open-access web application (https://pangeamodels.org). This tool enables users to explore and compare the performance of PANGEA 2.0 and the 20/2/20 model across all cohorts and within specific subgroups, thereby facilitating a nuanced understanding of model behavior in different clinical contexts.
In conclusion, we developed PANGEA-SMM using one of the largest cohorts identified to date, with 2,344 SMM cases and international validation cohorts from six distinct institutions. This international collaborative effort demonstrated that PANGEA-SMM outperforms current criteria for SMM stratification, including 20/2/20. PANGEA-SMM is accessible online as an easy-to-use tool that can be immediately integrated into clinical practice. PANGEA-SMM can be used both for baseline stratification and for serial monitoring under various schedules of visit frequency. For serial monitoring, PANGEA-SMM leverages evolving biomarkers to detect early patterns of progression, outperforming current criteria. The addition of BM and FISH variables is also critical but not necessary for the use of the PANGEA model, making it easy and available to use in all clinical practices.
PANGEA-SMM models are available as a free open-access web application, along with a user-driven validation analysis tool, available at https://pangeamodels.org.
Methods
Cohorts and patients
This study was approved by the Dana-Farber/Harvard Cancer Center institutional review board (no. 21-127) in accordance with the Declaration of Helsinki. The PANGEA project is based on a cohort of patients with precursor conditions for MM identified at the DFCI for which longitudinal follow-up data, including clinical and biological variables, were collected and curated between 25 March 2021 and 21 October 2024. The primary hypothesis of the PANGEA project was an improvement of prediction accuracy compared to the previous SMM stratification models determined by the inclusion of new features (that is, dynamic biomarker) of the individual clinical profile. Among this cohort, 1,031 patients diagnosed with SMM were included as a training cohort in this study. PANGEA is a long-term cohort study at the DFCI, and all eligible patients with SMM were included for model training. To our knowledge, this is the largest cohort used for characterizing the transition from SMM to MM. Model validation is based on five independent cohorts of patients with SMM from six international centers. Cohort 1 included 380 and 105 cases from the National and Kapodistrian University of Athens (Athens, Greece) and University College London (London, UK), respectively; cohort 2 included 447 cases from the Heidelberg University Hospital (UKHD, Heidelberg, Germany); cohort 3 included 240 cases from the University of Navarra (Pamplona, Spain); cohort 4 included 67 cases from the University of Milan (Milan, Italy); and cohort 5 included 74 cases from the University Hospital of Würzburg (Würzburg, Germany). The recruitment of the training cohort and validation cohort 1 was approved by the Dana-Farber/Harvard Cancer Center institutional review board (no. 21-127). In accordance with ethical guidelines, our study was granted a waiver of informed consent by the institutional review board because the information collected on this protocol was retrospective and, thus, involved no more than minimal risk to the included patients. For validation cohort 2, written informed consent was obtained from all patients individually. Approval of the cohort recruitment was granted in ethics approval S-578/2023 by the Heidelberg ethics committee. For validation cohort 3, the study protocol, including recruitment and informed consent form, was approved by the ethics committee of the University of Navarra (no. 2017.134), and informed consent was obtained from all participants. For validation cohort 4, data were acquired within a protocol approved by the institutional review board of Milan (no. 419 on 30 August 2021), and all patients signed an informed consent. For validation cohort 5, informed consent was obtained based on a local ethics vote (no. 08/21) from Würzburg University.
Clinical annotation
For the training cohort, we collected baseline characteristics of patients at the date of diagnosis of SMM, including age, race, ethnicity and sex (self-reported), height and immunofixation isotype. We collected follow-up data with a median of two visits per year starting from the date of diagnosis of SMM until the date any of the following events occurred first: progression to active MM defined by SLiM-CRAB criteria, last follow-up visit, start of precursor treatment or death. Charts were manually reviewed by a team of expert clinical data annotators to identify any evidence of MM as defined by SLiM-CRAB criteria throughout follow-up and to ensure that any transition to MM was accurately dated. According to current standard of care, patients who met SMM criteria prior to clear SLiM-CRAB confirmation were classified and managed as SMM. In the training cohort, 114 patients (49% of 231 progressors) progressed to overt MM during follow-up based on SLiM criteria only (BMPC >60%, FLC ratio >100 with absolute involved FLC >100 mg l−1 and/or at least two magnetic resonance imaging focal lesions >5 mm). To rule out any substantial misclassification bias in our training cohort, we examined the 2-year progression rates stratified by the IMWG 20/2/20 risk category, which were as follows: 5.1% (95% CI: 3.1–7.1%), 18.6% (95% CI: 12.6–24.2%) and 41.9% (95% CI: 28.9–52.5%) for low, intermediate and high risk, respectively. These rates are similar to those reported by Mateos et al.3 in 2020 in their 20/2/20 validation study (6.2%, 17.9% and 44.2%, for low-risk, intermediate-risk and high-risk patients with SMM, respectively). Follow-up data included patient information relevant for the diagnosis and follow-up of MM and precursor conditions, including the following blood/serum values: total protein, IgA, IgM, IgG, κ and λ FLCs, sFLC ratio, calcium, creatinine, albumin, hemoglobin, lactate dehydrogenase, β-2 microglobulin and M-protein(s) concentration. Other collected variables include imaging, weight and therapy (including bisphosphonate use). Data from all BM biopsies annotated for patients during this follow-up and extracted BMPC and FISH findings, when available, were collected. FISH data were structured into one of four categories: positive, negative, not tested or unavailable. The following aberrations were captured: translocations t(4;14), t(6;14), t(11;14), t(14;16), t(14;20) and t(14;18), −17/17p deletion, 6q deletion, 11q22 deletion, 1q gain, 8q24/MYC rearrangements, −13/13q deletion, +3/+7 hyperdiploid, +9/+15 hyperdiploid, trisomy 4, trisomy 12 and trisomy 18. Study data were collected and managed using Research Electronic Data Capture (REDCap) electronic data capture tools hosted at the DFCI34,35. REDCap is a secure, web-based software platform designed to support data capture for research studies, providing (1) an intuitive interface for validated data capture; (2) audit trails for tracking data manipulation and export procedures; (3) automated export procedures for seamless data downloads to common statistical packages; and (4) procedures for data integration and interoperability with external sources.
For the validation cohorts, we extracted the targeted outcomes, time to progression, censoring or death and the biological data required by PANGEA-SMM analysis at initial and follow-up visits.
Defining dynamic/evolving biomarkers
For each of the four biomarkers in Table 2 we defined binary (0/1) dynamic variables indicating if the biomarker has increased/decreased in a way that markedly elevates the risk of progression to MM, beyond simply knowing the current biomarker value. We considered seven different candidate definitions of these binary dynamic variables and various thresholds. Candidate definitions were as follows:
-
(1)
The biomarker has increased by at least X% compared to any of the previous values in the past Y months.
-
(2)
The biomarker has increased by at least X (absolute increase) compared to any previous value in the past Y months.
-
(3)
The biomarker has increased by at least X% compared to the previous value.
-
(4)
The biomarker has increased by at least X (absolute increase) compared to the previous value.
-
(5)
The biomarker has increased by at least X (absolute increase) compared to the previous value and is at least as high as 90% of the maximum of all previous values.
-
(6)
The average change (slope, based on ordinary least squares regression) of the biomarker over the past Y months is greater than X.
-
(7)
The average change (slope) of the biomarker over the last K observations is greater than X.
The complete list of candidate thresholds (with X, Y and K values) tested can be found in Supplementary Table 12. For dynamic hemoglobin, we considered decreases (not increases) in definitions 1−7.
To determine the definition of each biomarker’s dynamic feature, we used a systematic grid search to evaluate the improvement from adding each candidate binary feature to a ‘basic’ model including only current biomarkers.
The baseline model was a multivariate Cox regression with time-varying biomarkers trained only with four biomarkers (that is, latest values of M-protein, involved/uninvolved sFLC ratio, creatinine and BMPC). Then, each candidate time-varying dynamic indicator variable was added one at a time as a predictor in the baseline model. We computed the model’s C-statistic by five-fold cross-validation (using only the training dataset). The optimal candidate dynamic definition for each biomarker was the definition that gave the greatest increase in C-statistic over the baseline model.
Training PANGEA-SMM models
The PANGEA-SMM models are two multivariate Cox regression models with time-varying predictors, namely the ‘BM’ and ‘no-BM’ models. Both include effects for three biomarkers (M-protein, log involved/uninvolved FLC ratio and log creatinine) and age as well as dynamic M-protein trend, dynamic involved/uninvolved sFLC ratio trend, dynamic creatinine trend and dynamic hemoglobin trend. Other demographic variables, including race, ethnicity and sex, were not included as input variables in the model because a previous study demonstrated that including them did not improve predictions of disease progression28. The dynamic biomarkers take values of 0 or 1 and can vary over time. The BM model also includes BMPC as a predictor, whereas the no-BM model does not and can be used when recent BMPC is not available. The dynamic variables are defined to be zero (not missing) when patient history is not available, so the models can be used without patient history (see ‘Handling of missing biomarker history’ in the Methods section for discussion of an alternative approach). The models were estimated using the survival36 package (version 3.7-0) in R37 (version 4.4.2) and output risk scores defined as the probability of progressing to MM within 2 years of the latest visit. Death was treated as a censoring event and not a competing risk, due to rare frequency in our training and validation cohorts (<5%)38.
We also assessed whether cytogenetic markers measured by FISH improve the predictions of the PANGEA-SMM BM model. Due to sample size limitations, we analyzed each FISH probe separately, adding it as a single new predictor in the PANGEA-SMM BM Cox model.
Handling of missing biomarker history
One important feature of the PANGEA-SMM models is that they are simple to use even when biomarker histories are unavailable. This is achieved by defining the trajectory variables to be zero when the biomarker histories are not available.
We considered an alternative approach to handle missing biomarker histories using a flexible framework that switches between dynamic and static submodels depending on data availability. The dynamic submodels were versions of the PANGEA-SMM models that were trained only on the subset of the training data in which all biomarker histories are observed (3,805 observations on 717 patients for the BM model; 3,912 observations on 733 patients for the no-BM model). The static submodels were simplified versions of the PANGEA-SMM models that excluded trajectory variables and were trained on the full training dataset. For each patient, risk predictions were generated from the dynamic submodels when biomarker histories were available and from the static submodels otherwise. This approach produced highly similar risk predictions to PANGEA-SMM (correlations of 0.98 for BM models and 0.97 for no-BM models) and nearly identical predictive accuracy (concordance: 0.8334 versus 0.8403 for BM models and 0.8083 versus 0.8103 for no-BM models) in the training cohort. Given these minimal differences, we chose to keep the simpler PANGEA-SMM strategy, which sets trajectory variables to zero when biomarker histories are not available.
Validating PANGEA-SMM models
We evaluated the ranking accuracy, risk stratification and calibration of the PANGEA-SMM models and rolling 20/2/20 on each validation cohort. ‘Rolling 20/2/20’ refers to the low−intermediate−high risk categories based on Lakshman et al.2, computed using the patient’s latest biomarker measurements22. We also evaluated the two PANGEA-SMM models when risk predictions use only the latest biomarker information (that is, all dynamic variables set to zero), in order to assess predictive performance when patient history is not available. Overall ranking accuracy was assessed for each model by generalized C-statistics39 including all serial observations for each patient. Dynamic ranking accuracy for each model was assessed by computing C-statistics based only on each patient’s most recent visit (and biomarker trends) at 0.1, 1, 2, 3, 4 or 5 years after baseline. C-statistics were pooled across validation cohorts using the random-effects meta-analysis technique of Debray et al.40. C-statistic differences (PANGEA-SMM BM minus rolling 20/2/20) were pooled across validation cohorts using the random-effects meta-analysis technique of Raudenbush41, with standard errors for the differences based on the cohort-specific standard errors of the C-statistics and the correlation between the PANGEA-SMM BM and 20/2/20 C-statistics estimated in the training cohort via the bootstrap.
Although PANGEA-SMM produces personalized, continuous 2-year risk scores (that is, progression probabilities between 0% and 100%), we also assessed its ability to stratify patients into low-risk, intermediate-risk and high-risk groups. This stratification was based on PANGEA-SMM’s predicted risk of progression within 2 years, with ‘low’ risk patients having less than 10% predicted risk, ‘intermediate’ risk patients having between 10% and 40% predicted risk and ‘high’ risk patients having greater than 40% predicted risk. The thresholds were selected based on their clinical relevance and practical applicability while also ensuring that the resulting subgroups were sufficiently large within the training cohort to allow for meaningful analysis. These thresholds were defined prior to examining the relative proportions of the three groups in the validation cohorts, in order to preserve the integrity of the validation process. We then computed Kaplan−Meier progression curves stratified by these risk groups and compared results to 20/2/20. We pooled the progression curves for each risk group across cohorts using the random-effects meta-analysis method of Combescure et al.42 with a continuity correction of 0.05.
We also evaluated the dynamic value of PANGEA-SMM and 20/2/20 high-risk status as predictors of progression to MM within 2 years. This was done using standard inverse probability of censoring estimates of the time-dependent PPVs and NPVs43,44 based on each patient’s most recent visit at 0.1, 1, 2, 3, 4 or 5 years after baseline. The time-dependent PPVs and NPVs were pooled across cohorts using the random-effects meta-analysis method of Leeflang et al.45. The overall PPVs and NPVs and their differences (PANGEA-SMM BM minus 20/2/20) were pooled across validation cohorts using the random-effects meta-analysis technique of Raudenbush41, with standard errors based on the cohort-specific and time-specific standard errors of the predictive values and the correlation between these cohort-specific and time-specific predictive values estimated in the training cohort via the bootstrap.
Finally, we assessed calibration of the PANGEA-SMM models, which refers to the level of agreement between the predicted and observed progression rates. For each entire validation cohort and various subcohorts (stratified by low versus high biomarkers), we computed (1) the average 2-year PANGEA risk of progression and (2) the actual 2-year rate of progression (based on Kaplan−Meier analysis). We compared these progression rates to 20/2/20 (based on the reported 2-year progression rates in Mateos et al.3).
Validation results with less frequent observations
We constructed an alternative version of validation cohort 1 in which each patient has, at most, one observation per year. This was achieved by keeping only the first visit per year for each patient, starting from baseline. For example, for a patient who originally had visits at 1.2, 1.4, 2.7, 3.1 and 3.9 years after baseline, in this alternate version of the dataset we would keep only the visits at 1.2, 2.7 and 3.1 years after baseline. Time to final censoring or progression was kept the same.
Supplementary Tables 13–15 and Supplementary Figs. 1 and 2 below describe the descriptive statistics and model performance for this ‘low frequency’ version of validation cohort 1. Despite the median time between observations more than doubling (from 5.5 months to 12.8 months; Supplementary Table 13), the comparisons between PANGEA-SMM and 20/2/20 in terms of ranking accuracy, predictive value and calibration remain similar to the original results.
Open-science validation application
We developed an open-access web application to evaluate the performance of PANGEA-SMM and alternative models on our training data as well as a subset of the validation cohorts. Using the application, users can specify a dataset (DFCI or Greek/UK) and subpopulation of interest (for example, female patients at the DFCI over age 60 with sFLC ratio >20) and see the ranking accuracy, calibration and predictive value of PANGEA-SMM compared to 20/2/20. This application allows users to compare the performance of PANGEA-SMM (BM and no-BM models) with 20/2/20 in flexible and detailed populations, facilitating both decision-making about appropriate populations to use each model and future research on risk models for MM.
Clinical calculator
To easily use PANGEA-SMM in the clinic, we developed an open-access web application. The application allows entering the individual’s values (M-protein concentration, sFLC ratio, creatinine, hemoglobin and ±BMPC) along with dates of measurement. Based on the filled-in information, PANGEA-SMM automatically calculates the evolving dynamic biomarkers when past values are available and identifies the relevant model for analyzing the patient’s data (no-BM and BM). Accordingly, PANGEA-SMM determines the personalized risk of progression to MM for the patient and classifies them into groups of low, intermediate or high risk of progression to MM by comparing their personalized risk to the thresholds described above.
Statistics and reproducibility
This study was a retrospective observational analysis of longitudinal cohorts of patients with SMM assembled from participating institutions. No statistical method was used to predetermine sample size; sample sizes were determined by data availability, and the training cohort represents, to our knowledge, the largest assembled to date for modeling progression from SMM to MM. No data were excluded from the analyses beyond standard cohort eligibility criteria described in Methods.
The study involved no experimental interventions, and patients were managed according to standard of care at their respective institutions; therefore, randomization and blinding were not applicable. All statistical analyses were conducted using prespecified validation and modeling procedures, as detailed in Methods. Model development was performed in a single training cohort, and reproducibility and robustness were assessed through independent external validation across five international cohorts using predefined performance metrics.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Aggregate data (for example, performance metrics of PANGEA-SMM models and demographic statistics of the training cohort) can be shared with investigators following appropriate proposal to the corresponding author (irene_ghobrial@dfci.harvard.edu) and after approval by the study team. Requests will receive an initial answer within 3 months. Data will be shared only under agreements that prevent participant reidentification and accessed exclusively within a secure research environment to further safeguard participant privacy. Data from validation cohorts can be shared only upon request to the respective principal investigator or institution.
Code availability
PANGEA-SMM models are available as a free open-access web application at https://pangeamodels.org, and user-driven validation analyses can be conducted at https://pangeamodels.org. Code used to generate cohort-specific validation results and figures depicted in this paper is available as a public GitHub repository at https://github.com/pangea-study/pangea_2.0.
References
Rajkumar, S. V. et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 15, e538–e548 (2014).
Lakshman, A. et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood Cancer J. 8, 59 (2018).
Mateos, M. V. et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 10, 102 (2020).
Ravi, P. et al. Evolving changes in disease biomarkers and risk of early progression in smoldering multiple myeloma. Blood Cancer J. 6, e454 (2016).
Fernández de Larrea, C. et al. Evolving M-protein pattern in patients with smoldering multiple myeloma: impact on early progression. Leukemia 32, 1427–1434 (2018).
de Daniel, A. et al. The evolving pattern of the monoclonal protein improves the IMWG 2/20/20 classification for patients with smoldering multiple myeloma. Hemasphere 8, e76 (2024).
Bladé, J. et al. Smoldering (asymptomatic) multiple myeloma: current diagnostic criteria, new predictors of outcome, and follow-up recommendations. J. Clin. Oncol. 28, 690–697 (2010).
Atrash, S. et al. Evolving changes in M-protein and hemoglobin as predictors for progression of smoldering multiple myeloma. Blood Cancer J. 8, 107 (2018).
Gran, C. et al. Dynamic follow-up of smoldering multiple myeloma identifies a subset of patients at high risk of progression. Am. J. Hematol. 96, E63–E65 (2021).
Akhlaghi, T. et al. Evaluating the effect of evolving changes in serum biomarkers on the risk of progression in smoldering multiple myeloma. American Society of Hematology https://ash.confex.com/ash/2023/webprogram/Paper180315.html (2023).
Rosiñol, L. et al. Smoldering multiple myeloma: natural history and recognition of an evolving type. Br. J. Haematol. 123, 631–636 (2003).
Wu, V. et al. Risk stratification of smoldering multiple myeloma: predictive value of free light chains and group-based trajectory modeling. Blood Adv. 2, 1470–1479 (2018).
Hillengass, J. et al. International myeloma working group consensus recommendations on imaging in monoclonal plasma cell disorders. Lancet Oncol. 20, e302–e312 (2019).
Abdallah, N. H. et al. Mode of progression in smoldering multiple myeloma: a study of 406 patients. Blood Cancer J. 14, 9 (2024).
Werly, A. et al. Patterns of progression in a contemporary cohort of 447 patients with smoldering multiple myeloma. Blood Cancer J. 14, 176 (2024).
Cordas dos Santos, D. M. et al. New horizons in our understanding of precursor multiple myeloma and early interception. Nat. Rev. Cancer 24, 867–886 (2024).
Avet-Loiseau, H. & Bahlis, N. J. Smoldering multiple myeloma: taking the narrow over the wide path. Blood 143, 2025–2028 (2024).
Dimopoulos, M.-A. et al. Phase 3 randomized study of daratumumab monotherapy versus active monitoring in patients with high-risk smoldering multiple myeloma. American Society of Hematology https://ash.confex.com/ash/2024/webprogram/Paper201057.html (2023).
Landgren, O. et al. Efficacy and safety of daratumumab in intermediate/high-risk smoldering multiple myeloma: final analysis of CENTAURUS. Blood 145, 1658–1669 (2025).
Mateos, M. V. et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (QuiRedex): long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 17, 1127–1136 (2016).
Lonial, S. et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. J. Clin. Oncol. 38, 1126–1137 (2020).
Visram, A. et al. Assessing the prognostic utility of smoldering multiple myeloma risk stratification scores applied serially post diagnosis. Blood Cancer J. 11, 186 (2021).
Avet-Loiseau, H. et al. International Myeloma Society/International Myeloma Working Group consensus recommendations on the definition of high-risk multiple myeloma. J. Clin. Oncol. 43, 2739−2751 (2025).
Dimopoulos, M. A. et al. Subcutaneous daratumumab (DARA SC) versus active monitoring in patients (pts) with high-risk smoldering multiple myeloma (SMM): randomized, open-label, phase 3 AQUILA study. J. Clin. Oncol. 40, TPS8075 (2022).
Nadeem, O. et al. Immuno-PRISM: a randomized phase II platform study of bispecific antibodies in high-risk smoldering myeloma. Blood 142, 206 (2023).
Landgren, C. O. et al. Daratumumab monotherapy for patients with intermediate-risk or high-risk smoldering multiple myeloma: a randomized, open-label, multicenter, phase 2 study (CENTAURUS). Leukemia 34, 1840–1852 (2020).
Mellgard, G. et al. Risk stratification models overestimate progression risk in contemporary patients with smoldering multiple myeloma. Hemasphere 8, e61 (2024).
Cowan, A. et al. Personalised progression prediction in patients with monoclonal gammopathy of undetermined significance or smouldering multiple myeloma (PANGEA): a retrospective, multicohort study. Lancet Haematol. 10, e203–e212 (2023).
Malik, S. U. et al. Update on risk stratification model of smoldering multiple myeloma: a systematic review. Blood 134, 5512 (2019).
Vasco-Mogorrón, M. A. et al. Blood-based risk stratification for pre-malignant and symptomatic plasma cell neoplasms to improve patient management. Am. J. Cancer Res. 11, 2736–2753 (2021).
Jackson, D. et al. Relaxing the independent censoring assumption in the Cox proportional hazards model using multiple imputation. Stat. Med. 33, 4681–4694 (2014).
National Academies of Sciences, Engineering, and Medicine. Reproducibility and Replicability in Science (National Academies Press, 2019); https://www.nationalacademies.org/projects/DBASSE-BBCSS-17-03 .
Riester, M. et al. Risk prediction for late-stage ovarian cancer by meta-analysis of 1525 patient samples. J. Natl Cancer Inst. 106, dju048 (2014).
Harris, P. A. et al. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 42, 377–381 (2009).
Harris, P. A. et al. The REDCap consortium: building an international community of software platform partners. J. Biomed. Inform. 95, 103208 (2019).
Therneau, T. M. A package for survival analysis in R. https://cran.r-project.org/web/packages/survival/vignettes/survival.pdf (2026).
R Core Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022); https://www.R-project.org
Maradit Kremers, H. et al. Competing risk analysis: what does it mean and when do we need it in orthopedics research? J. Arthroplasty 36, 3362–3366 (2021).
Bansal, A. & Heagerty, P. J. A tutorial on evaluating the time-varying discrimination accuracy of survival models used in dynamic decision making. Med. Decis. Making 38, 904–916 (2018).
Debray, T. P. et al. A framework for meta-analysis of prediction model studies with binary and time-to-event outcomes. Stat. Methods Med. Res. 29, 2768–2787 (2019).
Raudenbush, S. W. in The Handbook of Research Synthesis and Meta-Analysis (eds Cooper H. et al.) 295–316 (Russell Sage Foundation, 2009).
Combescure, C., Foucher, Y. & Jackson, D. Meta-analysis of single-arm survival studies: a distribution-free approach for estimating summary survival curves with random effects. Stat. Med. 33, 2521–2537 (2014).
Blanche, P., Dartigues, J. F. & Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 32, 5381–5397 (2013).
Trevethan, R. Sensitivity, specificity, and predictive values: foundations, pliabilities, and pitfalls in research and practice. Front. Public Health 5, 307 (2017).
Leeflang, M. M. G., Deeks, J. J., Rutjes, A. W. S., Reitsma, J. B. & Bossuyt, P. M. M. Bivariate meta-analysis of predictive values of diagnostic tests can be an alternative to bivariate meta-analysis of sensitivity and specificity. J. Clin. Epidemiol. 65, 1088–1097 (2012).
Acknowledgements
We acknowledge J. Colchie and K. Padgett for their regulatory work and S. Abuhasan, R. Patel and M. Rahman for their contributions to data collection and quality control, including clinical annotations. This work was supported, in part, by the National Institutes of Health and the National Cancer Institute (R35CA263817 and U01CA271492 awarded to I.M.G.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, Instituto de Salud Carlos III (CB16/12/00369, FIS no. PI17/01243), the ERA-NET TRANSCAN-2 iMMunocell (AC17/00101), Cancer Research UK (C355/A26819), the FC AECC (INVES211176MART), AIRC under the Accelerator Award Program (EDITOR, AIRC-IG-24689), Fondazione Regionale per la Ricerca Biomedica/FRRB (Regione Lombardia), 2017 MMRF Immunotherapy Networks of Excellence, the CRIS Cancer Foundation (PR_EX_2020-02), the Leukemia & Lymphoma Society Translational Research Program grant, European Commission Mission Cancer grant 101097094 (ELMUMY) and the Riney Family Multiple Myeloma Research Program Fund. The collection and assembly of the UKHD cohort was supported by the Dietmar-Hopp Foundation (grant no. 1DH1811373). The authors thank S. Krippner and Y. Tamamushi for the collection and curation of the UKW cohort data. F.C. was supported by the Ligue Contre le Cancer Association. D.E.S. was supported by National Institutes of Health and National Cancer Institute grant T32CA009337. D.M.C.d.S. received the Walter Benjamin Fellowship from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, grant no. 525171148). N.B. was supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 817997). M.C.D.V. was funded by the Umberto Veronesi Foundation (grant tracking no. 5693) and by Pfizer Global Medical Grants (grant tracking no. 75340503).
Author information
Authors and Affiliations
Contributions
F.C., D.E.S., S.G. and E.K.M. contributed equally as first authors. L.T. and I.M.G. contributed equally as senior authors. I.M.G., L.T., F.C., D.E.S., E.K.M. and M.A.T. contributed to the design of the study. E.K.M., J.P., T.H., A.W., S.K.S., F.T., D.F., C.I.L., N.K., N.C.G., E.M.-S., R.T., J.W., S.J.C., L.A., M.C.D.V., C.D.M., L.P., V.P., P.C., C.T., S.P., J.L., M.-T.S. and M.D. contributed to the acquisition of the data. F.C., D.E.S., S.G., E.K.M., T.R.G., T.H., M.A.T. and L.T. contributed to the analysis of the data. F.C., D.E.S., S.G., T.R.G. and L.T. contributed to the software creation. F.C., D.E.S., E.K.M., L.T. and I.M.G. drafted the paper. All authors reviewed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
E.K.M. served an advisory board member for Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen-Cilag, Menarini/Stemline, Oncopeptides, Pfizer, Sanofi and Takeda; received honoraria from Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen-Cilag, Menarini/Stemline, Oncopeptides, Pfizer, Sanofi and Takeda; received research funding from Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen-Cilag, Sanofi and Takeda; and received travel expenses from Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen-Cilag, Menarini/Stemline, Pfizer, Sanofi and Takeda. D.M.C.d.S. received research funding from Regeneron. J.W. has consulted for Johnson & Johnson, Sanofi, Takeda, Pfizer, Stemline, Oncopeptides and Skyline Dx; received honoraria from Takeda, GlaxoSmithKline and BeiGene; and received research support from Bristol Myers Squibb. L.A. declares grants from the International Myeloma Society for travel and conference expenses. E.K. reports honoraria from Amgen, Janssen, Takeda, Genesis Pharma, Pfizer and GlaxoSmithKline; travel grants from Janssen; and being an advisory board member for Janssen and Prothena. M.A.D. declares honoraria from Amgen, Bristol Myers Squibb, Takeda and Janssen and is an advisory board member for Amgen, Bristol Myers Squibb, Takeda and Janssen. C.R.M. reports research funding from GRAIL. O.N. served on advisory boards for Johnson & Johnson, Bristol Myers Squibb, Sanofi, GPCR Therapeutics, Kite and AstraZeneca and received research funding from Bristol Myers Squibb, Johnson & Johnson and Takeda. N.B. received unrestricted research funding from Janssen; served on advisory boards for Takeda, Pfizer and Johnson & Johnson; and served on speakers bureaus for Johnson & Johnson, Sanofi, Amgen, Oncopeptides, Pfizer and GlaxoSmithKline. M.C.D.V. served on advisory boards for Takeda, Pfizer, Menarini/Stemline, Amgen and Johnson & Johnson and on speakers bureaus for Johnson & Johnson, Sanofi and GlaxoSmithKline. K.M.K. received honoraria and/or research support from AbbVie, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Novartis, Pfizer, Sanofi, Takeda and Stemline. H.E. has consulted for Bristol Myers Squibb, Johnson & Johnson, Amgen, Takeda, Sanofi, GlaxoSmithKline and Novartis; discloses research funding from Bristol Myers Squibb, Johnson & Johnson, Amgen, GlaxoSmithKline and Sanofi; has received honoraria from Bristol Myers Squibb, Johnson & Johnson, Amgen, Takeda, Sanofi, GlaxoSmithKline and Novartis; and received travel support from Bristol Myers Squibb, Johnson & Johnson, Amgen, Takeda, Novartis and Sanofi. J.F.S.-M. reports consultancy fees from AbbVie, Amgen, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Haemalogix, Janssen, Karyopharm Therapeutics, Merck Sharp & Dohme, Novartis, Regeneron, Roche, Sanofi, Secura Bio and Takeda. B.P. reports consultancy fees from Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Roche, Sanofi and Takeda; research funding from AstraZeneca, BeiGene, Bristol Myers Squibb, GlaxoSmithKline, Roche and Sanofi; and honoraria from Adaptive, Amgen, Becton Dickinson Biosciences, Bristol Myers Squibb/Celgene, GlaxoSmithKline, Janssen, Roche and Sanofi. I.M.G. has consulted for Bristol Myers Squibb, AstraZeneca, Amgen, Curio Science, Sanofi, Johnson & Johnson, Pfizer, Menarini Silicon Biosystems, Aptitude Health, GlaxoSmithKline, AbbVie, Adaptive Biotechnologies, Window Therapeutics and Regeneron. She has received honoraria or speaker fees from Vor Biopharma, Janssen, MJH Life Sciences, Novartis, Takeda, Amgen, Regeneron, Curio Science, Standard Biotools and Physicians’ Education Resource. She is a founder and executive board member of, and holds private equity in, Predicta Biosciences. Her spouse is the chief medical officer of, and holds private equity in, Disc Medicine. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Nizar Bahlis, Pieter Sonneveld and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Unstratified Kaplan-Meier estimates (and 95% pointwise confidence intervals) of non-progression probability for each cohort.
Sample sizes for each cohort are described in Table 1.
Extended Data Fig. 2 Risk stratification of the PANGEA-SMM dynamic model (BM) compared to rolling 20/2/20 for each of the validation cohorts.
Plots show Kaplan-Meier estimates (and 95% pointwise confidence intervals) of non-progression probability stratified by risk category (PANGEA-SMM or 20/2/20). Sample sizes for each cohort are described in Table 1.
Extended Data Fig. 3 Point estimates and 95% confidence intervals of the dynamic positive predictive value (PPV) for 20/2/20 and PANGEA-SMM BM high-risk status in each cohort.
The PPV at each time point (x-axis) is computed using only the most recent observation for each patient who is still being followed up that many years after diagnosis. It is the probability of progressing to MM within 2 years for high-risk patients. At some time points the PPV could not be computed because sample sizes (esp. the number of 20/2/20 or PANGEA-SMM high-risk cases) were too small. Results shown in panel “Validation Cohorts 1-5” are based on meta-analysis as described in the methods section. Overall sample sizes for each cohort are described in Table 1.
Extended Data Fig. 4 Point estimates and 95% confidence intervals of the dynamic negative predictive value (NPV) for 20/2/20 and PANGEA-SMM BM high-risk status in each cohort.
The NPV at each time point (x-axis) is computed using only the most recent observation for each patient who is still being followed up that many years after diagnosis. It is the probability of not progressing to MM within 2 years for patients classified as low- or intermediate-risk. At some time points the NPV could not be computed because sample sizes (esp. the number of 20/2/20 or PANGEA-SMM low- or intermediate-risk cases) were too small. Results shown in panel “Validation Cohorts 1-5” are based on meta-analysis as described in the methods section. Overall sample sizes for each cohort are described in Table 1.
Extended Data Fig. 5 Risk stratification of the PANGEA-SMM dynamic model (BM) compared to rolling 20/2/20 for the training cohort.
Plots show Kaplan-Meier estimates (and 95% pointwise confidence intervals) of non-progression probability stratified by risk category (PANGEA-SMM or IMWG). Figure includes the subset of patients with available data for IMWG FISH criteria: hyperdiploidy, 13q deletion, 1q gain, or translocation t(4; 14); (n = 732).
Extended Data Fig. 6 Illustration of follow-up using the rolling 20/2/20 and dynamic PANGEA-SMM models.
Predicted risk scores for (first row) three progressor patients with higher PANGEA-SMM risk and lower 20/2/20 risk and for (second row) three non-progressor patients with lower PANGEA-SMM risk and higher 20/2/20 risk.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1−15 and Figs. 1 and 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chabrun, F., Schwartz, D.E., Gentile, S. et al. Enhanced dynamic risk stratification of smoldering multiple myeloma. Nat Med (2026). https://doi.org/10.1038/s41591-026-04304-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41591-026-04304-x
