
Abstract
T cell receptors (TCRs) play a vital role in immune recognition by binding specific epitopes. Accurate prediction of TCR–epitope interactions is fundamental for advancing immunology research. Although numerous computational methods have been developed, a comprehensive evaluation of their performance remains lacking. Here we assessed 50 state-of-the-art TCR–epitope prediction models using 21 datasets covering 762 epitopes and hundreds of thousands binding TCRs. Our analysis revealed that the source of negative TCRs substantially impacts model accuracy, with external negatives potentially introducing uncontrolled confounders. Model performance generally improved with more TCRs per epitope, highlighting the importance of large and diverse datasets. Models incorporating multiple features typically outperformed those using only complementarity-determining region 3β information, yet all struggle to generalize to unseen epitopes. The use of independent test sets proved crucial for unbiased assessment on both seen and unseen epitopes. These insights will guide the development of more accurate and generalizable TCR–epitope prediction models for real-world applications.
Similar content being viewed by others


Main
T cell receptors (TCRs) are key components of the adaptive immune system, responsible for recognizing specific epitopes—short peptide fragments derived from pathogens or self-proteins—presented by major histocompatibility complex (MHC) molecules1. Approximately 95% of TCRs consist of one α chain and one β chain2, each containing three complementarity-determining regions (CDRs), in which CDR1 and CDR2 are well conserved and CDR3 is the primary region for antigen contact3,4,5. TCR–epitope interactions are pivotal for initiating immune responses against foreign invaders and tumor cells.
The high diversity of TCRs, coupled with the specificity of their interactions with epitopes, makes large-scale experimental determination of TCR–epitope interactions challenging. Traditionally, methods such as multimer-based assays6, in vitro stimulation7, peptide scanning8 and enzyme-linked immunospot assays9 are labor-intensive and low-throughput, necessitating the development of high-throughput ways to recognize TCR–epitope interactions10. Recent advancements in single-cell sequencing technologies have facilitated the identification of a growing number of TCR–epitope pairs. This surge in experimental data has driven rapid progress in computational prediction models of TCR–epitope interactions.
Nevertheless, several key challenges continue to impede progress in understanding and predicting TCR–epitope interactions. (1) The complexity and limited understanding of interactions present a major barrier. Current knowledge is largely based on a relatively small number of structural models, which fall short of providing comprehensive rules applicable to TCR–epitope interactions10,11. (2) The diversity of features that need consideration adds another layer of complexity5. These features include all six CDRs from α and β chains, MHC classes and allotypes, introducing high dimensionality and variability. (3) Many models struggle to predict interactions with unseen epitopes, hindering the application of real-world scenarios where new epitopes need to be rapidly identified12,13. (4) Labeled TCR–epitope data are notably scarce and available in substantial quantities for only a few epitopes, varying widely in terms of the features provided. (5) The choice of negative datasets, which consists of TCRs that do not bind to specific epitopes, can introduce biases into the models and affect their predictive power.
To assess the performance of TCR–epitope prediction models, several benchmarking studies13,14,15,16 have been conducted with focuses on model generalizability and data dependency, such as IMMREP2213 and IMMREP2314. These studies provided valuable insights into the strengths and weaknesses of different methods, highlighting advancements in performance when incorporating features beyond CDR3β and the challenges associated with generalizing predictions for unseen epitopes.
However, these studies often involve a limited number of evaluated models or focus primarily on specific aspects of evaluation. For instance, IMMREP22 focused on retraining and evaluating TCR–epitope prediction methods using paired αβ TCR sequence data, specifically targeting the seen-epitope scenario. IMMREP23 introduced a dataset comprising unpublished paired TCR data, aiming to address some of the gaps left by IMMREP22. However, it is reported that this test data contained potential target leakage, which would allow some models to exploit the test dataset structure, potentially inflating their performance metrics. In addition, previous studies did not define specific training data, complicating comparisons between different model architectures and training strategies.
To address these limitations, we conducted a comprehensive benchmark of 50 publicly available TCR–epitope prediction models (including variants) using well-curated data from 21 databases. Our multifaceted evaluation strategy included (1) comprehensive data collection: we gathered data from multiple sources to ensure diversity and representativeness across human epitopes and TCRs; (2) extensive prediction models: we assessed not only models that consider CDR3β-only feature but also those that incorporate additional features; (3) independent testing: we used fully independent test sets to evaluate reliability and generalization of models; (4) model retraining: we retrained available models to control for variations in implementation and data settings, allowing for a fair comparison under standardized conditions; (5) impact of TCR similarity and cross-reactivity: we applied a stringent evaluation by excluding similar TCR sequences between training and test sets, and assessed model robustness with and without cross-reactivity; (6) analysis of training set characteristics: we analyzed the effects of different training set compositions, including negative TCR source and P-to-N ratio; and (7) evaluation under different scenarios: we assessed original and retrained models on both seen- and unseen-epitope prediction to evaluate robustness and generalization capability of models.
Our comprehensive benchmarking study offers a valuable resource for both model developers and end users, facilitating informed decisions in selecting the most appropriate models for specific applications. This work lays the groundwork for future developments in TCR–epitope prediction models, contributing to our understanding of the immune system and aiding in the design of personalized immunotherapies.
Results
Data collection and study design
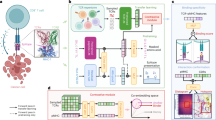
We designed a workflow integrating systematic data collection, model retrieval and multiple comparison strategies (Fig. 1a). TCR–epitope data were curated from 21 datasets: 19 with positive binding pairs and the remaining 2 with unbound TCRs for negatives (Supplementary Table 1). After rigorous filtering, such as preventing data leakage with CD-HIT17 and removing cross-reactive TCRs, we constructed the training, test and independent test sets for both original and retraining model evaluation. Negative datasets were constructed using antigen-specific (AS), patient-sourced (PS) and healthy-sourced (HS) TCRs. Importantly, we introduced a refined cross-matching-based AS strategy under immunologically relevant categories (Methods), which minimizes false-negative pairings. Dataset analysis confirmed the rarity of such cross-category matches, validating the reliability (Extended Data Fig. 1a–c).

a, Overview of the TCR–epitope binding, data collection, model training and evaluation. b, Framework for assessing TCR–epitope prediction models and evaluating key effect factors.
We focused on 50 models published in recent years, 46 of which provided accessible model/portal/code (including training data) for testing and 31 of which supplied complete code for retraining. The collection includes 7 traditional machine-learning and 43 deep-learning models, with focus on predicting both seen epitopes (in the training set) and unseen epitopes (not in the training set) interactions. Models are categorized based on training features as CDR3β-only models, which rely solely on CDR3β sequences and constitute most of the models; and CDR3β + others models, which incorporate additional features beyond CDR3β, such as MHC and CDR3α-derived features (Supplementary Table 2).
First, to ensure unbiased evaluation of original models, we constructed several independent test sets containing TCRs not present in any training data of the original models (Fig. 1b and Extended Data Fig. 1d), enabling assessment of performance on entirely new data. Given the large disparity in size of the available test data, CDR3β-only and CDR3β + others models were assessed separately for seen- and unseen-epitope prediction. Models trained exclusively on individual epitopes were excluded from unseen-epitope evaluation to ensure fairness.
Second, to ensure standardized evaluation, we retrained 31 models with accessible code under consistent conditions (Fig. 1b and Extended Data Fig. 1e), aiming to assess the methodological superiority of different model designs. Both categories of models were evaluated on test data (internal datasets, same sources as training data) and independent data (external datasets, different sources from training data) for seen- and unseen-epitope prediction. Our evaluations primarily focused on CDR3β-only models due to their larger data availability and representation, with key evaluations including the impact of negative sample sources and other factors (Supplementary Note 1).
The evaluations from multiple angles, using area under the precision–recall curve (AUPRC) as the primary metric, complemented by other metrics, such as accuracy, precision and recall, ensured that our analysis provided a robust and unbiased assessment of TCR–epitope prediction models.
Building test sets to evaluate the original models
We evaluated 46 published original TCR–epitope prediction models (31 CDR3β-only, 15 CDR3β + others) (Supplementary Note 2). During preprocessing, non-canonical TCR sequences were adjusted by adding ‘C’ and ‘F’ residues to increase predictive data coverage18 (Extended Data Fig. 2a,b). For CDR3β-only models, separated test sets were constructed for seen (S_Data1: 978 TCRs across 3 epitopes; Fig. 2a) and unseen (U_Data1: 345 TCRs across 40 epitopes; Fig. 2b) epitope scenarios. For CDR3β + others models, test sets were also built for seen (S_Data2: 239 TCRs across 2 epitopes; Fig. 2c) and unseen (U_Data2: 67 TCRs across 14 epitopes; Fig. 2d) epitope prediction. To reduce bias from inconsistent negative TCR sampling (Supplementary Table 3), CDR3β-only models were tested with AS, PS and HS negatives, whereas only AS negatives were used for CDR3β + others models.

a–d, Distribution of TCR and epitope counts in seen-epitope (a,c) and unseen-epitope (b,d) for CDR3β-only datasets and CDR3β + others datasets used to assess originally trained models. e,f, Performance of CDR3β-only models on seen-epitope (e) and unseen-epitope (f) evaluation. g, Performance of CDR3β + others models on seen-epitope evaluation. h, AUPRC comparison between CDR3β-only (n = 31) and CDR3β + others (n = 15) models on seen-epitope CDR3β + others test data. i, Performance of CDR3β + others models on unseen-epitope evaluation. j, AUPRC comparison between CDR3β-only (n = 28) and CDR3β + others (n = 10) models on unseen-epitope CDR3β + others test data. Heatmaps (e–g,i) show epitope-level AUPRC, with adjacent bar charts showing overall AUPRC. Box plots (h,j) show mean (center line), the first and third quartiles (box) and minimum and maximum values within 1.5 × interquartile range (whiskers). All P values were from two-sided Wilcoxon rank-sum tests.
Performance of original models with CDR3β-only feature
For CDR3β-only models using AS negatives, ATM-TCR achieved the highest AUPRC (0.70) in the seen-epitope scenario (S_Data1), followed by the TEIM (0.68) and TEPCAM (0.67), whereas models like PiTE-epiSplit, TITAN and TCRfinder performed near random (AUPRC of ~0.5) (Fig. 2e). Among higher-scoring models, only ATM-TCR demonstrated a relatively good trade-off between precision and recall, with an F1 score of 0.57 (Extended Data Fig. 2c and Supplementary Table 4). Other models like TEIM showed notably low recall values around 0.2, indicating they missed many true TCR–epitope binding pairs, despite maintaining high precision and specificity under the fixed threshold of 0.5. Conversely, models like epiTCR and AttnTAP-vdj exhibited high recall (>0.8) but low precision (~0.5), reflecting a more aggressive strategy that increases positive predictions at the cost of misclassifying many non-binding pairs.
In the unseen-epitope scenario (U_Data1), overall performance decreased compared with the seen-epitope case (Fig. 2e,f). ImRex achieved the highest AUPRC value of just 0.55, followed by ATM-TCR and others at 0.52 (Fig. 2f). Notably, 13 out of 28 models (46.4%) exhibited AUPRC ≤ 0.5, suggesting that these models failed to effectively learn the underlying TCR–epitope binding pattern. For fixed-threshold metrics, ImRex maintained a relatively better specificity–recall trade-off. However, most models showed unbalanced performance, with occasional high values on individual metrics likely due to extreme predictions rather than consistent, generalizable performance (Extended Data Fig. 2d and Supplementary Table 4).
When using PS and HS negatives, the overall model rankings were similar to those obtained using AS negatives (with a correlation of 0.94 and 0.92 for PS and HS, respectively) in the seen-epitope scenario (Extended Data Fig. 2e–i). For instance, TEIM (AUPRC of 0.70 and 0.74 for PS and HS, respectively) and ATM-TCR (0.68 and 0.67 for PS and HS, respectively) remained the top-ranked performers (Extended Data Fig. 2e,f). Additionally, the averaged prediction performances of AS-, PS- and HS-based methods were consistent (Extended Data Fig. 2g), likely because most models were originally trained with the stringent AS-based method, leading to potentially in-depth learning of TCR–epitope binding features and more robust handling of different negative sources.
In the unseen-epitope scenario, similar to the results of AS-based strategy, using PS and HS negatives produced AUPRC near 0.5 for the majority of models (Extended Data Fig. 2j–l and Supplementary Table 4). This random-resemble performance diminished the interpretability of relative model rankings and showed low correlation in overall rankings across different negative types (Extended Data Fig. 2m,n), highlighting the weak generalization to new epitopes.
Model performance variability for both seen and unseen epitopes may arise from a combination of factors including model architecture, training data sizes and epitope-specific capabilities. We counted the total number of TCRs used by models and the number of TCRs that matched our three tested seen epitopes (Supplementary Tables 5 and 6) in the training data: although models trained with larger numbers of TCRs, such as ATM-TCR, tended to perform better, this does not fully account for all results. Standardized retraining and evaluation are essential to accurately assess intrinsic model performance.
Performance of original models with CDR3β and other features
In the seen-epitope scenario, CDR3β + others models overall underperformed compared to CDR3β-only models, likely due to limited multifeature data (Fig. 2a,c). vibtcr-AB demonstrated the top performance with AUPRC of just 0.59, followed by PISTE-reftcr (0.57) and TCRconv-large (0.55) (Fig. 2g). Some models showed relatively high precision (≥0.7) but poor specificity–recall balance (Extended Data Fig. 3a and Supplementary Table 4). When CDR3β-only models were applied to the multifeature dataset (S_Data2), results were similarly modest (AUPRC ranging from 0.45 to 0.57) (Extended Data Fig. 3b,c and Supplementary Table 4). In this context, CDR3β + others models generally performed better, although the improvement was not statistically significant (Fig. 2h). Notably, several models that were designed to accept both CDR3β-only and CDR3β + others features, such as vibtcr, benefited from incorporating additional features beyond CDR3β.
In the unseen-epitope scenario, model performance remained around 0.5, consistent with CDR3β-only models (Fig. 2f,i). ERGOII-vdj achieved the top performance with an AUPRC of only 0.58. Most models showed poor specificity–recall balance, often making extreme predictions (Extended Data Fig. 3d and Supplementary Table 4). When CDR3β-only models were again tested on the multifeature dataset (U_Data2) using only CDR3β input, models integrating additional features still showed modest gains (Fig. 2j, Extended Data Fig. 3e,f and Supplementary Table 4). For instance, epiTCR, ERGO-vdj and vibtcr exhibited improved predictive performance when incorporating additional features.
Overall, originally trained models obviously perform better on seen than unseen epitopes, especially among CDR3β-only models (Extended Data Fig. 3g), highlighting generalization challenges with unseen epitopes. It is important to note that the small size of multifeature tested data might influence the robustness of overall performance of models. Consistent training and reliable data are needed to better assess performance and influencing factors.
Building standardized datasets to retrain the models
To impartially evaluate TCR–epitope prediction models, we retrained 31 available models (24 CDR3β-only, 7 CDR3β + others) on integrated datasets (Fig. 3a and Supplementary Note 3). For CDR3β-only models, the dataset contained 600 epitopes and 98,846 binding TCRs (Extended Data Fig. 4a). Data were split under stratified sampling into (1) cross-validation training and intradatabase-sourced seen-epitope testing (389 epitopes, 94,361 TCRs; Extended Data Fig. 4b,c) sets, and (2) independent test sets for seen (80 epitopes, 2,941 TCRs) and unseen (211 epitopes, 1,581 TCRs; Extended Data Fig. 4d–g) epitopes. Similarly, the CDR3β + others dataset (249 epitopes, 5,294 TCRs; Extended Data Fig. 4h) was divided for cross-validation (57 epitopes, 4,292 TCRs; Extended Data Fig. 4i,j) and independent testing for seen (18 epitopes, 313 TCRs) and unseen (192 epitopes, 689 TCRs; Extended Data Fig. 4k–n) epitopes.

a, Epitope (and corresponding TCR) counts across antigen groups from different datasets. YFV, yellow fever virus; HBV, hepatitis B virus; HHV, human herpesvirus; HIV, human immunodeficiency virus. b, Performance of retrained CDR3β-only models on seen-epitope test. c, AUPRC comparison of models (n = 24) from b across AS/PS/HS negatives. d, Performance of retrained CDR3β-only models on seen-epitope independent test. e, AUPRC comparison of models (n = 24) from d across AS/PS/HS negatives. f, Performance of retrained CDR3β-only models in unseen-epitope test. g, AUPRC comparison of models (n = 21) from f across AS/PS/HS negatives. h,i, Performance on seen-epitope CDR3β + others test data of retrained CDR3β + others models (h) and CDR3β-only models (i). j, AUPRC comparison between CDR3β-only (n = 24, i) and CDR3β + others (n = 7, h) models. k,l, Performance on seen-epitope CDR3β + others independent test data of retrained CDR3β + others models (k) and CDR3β-only models (l). m, AUPRC comparison between CDR3β-only (n = 24, l) and CDR3β + others (n = 7, k) models. n,o, Performance on unseen-epitope CDR3β + others independent test data of retrained CDR3β + others models (n) and CDR3β-only models (o). p, AUPRC comparison between CDR3β-only (n = 21, o) and CDR3β + others (n = 3, n) models. Dot plots (b,d,h,i,k l) show per-antigen AUPRC, with adjacent heatmaps showing overall AUPRC, ordered by AS-based AUPRC. Heatmaps (f,n,o) show epitope-level AUPRC, categorized by antigen group and ordered by AS-based AUPRC. Colored dots (c,e,g) represent individual model AUPRC, black dots indicate the mean and error bars represent the mean ± s.d. Box plots (j,m,p) show mean (center line), first and third quartiles (box) and minimum and maximum values within 1.5 × interquartile range (whiskers). All P values were from two-sided Wilcoxon rank-sum tests.
Performance of retrained models with only CDR3β feature
In the seen-epitope scenario, models using AS negatives generally achieved lower AUPRC than those using PS or HS negatives (Fig. 3b,c). With AS-based strategy, epiTCR (0.83) and TEPCAM (0.82) achieved the highest AUPRC. Top-performing models exhibited relatively balanced AUPRC, precision, recall, specificity and F1 score, indicating their capability to distinguish positive and negative samples. In contrast, lower-ranked models often showed extreme predictions, such as high specificity with low recall (for example, TITAN and MCMC) or vice versa (for example, ERGO-lstm) (Extended Data Fig. 5a and Supplementary Table 7).
For the results from the independent test, which was considered a more stringent test compared to the initial test dataset, top-ranked models like epiTCR, TCRGP, TEIM, TCR-BERT and TEPCAM remained consistent with the initial test results but showed AUPRC declines of up to 0.23, along with similar drops in other metrics (Fig. 3d,e and Extended Data Fig. 5b). Taking TEPCAM with AS negatives as an example, AUPRC fell from 0.82 (test) to 0.59 (independent test) (Fig. 3b,d and Supplementary Table 7), indicating common challenges of overfitting or differences in intradata distribution.
In the unseen-epitope scenario, AS-based models like TCR-H, TEIM and NetTCR ranked higher, with relatively balanced metrics, but only achieved mean AUPRC of 0.52–0.53 (Fig. 3f and Extended Data Fig. 5c). Although some top-ranked models in the seen-epitope scenario, such as TEIM and ATM-TCR, also performed relatively better in unseen-epitope prediction, overall performance declined sharply (Fig. 3b,d,f). For instance, the AUPRC of epiTCR declined from 0.7 in the seen-epitope independent test (Fig. 3d) to 0.51 in unseen-epitope prediction (Fig. 3f). Models like TPBTE, MCMC and ERGO-lstm continued to display extreme one-class bias (Extended Data Fig. 5c and Supplementary Table 7), underscoring poor generalization to external datasets and unseen-epitope predictions.
Overall, across both test and independent datasets in seen- and unseen-epitope scenarios, models trained with PS or HS negatives consistently outperformed those using AS negatives, except for DLpTCR-series models (Fig. 3b–g). Models like vibtcr, ERGO-lstm, AttnTAP and TEINet showed unusually high gains in AUPRC with PS/HS negatives, suggesting potential model-specific sensitivities to negative data composition. Despite this overall advantage, it remains unclear whether PS or HS TCRs are superior as negative controls, as models were retrained and tested on matching negative source—such as HS-trained models being used exclusively on HS-test data—and their use may introduce confounding biases19,20.
Performance of retrained models with CDR3β and other features
When retraining models using the CDR3β + others dataset, only AS negatives were applied because PS and HS TCRs rarely contain additional information beyond CDR3β. In the seen-epitope scenario, three TCRconv models exhibited top-ranked AUPRC (0.76, 0.71 and 0.71) but suffered from low recall (≤0.44). Other CDR3β + others models showed balanced but poor performance across all metrics (Fig. 3h, Extended Data Fig. 5d and Supplementary Table 7).
To fairly assess the value of additional features, we also retrained CDR3β-only models using the multifeature dataset but relying solely on CDR3β input (Fig. 3i and Extended Data Fig. 5e). Their performance rankings remained highly consistent with those retrained on the standard CDR3β-only dataset (Fig. 3b), although the average performance was lower, likely due to the substantial difference in training data size (Extended Data Fig. 4b,i). Independent test results showed a similar trend (Fig. 3k,l), with top-ranked models performance consistent across CDR3β-only and CDR3β + others datasets (Fig. 3d,l, Extended Data Fig. 5f,g and Supplementary Table 7).
Overall, CDR3β + others models generally outperformed CDR3β-only models when retrained and tested under the same data conditions, although not significantly (Fig. 3j,m). Among four models supporting both CDR3β-only and CDR3β + others features (DeepTCR, NetTCR, TCRGP and vibtcr), two improved and one performed comparably with added features (Fig. 3j,m).
In the unseen-epitope scenario, only three CDR3β + others models were tested, with TCRconv and TCRGP excluded as they cannot predict unseen epitopes. All models performed close to random prediction (AUPRC around 0.5), with DeepTCR-ABVJ showing extreme class bias (Fig. 3n–p, Extended Data Fig. 5h,i and Supplementary Table 7). These results again highlight the need to develop specialized models to improve unseen-epitope prediction in real-world applications.
Source effects of negative TCR data on retrained models
To evaluate whether key factors, including data leakage and negative sample sources, affect TCR–epitope prediction, we focused on CDR3β-only models for their larger training data and broader representation. Using CD-HIT17 to remove similar TCR sequences and prevent data leakage, we found that AUPRC values for models trained on AS/PS negatives remained stable, whereas those using HS negatives decreased, suggesting that HS-based sampling may introduce confounders and overfitting, whereas AS/PS negatives offer more robust predictions in the context of TCR sequence similarity (Extended Data Fig. 6a–c and Supplementary Note 4).
When models retrained on PS or HS negatives were evaluated on rigorous AS-based test and independent sets, performance dropped significantly in both seen- and unseen-epitope scenarios, despite the models performing well on their own internal testing (Fig. 4a,b, Extended Data Fig. 6d and Supplementary Table 8). Specifically, PS–AS and HS–AS training–test pairs exhibited substantially lower performance compared to PS–PS and HS–HS strategies in both test and independent sets, suggesting that the models trained with external HS/PS negatives may learn dataset-specific artifacts rather than true binding patterns. Interestingly, whereas PS–AS and HS–AS testing performed worse than AS–AS on internal test sets, their performance aligned closely with AS–AS on external independent data. This indicates that although AS-based training is a stringent approach, it may still be preferentially influenced by internal dataset-specific structures. Conversely, when AS-trained models were evaluated on PS or HS test sets, performance declined in the seen-epitope scenario. However, on independent test sets for both seen and unseen epitopes, their performance remained consistent with that of AS–AS testing (Fig. 4c,d, Extended Data Fig. 6e and Supplementary Table 8).

a,b, Performance of PS- and HS-based retrained models evaluated on AS-based seen-epitope (a, n = 24) and unseen-epitope (b, n = 21) test data using CDR3β-only features. c,d, Performance of AS-based retrained models on PS- and HS-based seen-epitope (c, n = 24) and unseen-epitope (d, n = 21) test data with CDR3β-only features. Lines connect the same models across evaluation settings. All P values were from two-sided Wilcoxon signed-rank tests with Benjamini–Hochberg correction.
These findings highlight that using external PS or HS negatives may artificially inflate internal validation performance by leveraging systematic biases. In contrast, the AS-based reshuffling strategy—aligned with immunological context—enables more reliable learning of biologically meaningful TCR–epitope binding patterns. Despite its advantages, AS-based training still benefits from independently sourced test sets to ensure objective assessment.
Cross and low-prevalence effects of TCRs on retrained models
In generating negative samples using the AS-based approach, cross-reactive TCRs—those that bind multiple epitopes—are likely to introduce false negatives (FNs). In our dataset, about 10.5% of positive TCRs were cross-reactive (Extended Data Fig. 7a). Although these TCRs were excluded by default to reduce noise, we reintroduced them into all data splits to evaluate their effect on model performance.
We first compared models retrained and evaluated with and without cross-reactive TCRs using the AS negatives. Overall, including cross-reactive TCRs did not significantly alter model performance on both test and independent test sets (Extended Data Fig. 7b,c and Supplementary Table 8). Additionally, we evaluated a traditional random reshuffling method (defined as AS-Rand), which is commonly used in model training, as a control, confirming minimal performance differences between models trained with or without cross-reactive TCRs (Extended Data Fig. 7d). These results suggest that with a relatively low cross-reactivity rate, which may introduce FNs, model predictability for both seen and unseen peptides remains stable.
We further specifically compared the model performance between AS and AS-Rand methods, using training data that included cross-reactive TCRs (Extended Data Fig. 7e,f). The AS method outperformed AS-Rand in seen-epitope scenarios when test data originated from the same databases as training data, highlighting that the AS method improves model performance and mitigates the potential risk of FNs caused by cross-reactivity. However, for external independent test data, performance differences between AS and AS-Rand groups were negligible in both seen and unseen-epitope scenarios. These findings indicate that the AS-based method could mitigate some issues related to cross-reactivity within internal datasets. Nonetheless, it does not substantially enhance the model’s generalization for external data compared to its ability to learn from internal data.
To evaluate TCR–epitope binding prediction under realistic low-prevalence conditions (as low as 0.1%), we systematically tested multiple models using downsampled datasets. In both seen and unseen-epitope scenarios, nearly all models exhibited a sharp decline in precision as prevalence decreased (Extended Data Fig. 8a–c and Supplementary Note 5). These results indicate that despite balanced training, current models perform poorly in real-world scenarios with rare bindings, highlighting a critical limitation in their practical applicability.
Performance of retrained models under different sample sizes
To explore the effect of sample size on model performance, we constructed multiple subsets of training and test sets with varying numbers of TCRs per epitope. Results reveal that the average AUPRC of all models declines as the number of TCRs per epitope decreases during training, with 15 out of 24 models exhibiting a general decline in AUPRC with fewer training TCRs (Fig. 5a,b and Supplementary Table 9), highlighting the importance of sufficient data availability for improving predictive performance.

a, Performance of models trained with different TCR counts per epitope in seen-epitope prediction. Dot plots show per-antigen AUPRC, with adjacent heatmaps showing overall AUPRC. b, AUPRC comparison of models (n = 24) from a across TCR count groups. Colored dots represent individual model AUPRC, black dots indicate the mean and error bars represent the mean ± s.d. c, Correlation between epitope-associated TCR counts and AUPRC for the top three models from a of the >300 group. Dots represent epitopes, colored by antigen group. P values were from two-sided t-tests (n = 53). d, Performance saturation analysis on the top three models using five epitopes with the greatest TCR counts, showing per-epitope AUPRC and mean performance (red line).
Under the tests from the subset where TCR count per epitope exceeded 300, a generally positive correlation between the number of TCRs per epitope and model performance was observed for some well-performing models, such as epiTCR, TCRGP and TEPCAM (Fig. 5c and Supplementary Table 9). The results of other models were also positively correlated, with the exception of TPBTE, TITAN, DeepTCR, MCMC, DLpTCR-RESNET, DLpTCR-CNN and DLpTCR-FULL, which showed relatively poor performance in prediction (Supplementary Fig. 1). These findings indicate that in most cases, epitopes with a larger number of associated TCRs may help enhance model performance. However, certain models were still capable of achieving high AUPRC on epitopes with relatively few TCRs, indicating that sample size is not the sole determining factor. Although the number of TCRs appears to play a role, the task of predicting TCR–epitope binding likely depends on multiple factors, including the type of features used and model architecture. For instance, beyond sequence-based features, incorporating structural features of TCRs during training has been shown to improve prediction accuracy21.
Although increasing the number of training samples could enhance model performance, experimentally reliable TCR–epitope pairs are typically limited. We also compared multimer-based and in vitro stimulation-derived datasets. Using consensus predictions from top models and cross-validation on high-confidence external data, we found that in vitro stimulation data exhibited a relatively lower false-positive (FP) rate, but further experimental validation remains essential for conclusive reliability assessment (Supplementary Fig. 2a,b and Supplementary Note 6).
To further evaluate model predictive capability across different sample sizes for the same epitope, we retrained the top 10 models (identified in Fig. 3d) on datasets of varying TCR sizes for the five epitopes with the most TCRs, with hyperparameter tuning to ensure optimal performance (Supplementary Fig. 3). Using the top three models as examples, epiTCR, TCRGP and TEPCAM showed marked performance improvements as the number of TCRs increased, plateauing when the number of TCRs exceeded around 1,000 (Fig. 5d). Most models followed this trend, although PiTE showed continuous improvement (Extended Data Fig. 9a,b and Supplementary Table 9). This saturation may be attributed to the diminishing novel patterns available for model learning or the increasing TCR heterogeneity. Growth rate analysis confirmed substantial improvements when testing with fewer than 1,000 TCRs, with marginal gains beyond this point (Extended Data Fig. 9c). Across all five epitopes, well-performing models like epiTCR tended to maintain relatively high performance even when trained on smaller TCR datasets and consistently improved with additional TCRs (Extended Data Fig. 9b). Additionally, nearly all these top models consistently exhibited a negative correlation between prediction performance and TCR sequence dissimilarity (Extended Data Fig. 9d,e and Supplementary Note 7). Overall, our findings indicate that predictive performance generally improves with larger positive datasets and higher sequence similarity among TCRs targeting the same epitope.
Performance of retrained models with different positive-to-negative ratios
The number of TCRs with unknown epitopes far exceeds those with known bindings, implying a larger pool of potential negative samples compared to positive samples. Published studies vary in their use of positive-to-negative (P-to-N) ratios for model training. To explore how this factor impacts model performance, we retrained models with different P-to-N ratios.
In the seen-epitope test, most models showed improved performance as negative samples increased, with performance stabilizing at a P-to-N ratio of approximately 1:1 (Fig. 6a,b and Supplementary Table 10). Top-ranked models like epiTCR, TEPCAM, TEIM and TCR-BERT particularly benefited from this moderate increase in negative samples, indicating that balanced training enhances performance up to a certain point beyond which additional negative samples offer little further improvement, likely due to a lack of novel patterns for the models to learn. In contrast, ATM-TCR and TEINet showed declining performance at higher ratios (Fig. 6a), suggesting limited tolerance to large-scale class imbalance. Similar trends were observed on independent test sets, although overall performance was lower (Fig. 6c,d and Supplementary Table 10).

a, Performance of models in the seen-epitope test across P-to-N ratios. b, AUPRC comparison across ratios based on the results from a (n = 23). c, Performance of models in the seen-epitope independent test across different ratios. d, AUPRC comparison across different ratios based on the results from c (n = 23). e, Performance of models in the unseen-epitope independent test across different ratios. f, AUPRC comparison across ratios based on the results from e (n = 21). Colored dots (b,d,f) represent individual model AUPRC, black dots indicate the mean and error bars represent the mean ± s.d.
In unseen-epitope prediction, model performance was obviously reduced. Nevertheless, a slight increase in average AUPRC also occurred up to a P-to-N ratio of 1:1 (Fig. 6e,f and Supplementary Table 10). Only epiTCR showed noticeable improvement with the addition of negative samples before stabilizing, whereas other models were almost unaffected by P-to-N ratio changes. Overall, balancing positive and negative data (~1:1) optimizes performance for both seen and unseen epitopes, whereas excess negatives offer little gain in generalization and may harm performance or increase computational cost.
Comparison of computational efficiency of models
We evaluated time and memory usage across dataset sizes under uniform hardware. Although training time and memory increases were relatively small on smaller datasets, they rose sharply with scale. At 1 million samples, TCR-H, TCRconv and TCR-BERT required more than 50 hours for training, whereas epiTCR, DeepTCR and DLpTCR-FULL were the fastest. TITAN used the least memory, and VitTCR consumed the most memory (Extended Data Fig. 10a,b and Supplementary Table 10). TCRGP and TCRGP-AB failed at 100,000 samples due to memory overflow. During testing, runtime and memory usage were generally lower than during training. DeepTCR, DeepTCR-ABVJ, NetTCR and epiTCR had relatively short testing durations, whereas TCR-H, TCRconv and TCR-BERT required considerably longer. TCRGP and TCR-BERT exhibited unstable memory usage, whereas AttnTAP and TEINet were memory efficient (Extended Data Fig. 10c,d and Supplementary Table 10). This assessment offers practical insights for researchers selecting models for large-scale TCR–epitope prediction tasks.
Discussion
In this study, we conducted a comprehensive benchmarking of TCR–epitope prediction models, systematically evaluating their performance in both seen- and unseen-epitope scenarios. Beyond comparing originally trained models, we established a unified retraining and evaluation framework with standardized datasets to ensure fair and reproducible comparisons. In addition, our analysis extends beyond model architectures to explore the influence of several biological and methodological factors—including the integration of MHC class and paired αβ TCR chains, negative sampling strategies, cross-reactivity, low prevalence of true binders, potential FPs of different experiment methods and data imbalance.
Our results indicate that several models perform relatively well in predicting seen epitopes. Recent studies13,14,22,23 identified IMW DETECT14 (code not available), MixTCRpred24 and NetTCR25 as effective models for seen-epitope prediction. Consistently, both MixTCRpred and NetTCR ranked among the top 10 performing models in our assessment. However, when faced with unseen epitopes, even the top-performing models exhibit a dramatic decline in performance, often approaching levels akin to random guessing. This observation is consistent with prior studies such as IMMREP2213, IMMREP2314 and ref. 12 and highlights a fundamental limitation of current modeling strategies.
Our analysis reaffirmed earlier observations from IMMREP23 regarding the overestimation of model performance when using intradataset test sets. We found that performance on independent test sets was consistently lower across almost all models, underscoring the critical need for rigorous external validation and raising concerns about model generalizability in real-world applications. Another crucial finding was the benefit of incorporating additional biological features. Models that included MHC class and αβ TCR information generally outperformed those trained on CDR3β sequences alone, consistent with IMMREP2213.
A key focus of our analysis was the impact of negative control sampling strategies. In retrained models, we compared AS, PS and HS negatives and found that incorporating external PS or HS TCRs would introduce batch-like confounders, causing models to learn dataset-specific artifacts rather than true TCR–epitope binding signals. This finding aligns with previous studies12,15. Regarding data leakage, IMMREP23 employed a Levenshtein-distance-based strategy to avoid FNs during reshuffling, which would result in target leakage during random reshuffling due to repeated TCR reuse. In contrast, we applied a refined AS strategy that could minimize FNs and prevent repeated sampling of cross-matched TCRs, thereby reducing bias and enabling models to learn more robust and biologically realistic binding patterns.
Cross-reactivity remains a challenging issue in TCR–epitope modeling. Although some studies suggest that random reshuffling for negative sampling may introduce FNs from cross-reactive TCRs, potentially biasing model learning26, our evaluation comparing models with and without cross-reactivity revealed minimal impact on performance. Furthermore, implementing the proposed refined AS reshuffling strategy would mitigate this concern, allowing the inclusion of cross-reactive TCRs without significantly degrading model performance.
Although we designed this analysis from multiple aspects, it still has several limitations. (1) Input sequence length restrictions imposed by many models reduced the number of usable TCR–epitope pairs. This is particularly problematic for models trained with CDR3β + others features in unseen scenarios, where limited available test data might introduce performance fluctuations. (2) Current models predominantly focus on CDR3β-only feature because most available data provide only CDR3β information. This restricts the full performance potential of models incorporating CDR3β + others features due to limited data availability for retraining. (3) Although we applied a refined AS-based TCR reshuffling approach to increase the likelihood of true negatives (TNs), this method does not guarantee that they are ground-truth non-binders. (4) To ensure sufficient data for evaluation, we used high-confidence pairs when scores were available and included all pairs from datasets without such scores. Although we computationally estimated FP rates across antigen identification methods, experimental confirmation is still required. (5) This study primarily focused on supervised sequence-based models, as a majority of developed tools adopt this strategy. Unsupervised models, such as TULIP27 and TCRdock28, which do not consider negative samples, and models like TCRen29, which require experimentally resolved TCR-pMHC structures, were not included.
To advance the field, future efforts would prioritize several key areas. (1) Expanding high confidential TCR–epitope data is crucial to minimize FPs. Beyond experimentally generating reliable unpaired TCR–epitope data, incorporating MHC class, antigen specificity and other biologically relevant information may help construct credible non-binding datasets. (2) Our analysis indicates that incorporating multiple features generally improves model performance. Cross-modal learning frameworks that combine sequence, structural and contextual information represent a promising direction for more effective model development. (3) Our findings highlight the limited performance of current models on novel epitopes, underscoring the need for innovative architectures capable of capturing broader binding patterns. In parallel, curating training datasets with extensive diversity in both TCRs and epitopes is essential to support real-world applicability. (4) Accurate assessment of model generalization requires the use of independent external test sets, rather than relying only on internal training data-derived test sets. This approach ensures a more realistic performance evaluation.
In summary, our benchmarking study not only compares the performance of current models but also analyzes the methodological choices that most impact predictive success. It would serve as a valuable guide for model developers and end users, offering a foundation upon which more robust, interpretable and generalizable models can be developed to accelerate immunological research and applications.
Methods
Workflow of model evaluation
Our evaluation involves collecting and preprocessing data from various sources, preparing models (both originally trained and retrained) and conducting testing and independent testing with external datasets (Fig. 1b). The assessment process considers several factors: the impact of different negative TCR sources (AS, PS and HS), the impact of cross-reactivity, the influence of training data size (number of samples, P-to-N sample ratios, dataset size for model saturation and the correlation between epitope-associated TCR numbers and model performance) and the effects of epitope type (seen versus unseen epitopes) on model predictions.
Data collection of TCRs and epitopes for model evaluation
To ensure a robust and comprehensive evaluation of TCR–epitope binding prediction models, we systematically gathered data from a total of 21 authoritative databases and scholarly articles14,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49. Detailed information on these data sources is provided in Supplementary Table 1 and Supplementary Note 8. These databases and studies collectively provide a comprehensive set of TCR–epitope bindings, ensuring a robust data foundation for the objective and accurate evaluation of TCR–epitope prediction models.
Model collection for TCR–epitope binding prediction
This study comprehensively collected 54 original and derived TCR–epitope binding prediction models published before October 2024 (Supplementary Table 2). Of these, 50 models12,21,23,24,25,43,47,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73 were evaluated, and the remaining 4 were excluded due to data requirements or lack of open-source implementation. These models encompass a wide range of methodologies to ensure a holistic evaluation framework. The collected models exhibit the following characteristics: (1) they employ traditional machine-learning approaches or cutting-edge deep-learning techniques that leverage large datasets; (2) some models are designed to predict only seen epitopes, whereas others can handle both seen and unseen epitopes; (3) the models vary in their use of features for training. Some consider only the CDR3β feature, whereas others incorporate additional features such as MHC classes and both α and β TCR chains. The brief summary for each model included in our benchmark study is provided in Supplementary Note 9.
Preprocessing of TCR and epitope sequence data
Positive data obtained from 19 data sources (listed in Supplementary Table 1) were initially preprocessed separately for the original model testing task and the model retraining task. Given the limited availability of data for testing the original models, we retained all available data when constructing the test set. We noted that a great number of TCR sequences within the IEDB database deviated from established research findings, which indicate that the CDR3 region of TCRs typically begins with a conserved cysteine (‘C’) and ends with phenylalanine (‘F’). Upon aligning the sequence lengths to a uniform format, we observed that the first amino acid of these aberrant sequences matched the second position of the normal sequence, and the last amino acid aligned with the penultimate position. To rectify the format of the TCR sequences in the IEDB, we prefixed a ‘C’ and appended an ‘F’ to these aberrant sequences. In addition, TCR–epitope pairs belonging to the MHC-II class were excluded from original model testing because the majority of models were trained using only MHC-I-class data. For the retraining of models, given the sufficient volume of data available for both training and testing phases, we directly filtered out TCR sequences that did not start with ‘C’ and end with ‘F’. Both MHC-I and MHC-II class data were retained in retraining for assessing models comprehensively.
Subsequently, for both the dataset intended for model retraining and assessment as well as the test set used for the evaluation of the original models, we implemented the following sequence procedures:
-
(1)
Standard amino acid consideration: Because most feature-encoding methods consider only the standard 20 amino acids, we deleted sequences of TCRs or epitopes that contained special symbols, lowercase letters and uncommon amino acids to ensure the accuracy of feature encoding.
-
(2)
Sequence length criteria: Considering the consensus criteria of all collected models, for the original model testing, we retained epitopes with a length of 9 amino acids and TCR sequences ranging from 10 to 18 amino acids. However, in model retraining, we increased the length scale of epitopes to 8–15 amino acids to build a larger retraining dataset.
-
(3)
Binding confidence: We removed sequences with low TCR–epitope binding confidence. In the VDJdb database, sequences are assigned confidence scores ranging from 0 to 3 based on specificity and credibility. We excluded all TCR sequences with a confidence score of 0 to maintain high-quality data. From the dbPepNeo2.0 database, only high-confidence neoantigen entries validated by specific TCR recognition assays were retained. In the case of the MIRA database, we included only statistically inferred high-confidence TCR–epitope pairs with a posterior probability greater than 0.9 of being associated with a specific query antigen.
-
(4)
Unique TCR–epitope pairs: The raw data contain a large proportion of TCRs that do not bind to unique epitopes, a phenomenon referred to as cross-reactivity. Although genuine cross-reactivity does exist biologically, in certain experimental contexts such patterns may arise from technical limitations or annotation errors, potentially introducing FPs. Specifically, in the MIRA dataset, cross-reactive TCRs account for up to 66% of the entries within the high-confident annotated subset. This method likely overestimates the actual degree of cross-reactivity, as it may be influenced by methodological limitations rather than genuine TCR–epitope recognition. To minimize redundancy, reduce noise and ensure the uniqueness of TCR–epitope interactions in our benchmark, we excluded entries in which a single TCR was linked to more than one epitope.
-
(5)
Feature categories for TCR–epitope pairs: We considered the following two scenarios to filter data according to feature availability: (1) the CDR3 sequence of the TCR β-chain is provided and (2) additional features beyond the CDR3β sequence are available, including CDR3α, MHC type and V(D)J genes. Thus, we generate two datasets (‘CDR3β-only’ and ‘CDR3β + others’ datasets) for both original model testing and retraining model assessment.
For negative sequence data, we applied the TCR filtering conditions mentioned above to ensure consistency across all data. This approach ensures that the datasets used for training and testing are of high quality and consistency, thereby enhancing the reliability of the subsequent model evaluations.
Generation of negative data
We evaluated the models using three different sources of negative data: AS, PS and HS TCRs. Regarding the size of negative data, for our default setting, we maintained a 1:1 ratio between the positive and negative datasets. This balanced ratio was used unless we were specifically investigating the effects of varying the P-to-N ratio.
The approach of AS TCRs (set as the default) is a commonly used and stringent method to construct negative data by randomly reshuffling positive TCR–epitope pairs, but it would have introduced FNs caused by probable cross-reactivity. To mitigate this effect, we employed a refined approach under immunologically relevant categories, which considers the cross-matching of MHC classes, MHC alleles and antigen groups rather than relying solely on random shuffling. This approach is based on the following assumptions: (1) the probability of cross-reactivity between different MHC alleles is lower than within the same allele, (2) MHC-II restricted TCRs have a lower likelihood of binding to MHC-I restricted peptides and (3) the probability of TCR binding to epitopes within one type of antigen is greater than for other types of antigens.
Given that the number of MHC-I restricted TCR–epitope pairs is substantially larger than those restricted by MHC-II, and there is dominance of certain MHC alleles (for example, HLA-A*02:01) and antigens (for example, SARS-CoV-2) of positive TCR–epitope pairs compared with alleles and antigens, it is impractical to rely exclusively on MHC class, MHC allele or antigen information to construct the entire negative dataset. Therefore, we adopted a stepwise cross-matching method. Specifically, for both seen-epitope and unseen-epitope scenarios, we first created negative pairs using cross-matched MHC information when both MHC-I and MHC-II classes were present. In this process, MHC-II restricted TCRs served as negative controls for MHC-I restricted positive TCR–epitope pairs and vice versa. If any MHC-I data remained, we then employed MHC-I restricted TCRs specific to different alleles as negative controls. For any remaining MHC-I data with the same allele information, we created negative pairs between different antigen types. Finally, if there were remaining data that could not be cross-matched, we resorted to random reshuffling.
To generate HS and PS negative data, we obtained TCR sequences from two sources: the Dean-2015 dataset for healthy individuals and the TCRdb database for patients. When generating negative samples from HS TCRs, we excluded CMV-positive samples to avoid FNs. For PS TCRs, we focused on clonally expanded TCRs, which have a high probability of being disease-associated. For both seen-epitope and unseen-epitope scenarios, we generated negative samples by randomly sampling TCRs from either healthy or patient individuals, ensuring the sampling size matched the number of TCRs in the positive dataset. These sampled TCRs were then combined with the preprocessed epitopes to create a set of negative data.
Construction of consensus test sets for original model evaluation
To construct test sets for evaluating the original models, we followed a systematic process. We first merged the 19 preprocessed positive datasets and removed any duplicate data. For the seen-epitope scenario, we retained only the epitopes commonly used by all models and deleted the TCR sequences corresponding to these epitopes that had already been used in model training. The remaining TCR–epitope pairings were used as positive samples for the seen-epitope test set. For the unseen-epitope scenario, we removed all epitopes and TCRs used by the original models. The remaining TCR–epitope pairings formed the positive samples for the unseen-epitope test set. Then, negative samples were generated using the above-described negative data generation method for three types of negative data sources (AS, PS and HS).
In the original publications of the epiTCR, epiTCR-BH and NetTCR models, the cysteine (‘C’) and phenylalanine (‘F’) amino acids at the beginning and end of TCR sequences were removed during training. To ensure consistency between the test data and the training data for these models, we also artificially removed these amino acids when using these models for prediction.
By following these steps, we ensured that the test sets accurately reflected the requirements for evaluating the original models in both seen-epitope and unseen-epitope scenarios.
To prevent data leakage, we used CD-HIT17 to exclude highly similar sequences (>95% similarity) between the training and test sets. Specifically, after integrating the positive samples with the generated negative samples for each data group, CD-HIT was applied to eliminate these highly similar TCR sequences, ensuring robust and unbiased evaluation of the models.
Construction of training, test and independent sets for model retraining
To construct training, test and independent sets for model retraining, we followed a systematic approach. Initially, we removed all duplicate TCR–epitope pairings derived from 19 data sources (Supplementary Table 1). Positive samples for the seen-epitope and unseen-epitope independent test sets were sourced from IMMREP23, McPAS-TCR and VDJdb, and positive samples from the remaining 16 databases were used for model retraining and testing.
To guarantee complete separation between the independent sets and the training/test sets, we excluded any samples from the training and test data sources that overlapped with those in the independent data sources (IMMREP23, McPAS-TCR and VDJdb). For the unseen independent set, we retrained only the epitopes that did not appear in the training sets.
Subsequently, we employed a 5-fold cross-validation strategy to generate five groups of training and test sets. A stratified sampling method was applied to ensure uniform distribution of epitopes across each fold. For the seen-epitope scenario, we further filtered the candidate training and test samples by retaining only positive samples with five or more TCRs corresponding to an epitope. For each set of positive samples, we matched the epitopes with TCRs from three data sources (AS, PS and HS) to create negative samples.
In model retraining, we also used CD-HIT to exclude TCR sequences with greater than 95% similarity between the training and test sets, and between the training set and the independent test sets. This procedure ensured the removal of highly similar sequences, thereby enhancing the robustness and fairness of retraining model evaluation.
Evaluation of the impact of cross-reactivity on model performance
Cross-reactivity poses a challenge in analyzing TCR–epitope binding data. When negative data are generated using the AS-based reshuffling approach, cross-reactive TCRs could result in FNs. To systematically assess the impact of cross-reactivity on model performance, we conducted an analysis by incorporating cross-reactive data into our model evaluation framework, which initially excluded cross-reactive TCRs. We identified 11,667 cross-reactive TCR–epitope entries (cross-reactive data from MIRA dataset were not included due to an unusually high ratio of cross-reactive TCRs). After applying CD-HIT to eliminate sequences with high similarity in both test and independent test sets, 11,083 unique cross-reactive entries were added in this evaluation. Specifically, 9,104 of these entries were evenly assigned across training and test sets within a 5-fold cross-validation scheme. Additionally, 971 cross-reactive samples were included in the seen-epitope independent test set, and 1,008 were included in the unseen-epitope independent test set.
The performance of models trained both with and without cross-reactive TCRs was then evaluated by predicting the test and independent test datasets comprising both cross-reactive and non-cross-reactive entries. This comparison provides insights into the extent to which cross-reactivity influences predictive accuracy and model generalizability.
Evaluation of AS TCR identification methods on retrained models
The quality of AS TCRs directly impacts the reliability of TCR–epitope binding prediction models. In this study, we leveraged a large dataset of TCRs to evaluate model performance and implement various data filtering strategies to ensure data quality. However, challenges arising from the AS TCR identification methods themselves cannot be fully addressed through preprocessing alone.
To investigate the quality of data derived from different AS TCR identification methods, we examined the annotation information across our datasets. We found that the majority of samples lacked explicit labeling of experimental methods, whereas the clearly annotated entries primarily fell into two categories of well-established methods: (1) multimer-related assays and (2) in vitro stimulation-related assays. Accordingly, we focused our comparative analysis on these two classical methods.
We conducted two key analyses. First, we applied the 8 retrained models—selected from the top 10 performers in our benchmark (Fig. 3d) and capable of predicting unseen epitopes—to predict samples from each group and estimate their FP rates. This analysis assumes that the top-performing models have adequate discriminative power and that consensus predictions across multiple models can act as an indirect measure of data quality.
Second, we trained models independently on datasets generated by each method and evaluated their performance on the same high-confidence test sets. This approach assumes that model performance reflects the reliability of the training data. To ensure a fair comparison, we standardized the training set size by aligning it with the method that yielded fewer samples: in vitro stimulation. Specifically, both training sets were limited to 1,409 TCRs, matching the sample size of the in vitro stimulation group. A shared high-confidence test set containing 274 TCRs was used for evaluation. To mitigate the effects of random sampling and ensure robust comparison, we downsampled the positive samples from the multimer datasets and repeated the model training and evaluation process 10 times.
Evaluation of the size effects of TCR–epitope pairs in model retraining
To examine the impact of TCR numbers on model performance, we created several groups of training and test sets by varying the number of TCRs associated with each epitope. This process was based on the five standardized training and test splits used for model retraining when using AS TCRs as a negative data source. For epitopes with more than 300 associated TCRs, we retained all TCR–epitope pairings where the TCR count exceeded 300 in both the five training sets and the corresponding five test sets. Subsequently, for specific TCR count thresholds of 300, 200, 100 and 10, we constructed training sets by selecting TCR–epitope pairings in which the TCR count per epitope equaled exactly 300, 200, 100 or 10 within the five training splits. For all these training configurations, the same five test sets—originally generated for the group with TCR counts exceeding 300—were consistently used for evaluation, ensuring comparability across different TCR count settings.
To test the required TCR number for different models to reach optimal performance, we extracted epitopes whose TCR numbers ranked among the top five from all databases and validated how TCR number impacted model performance with samples grouped by epitope. For the training data of each epitope, we created multiple training sets with 16 different TCR sizes, ranging from 50 to 3,000, with each size repeated five times. For test data, we randomly extracted 500 binding TCRs for each epitope to construct positive samples and repeated five times. To ensure balanced datasets, an equal number of negative samples were generated for each training or test set using the refined AS-based negative-data-creation strategy. A separate dataset was constructed for each epitope, where negative samples were created by pairing the given epitopes with TCRs not included in the corresponding positive set. Thus, for each epitope, we obtained five training sets and test sets by combining positive and negative samples. The top 10 models identified in Fig. 3d, which previously demonstrated strong generalization to the seen-epitope independent test set, were retrained for this evaluation.
To assess whether TCR sequence heterogeneity within the same epitope affected model performance, we used the data from one of the five training–test splits generated through 5-fold cross-validation during model retraining, corresponding to the results shown in Fig. 3b. For each epitope, we calculated the pairwise Levenshtein distances among all associated TCR CDR3β sequences and used the average distance as a measure of TCR heterogeneity. We then computed the Pearson correlation coefficient between TCR heterogeneity and model performance (measured by AUPRC) for each model across epitopes. To evaluate statistical differences in correlation strength between models, we performed pairwise comparisons using Fisher’s r-to-z transformation and calculated the corresponding P values. To account for multiple comparisons and reduce the likelihood of FP findings, we applied the Benjamini–Hochberg correction to the resulting P values. The same top 10 models identified in Fig. 3d were also used in this analysis.
Evaluation of the effects of P-to-N ratios in model retraining
When exploring the model’s performance under varying degrees of data imbalance, we constructed seven groups of training sets with P-to-N sample ratios of 9:1, 6:1, 3:1, 1:1, 1:3, 1:6 and 1:9 based on the positive samples used for model retraining. This evaluation used AS TCRs as the negative data source. It is worth noting that the TCRGP model could not be trained at the 1:3 ratio due to excessive data volume, and thus its results are not included in this part.
To generate the most imbalanced dataset (1:9 P-to-N ratio), we employed the refined AS-based reshuffling strategy with repetition applied seven times, creating the maximum possible number of synthetic TCR–epitope pairs based on the available positive samples. Only epitopes with a sufficient number of corresponding negative matches were retained.
This 1:9 dataset was then used to generate five training–test splits via 5-fold cross-validation, employing stratified sampling to ensure an even distribution of epitopes across all folds. The five training–test splits under other P-to-N ratios were derived by downsampling the negative samples accordingly while keeping the positive samples consistent across all datasets.
To ensure a fair comparison of prediction performance across different P-to-N ratios, we used the test sets from the 1:1 ratio configuration for evaluation in all cases. Finally, the models shown in Fig. 3b were retrained using each dataset to assess the impact of different P-to-N sample ratios.
To evaluate generalizability, we built seen- and unseen-epitope independent test sets (1:1 P-to-N ratio) using IMMREP23, McPAS-TCR and VDJdb. The seen-epitope set shared epitopes with training data, and unseen-epitope sets contained the remaining epitopes.
Evaluation of time and resource consumption in model training and testing
To evaluate the computational demands of various models, we created datasets with 1,000, 5,000, 10,000, 100,000 and 1,000,000 samples by randomly selecting TCRs and epitopes. Each dataset was used for both training and testing to record runtime and memory usage. For each run, we allocated the same amount of memory and number of CPU cores, and deep-learning models were executed on a GPU with uniform settings. All experiments were performed on a computing server with the following hardware configuration: Intel Xeon Gold 6342 CPU (2.8 GHz, 48 cores) with 1,024 GB of RAM and NVIDIA A100-PCIE GPU with 80 GB of VRAM.
Model preparation and tuning
The original models utilized in the evaluation were primarily the versions released on GitHub. Models that were not available were trained using the original training dataset and default settings as specified in the respective articles (Supplementary Table 2). In the unseen-epitope prediction scenario, we excluded several models and their variants (if available)—TCRGP, TCR-BERT, SETE, MixTCRpred, DeepTCR, TCRconv and TCR-H—for the following reasons: DeepTCR was originally trained on non-human data; TCR-H did not provide access to its exact training data or pretrained model; and the remaining models generate separate models for each epitope, making them unsuitable for predicting unseen epitopes. In comparing the impact of different P-to-N ratios of samples on model performance, we excluded TCRGP because it failed to run properly when the ratio reached 1:3 due to the limitations of TensorFlow, which cannot handle tensors larger than 2 GB.
During the retraining process, we examined the effects of tuning key hyperparameters for models. However, the observed performance differences were minimal, and in most cases, the default or recommended settings yielded comparable or superior results. Therefore, we adopted the default configurations or those suggested in the original publications for consistency and reproducibility. When evaluating the impact of data size on model performance, the number of epochs was a factor influencing the convergence of deep-learning models. We tested model performance under five different epoch settings and used the best results for comparison.
Metrics for model evaluation
When evaluating model performance, a large portion of the outputs represent the binding probability or binding affinity between TCRs and epitopes, which does not clearly indicate whether binding will occur. Most models consider a binding likelihood greater than 0.5 as a positive prediction. However, the binding relationship between TCRs and epitopes is complex, making it challenging to establish a precise binding threshold.
In classification models, predictions fall into four categories: true positives (TPs), where the model correctly predicts positive samples; FPs, where negative samples are incorrectly predicted as positive; TNs, where negative samples are correctly identified; and FNs, where positive samples are incorrectly predicted as negative.
In our evaluation, the primary metric we adopted was AUPRC, which quantifies the trade-off between precision and recall across all possible classification thresholds. AUPRC is widely recognized as a robust evaluation metric for imbalanced classification tasks, as it reflects a model’s ability to rank TPs—such as high-affinity TCR–epitope pairs—above FPs. We calculated AUPRC using the precrec package, as recommended in the literature74.
In addition, we evaluated the models using a comprehensive set of performance metrics including area under the receiver operating characteristic curve for all models. Other metrics, including accuracy, precision, recall, specificity, Matthews correlation coefficient (MCC) and F1 score, discussed in specific sections, offer threshold-specific insights that are intuitive for fixed thresholds (with 0.5 set as the default to distinguish true from false). These additional metrics offer targeted evaluations but may be influenced by the chosen threshold.
Accuracy measures the overall correctness of classifications, defined as
Recall assesses models’ sensitivity in identifying TPs from actual positives, defined as
Precision evaluates the proportion of TP predictions among all positive predictions, defined as
Specificity quantifies models’ ability to correctly identify negative instances, defined as
MCC provides a balanced assessment of model performance, taking into account both true and false positives and negatives, defined as
Finally, the F1 score offers a harmonic mean of precision and recall, reflecting a balance between these two metrics, defined as
These metrics collectively provide a robust framework for evaluating the effectiveness and reliability of the models across various aspects of their performance. For models like MixTCRpred and pMTnet, which generate relative binding affinity scores rather than probability thresholds or binary classifications, only AUPRC is calculated because other metrics requiring fixed cutoffs are not applicable. The detailed results for each metric are presented in the Supplementary Tables.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data were obtained from publicly accessible databases and scholarly articles, including VDJdb30, McPAS-TCR31, IEDB32, TBAdb33, dbPepNeo2.034, MIRA35, Glanville-201736, Tsuruta-201837, Luo-201838, TetTCR-201839, Huth-201940, TetTCRHD-202141, Francis-202242, pMTnet-202143, Ishigaki-202244, Minervina-202245, Mudd-202246, PISTE-202447, IMMREP2314, TCRdb2.048 and Dean-201549, with web links provided in Supplementary Table 1. The processed data employed to generate the results are available via figshare at https://doi.org/10.6084/m9.figshare.27020455 (ref. 75). Source data are provided with this paper.
Code availability
The source codes of the TCR–epitope binding prediction models evaluated in this paper are publicly available via GitHub at https://github.com/SuoLab-GZLab/TCREpitopeBenchmark.
References
Pishesha, N., Harmand, T. J. & Ploegh, H. L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 22, 751–764 (2022).
Kearse, K. P., Roberts, J. P., Wiest, D. L. & Singer, A. Developmental regulation of alpha beta T cell antigen receptor assembly in immature CD4+CD8+ thymocytes. Bioessays 17, 1049–1054 (1995).
Nikolich-Zugich, J., Slifka, M. K. & Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 4, 123–132 (2004).
Garcia, K. C. & Adams, E. J. How the T cell receptor sees antigen–a structural view. Cell 122, 333–336 (2005).
La Gruta, N. L., Gras, S., Daley, S. R., Thomas, P. G. & Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 18, 467–478 (2018).
Altman, J. D. et al. Phenotypic analysis of antigen-specific T lymphocytes. Science 274, 94–96 (1996).
Huang, H. et al. Select sequencing of clonally expanded CD8(+) T cells reveals limits to clonal expansion. Proc. Natl Acad. Sci. USA 116, 8995–9001 (2019).
Jamieson, A. G., Boutard, N., Sabatino, D. & Lubell, W. D. Peptide scanning for studying structure-activity relationships in drug discovery. Chem. Biol. Drug Des. 81, 148–165 (2013).
Patton, K. et al. Enzyme-linked immunospot assay for detection of human respiratory syncytial virus f protein-specific gamma interferon-producing T cells. Clin. Vaccin. Immunol. 21, 628–635 (2014).
Hudson, D., Fernandes, R. A., Basham, M., Ogg, G. & Koohy, H. Can we predict T cell specificity with digital biology and machine learning?. Nat. Rev. Immunol. 23, 511–521 (2023).
Teraguchi, S. et al. Methods for sequence and structural analysis of B and T cell receptor repertoires. Comput. Struct. Biotechnol. J. 18, 2000–2011 (2020).
Moris, P. et al. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief. Bioinform. 22, 1–12 (2021).
Meysman, P. et al. Benchmarking solutions to the T-cell receptor epitope prediction problem: IMMREP22 workshop report. ImmunoInformatics 9, 100024 (2023).
Nielsen, M. et al. Lessons learned from the IMMREP23 TCR–epitope prediction challenge. ImmunoInformatics 16, 100045 (2024).
Grazioli, F. et al. On TCR binding predictors failing to generalize to unseen peptides. Front. Immunol. 13, 1014256 (2022).
Deng, L. et al. Performance comparison of TCR-pMHC prediction tools reveals a strong data dependency. Front. Immunol. 14, 1128326 (2023).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Lefranc, M. P. et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 27, 55–77 (2003).
Dens, C., Laukens, K., Bittremieux, W. & Meysman, P. The pitfalls of negative data bias for the T-cell epitope specificity challenge. Nat. Mach. Intell. 5, 1060–1062 (2023).
Culka, M. et al. Predicting specificity of TCR-pMHC interactions using machine learning and biophysical models. Preprint at bioRxiv https://doi.org/10.1101/2025.04.04.647165(2025).
Zhao, Y. et al. DeepAIR: a deep learning framework for effective integration of sequence and 3D structure to enable adaptive immune receptor analysis. Sci. Adv. 9, eabo5128 (2023).
Salles, R., Pacitti, E., Bezerra, E., Porto, F. & Ogasawara, E. TSPred: a framework for nonstationary time series prediction. Neurocomputing 467, 197–202 (2022).
Chen, J. et al. TEPCAM: prediction of T-cell receptor-epitope binding specificity via interpretable deep learning. Protein Sci. 33, e4841 (2024).
Croce, G. et al. Deep learning predictions of TCR–epitope interactions reveal epitope-specific chains in dual alpha T cells. Nat. Commun. 15, 3211 (2024).
Montemurro, A. et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRalpha and beta sequence data. Commun. Biol. 4, 1060 (2021).
Gao, Y., Gao, Y., Dong, K., Wu, S. & Liu, Q. Reply to: The pitfalls of negative data bias for the T-cell epitope specificity challenge. Nat. Mach. Intell. 5, 1063–1065 (2023).
Meynard-Piganeau, B., Feinauer, C., Weigt, M., Walczak, A. M. & Mora, T. TULIP: a transformer-based unsupervised language model for interacting peptides and T cell receptors that generalizes to unseen epitopes. Proc. Natl Acad. Sci. USA 121, e2316401121 (2024).
Bradley, P. Structure-based prediction of T cell receptor:peptide-MHC interactions. eLife 12, e82813 (2023).
Karnaukhov, V. K. et al. Structure-based prediction of T cell receptor recognition of unseen epitopes using TCRen. Nat. Comput. Sci. 4, 510–521 (2024).
Shugay, M. et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res. 46, D419–D427 (2018).
Tickotsky, N., Sagiv, T., Prilusky, J., Shifrut, E. & Friedman, N. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929 (2017).
Vita, R. et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Zhang, W. et al. PIRD: pan immune repertoire database. Bioinformatics 36, 897–903 (2020).
Lu, M. et al. dbPepNeo2.0: a database for human tumor neoantigen peptides from mass spectrometry and TCR recognition. Front. Immunol. 13, 855976 (2022).
Nolan, S. et al. A large-scale database of T-cell receptor beta sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Front. Immunol. 16, 1488851 (2025).
Glanville, J. et al. Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017).
Tsuruta, M. et al. Bladder cancer-associated cancer-testis antigen-derived long peptides encompassing both CTL and promiscuous HLA class II-restricted Th cell epitopes induced CD4( + ) T cells expressing converged T-cell receptor genes in vitro. Oncoimmunology 7, e1415687 (2018).
Luo, G. et al. Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl Acad. Sci. USA 115, E12323–E12332 (2018).
Zhang, S. Q. et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol. 36, 1156–1159 (2018).
Huth, A., Liang, X., Krebs, S., Blum, H. & Moosmann, A. Antigen-specific TCR signatures of cytomegalovirus infection. J. Immunol. 202, 979–990 (2019).
Ma, K. Y. et al. High-throughput and high-dimensional single-cell analysis of antigen-specific CD8(+) T cells. Nat. Immunol. 22, 1590–1598 (2021).
Francis, J. M. et al. Allelic variation in class I HLA determines CD8(+) T cell repertoire shape and cross-reactive memory responses to SARS-CoV-2. Sci. Immunol. 7, eabk3070 (2022).
Lu, T. et al. Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat. Mach. Intell. 3, 864–875 (2021).
Ishigaki, K. et al. HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors. Nat. Genet. 54, 393–402 (2022).
Minervina, A. A. et al. SARS-CoV-2 antigen exposure history shapes phenotypes and specificity of memory CD8(+) T cells. Nat. Immunol. 23, 781–790 (2022).
Mudd, P. A. et al. SARS-CoV-2 mRNA vaccination elicits a robust and persistent T follicular helper cell response in humans. Cell 185, 603–613.e615 (2022).
Feng, Z. et al. Sliding-attention transformer neural architecture for predicting T cell receptor–antigen–human leucocyte antigen binding. Nat. Mach. Intell. 6, 1216–1230 (2024).
Yue, T. et al. TCRdb 2.0: an updated T-cell receptor sequence database. Nucleic Acids Res. 53, gkaf876 (2025).
Dean, J. et al. Annotation of pseudogenic gene segments by massively parallel sequencing of rearranged lymphocyte receptor loci. Genome Med. 7, 123 (2015).
Cai, M., Bang, S., Zhang, P. & Lee, H. ATM-TCR: TCR–epitope binding affinity prediction using a multi-head self-attention model. Front. Immunol. 13, 893247 (2022).
Xu, Y. et al. AttnTAP: A dual-input framework incorporating the attention mechanism for accurately predicting TCR-peptide binding. Front. Genet. 13, 942491 (2022).
Sidhom, J. W., Larman, H. B., Pardoll, D. M. & Baras, A. S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 12, 1605 (2021).
Xu, Z. et al. DLpTCR: an ensemble deep learning framework for predicting immunogenic peptide recognized by T cell receptor. Brief. Bioinform. 22, 1–13 (2021).
Pham, M. N. et al. epiTCR: a highly sensitive predictor for TCR-peptide binding. Bioinformatics 39, btad284 (2023).
Springer, I., Besser, H., Tickotsky-Moskovitz, N., Dvorkin, S. & Louzoun, Y. Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. Front. Immunol. 11, 1803 (2020).
Springer, I., Tickotsky, N. & Louzoun, Y. Contribution of T cell receptor alpha and beta CDR3, MHC typing, V and J genes to peptide binding prediction. Front. Immunol. 12, 664514 (2021).
Zhang, Y. et al. iTCep: a deep learning framework for identification of T cell epitopes by harnessing fusion features. Front. Genet. 14, 1141535 (2023).
Luu, A. M., Leistico, J. R., Miller, T., Kim, S. & Song, J. S. Predicting TCR–epitope binding specificity using deep metric learning and multimodal learning. Genes (Basel) 12, 572 (2021).
Gao, Y. et al. Pan-peptide meta learning for T-cell receptor–antigen binding recognition. Nat. Mach. Intell. 5, 236–249 (2023).
Zhang, P., Bang, S. & Lee, H. PiTE: TCR–epitope binding affinity prediction pipeline using transformer-based sequence encoder. Pac. Symp. Biocomput. 28, 347–358 (2023).
Yi, H., Yuqiu, Y., Yanhua, T., Fattah, F. J. & Itzstein, M. S. V. pan-MHC and cross-species prediction of T cell receptor-antigen binding. Preprint at bioRxiv https://doi.org/10.1101/2023.12.01.569599 (2023).
Tong, Y. et al. SETE: Sequence-based ensemble learning approach for TCR epitope binding prediction. Comput. Biol. Chem. 87, 107281 (2020).
Wu, K. E. et al. TCR-BERT: learning the grammar of T-cell receptors for flexible antigen-binding analyses. Proc. Mach. Learn. Comput. Biol. 240, 194–229 (2024).
Jokinen, E. et al. TCRconv: predicting recognition between T cell receptors and epitopes using contextualized motifs. Bioinformatics 39, 1–8 (2023).
Li, Y., Zhang, C., Zhang, X. & Zhang, Y. TCRfinder: improved TCR virtual screening for novel antigenic peptides with tailored language models. Preprint at bioRxiv https://doi.org/10.1101/2024.06.27.601008 (2024).
Tatikonda, R. R., Demerdash, O. N. A. & Smith, J. C. TCR-H: explainable machine learning prediction of T-cell receptor epitope binding on unseen datasets. Front. Immunol. 15, 1426173 (2024).
Jokinen, E., Huuhtanen, J., Mustjoki, S., Heinonen, M. & Lahdesmaki, H. Predicting recognition between T cell receptors and epitopes with TCRGP. PLoS Comput. Biol. 17, e1008814 (2021).
Peng, X. et al. Characterizing the interaction conformation between T-cell receptors and epitopes with deep learning. Nat. Mach. Intell. 5, 395–407 (2023).
Jiang, Y., Huo, M. & Cheng Li, S. TEINet: a deep learning framework for prediction of TCR–epitope binding specificity. Brief. Bioinform. 24, 1–10 (2023).
Weber, A., Born, J. & Rodriguez Martinez, M. TITAN: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 37, i237–i244 (2021).
Wu, J., Qi, M., Zhang, F. & Zheng, Y. TPBTE: a model based on convolutional transformer for predicting the binding of TCR to epitope. Mol. Immunol. 157, 30–41 (2023).
Grazioli, F. et al. Attentive variational information bottleneck for TCR-peptide interaction prediction. Bioinformatics 39, btac820 (2023).
Jiang, M., Yu, Z. & Lan, X. VitTCR: a deep learning method for peptide recognition prediction. iScience 27, 109770 (2024).
Chen, W. et al. Commonly used software tools produce conflicting and overly-optimistic AUPRC values. Genome Biol. 25, 118 (2024).
Lu, Y. et al. Assessment of computational methods in predicting TCR–epitope binding recognition. figshare https://doi.org/10.6084/m9.figshare.27020455 (2025).
Acknowledgements
We acknowledge the Bioinformation Center for Guangdong-Hong Kong-Macao Greater Bay Area for providing computational support. This work was supported in part by Major Project of Guangzhou National Laboratory (grant no. GZNL2023A02007 to S.S., grant no. GZNL2023A03005 to S.S.), National Key Research and Development Program (grant no. 2024YFF0509000 to S.S.), National Natural Science Foundation of China (grant no. 32370972 to S.S., grant no. 32300528 to H.X.), Guangdong Basic and Applied Basic Research Foundation (grant no. 2024B1515020052 to S.S., grant no. 2023A1515011783 to S.S.), the Union Project from Guangzhou National Laboratory and State Key Laboratory of Respiratory Disease, Guangzhou Medical University (grant no. GZNL2024B01004 to S.S.) and the Excellent Youth Foundation of Hunan Scientific Committee (grant no. 2024JJ2084 to H.X.).
Author information
Authors and Affiliations
Contributions
S.S. conceptualized and supervised the project. Y.L., Y.W. and B.X. designed the benchmarking study with the guidance from H.X. and S.S. Y.L. and Y.Y. performed the data collection and model preparation. Y.L., Y.W. and M.X. analyzed the model evaluation results. Y.L. and Y.W. prepared the figures and tables. Y.L., Y.W. and S.S. wrote the paper. All authors reviewed and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Justin Barton, William Lees and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Madhura Mukhopadhyay, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Proportional distribution of TCR-epitope pairings matching different immunologically relevant categories and study design for original and retraining model evaluations.
a-c, Proportional distribution of TCR-epitope pairings matching across different MHC classes (a), alleles (b) and antigens (c). d, Experimental design for original model evaluations. The evaluations were conducted separately for CDR3β-only models and CDR3β+others models. We constructed two groups of seen- and unseen-epitope test sets by excluding the training data of all original models from our collected databases: one group contains only CDR3β and epitope sequences, and the other group contains additional features other than CDR3β and epitope sequences (such as MHC classes, CDR3α sequences). e, Experimental design for retraining model evaluations. The evaluations were conducted separately for CDR3β-only models and CDR3β+others models. We constructed two groups of seen-epitope tests together with seen- and unseen-epitope independent test sets based on our collected 21 databases: one group contains only CDR3β and epitope sequences, and the other group contains additional features other than CDR3β and epitope sequences (such as MHC classes, CDR3α sequences). In retraining, CDR3β-only models were further tested for the impact of multiple factors, including TCR similarity, negative TCR sources, cross-reactive TCRs, the refined AS method, low prevalence of true bindings and training data size. Across both experimental designs (d, e), CDR3β-only models were evaluated using three types of negative data sources: AS, PS and HS TCRs, whereas CDR3β+others models were tested only with AS negatives, as PS and HS TCRs rarely contain additional information except for CDR3β. Additionally, the CDR3β-only models were also evaluated with CDR3β+others data to assess the impact of feature enrichment on model performance. For all tests, TCRs highly similar to training sequences were excluded from test sets to avoid data leakage.
Extended Data Fig. 2 Performance evaluation of originally trained CDR3β-Only models on seen- and unseen-epitope predictions based on CDR3β-only data.
a-b, Amino acid distribution of CDR3β sequences starting with C and ending with F (a) and of CDR3β sequences not starting with C and ending with F (b). c-d, Performance of original CDR3β-only models in seen-epitope (c) and unseen-epitope test (d) using AS negatives based on CDR3β-only data in terms of multiple metrics: AUPRC, Precision, Specificity, Recall, F1. e-f, Performance of CDR3β-only models on three seen epitopes using PS negatives (e) and HS negatives (f). g, AUPRC comparison of originally trained CDR3β-only models (n = 31) using AS/PS/HS negatives in seen-epitope test. h-i, AUPRC correlation between the seen-epitope test results of original CDR3β-only models (n = 31) obtained using AS and PS negatives (h) and using AS and HS negatives (i). j-k, Performance of CDR3β-only models on unseen epitopes using PS negatives (j) and HS negatives (k). l, AUPRC comparison of originally trained CDR3β-only models (n = 28) using AS/PS/HS negatives in unseen-epitope test. m-n, AUPRC correlation between the unseen-epitope test results of original CDR3β-only models (n = 28) obtained using AS and PS negatives (m) and using AS and HS negatives (n). Heatmaps (e, f, j, k) show epitope-level AUPRC, with adjacent bar charts showing overall AUPRC. Colored dots (g, l) represent individual model AUPRC, black dots indicate mean, error bars represent the mean ± SD. P-values of Pearson correlations (h, i, m, n) were from two-sided t-test.
Extended Data Fig. 3 Performance evaluation of originally trained CDR3β-only and CDR3β+others models on seen- and unseen-epitope predictions based on CDR3β+Others data in terms of multiple metrics.
a, Performance of original CDR3β+others models in seen-epitope test using AS negatives based on CDR3β+others data. b, AUPRC of CDR3β-only models on two seen epitopes of CDR3β+others data using AS negatives. c-d, Performance of original CDR3β-only models in seen-epitope test (c) and original CDR3β+others models in unseen-epitope test (d) using AS negatives based on CDR3β+others data. e, Performance of CDR3β-only models on unseen epitopes of CDR3β+others data using AS negatives. f, Performance of original CDR3β-only models in unseen-epitope test using AS negatives based on CDR3β+others data. g, AUPRC comparison of original CDR3β-only models (left) and CDR3β+others models (right) using AS negatives on seen- and unseen-epitope test (for the CDR3β-only models, n = 31 for the seen test and n = 28 for the unseen test; for the CDR3β+others models, n = 15 for the seen test and n = 10 for the unseen test); box plots display mean (center line), the first and third quartiles (box), minimum and maximum values within 1.5×interquartile range (whiskers). P-values are from two-sided Wilcoxon signed-rank tests. Heatmaps (a, c, d, f) show results of multiple metrics: AUPRC, Precision, Specificity, Recall, and F1. Heatmaps (b, e) show epitope-level AUPRC, with adjacent bar charts showing overall AUPRC.
Extended Data Fig. 4 Distribution of training, test and independent test data for retrained model evaluation using the CDR3β-only and CDR3β+others datasets.
a, Distribution of TCR length in the CDR3β-only dataset. b, Distribution of data used by retrained CDR3β-only models. c, Percentage and number of TCRs in the stratified sampling of 5 times for constructing training and test sets within the CDR3β-only dataset. d, Distribution of antigen types and epitopes in the seen-epitope independent test set of CDR3β-only data. e, Number of epitopes that correspond to different TCR numbers in the seen-epitope independent test set of CDR3β-only data. f, Distribution of antigen types and epitopes in the unseen-epitope independent test set of CDR3β-only data. g, Number of epitopes that correspond to different TCR numbers in the unseen-epitope independent test set of CDR3β-only data. h, Distribution of TCR length in the CDR3β+others dataset. i, Distribution of data used by retrained CDR3β+others models. j, Percentage and number of TCRs in the stratified sampling of 5 times for constructing training and test sets within the CDR3β+others dataset. k, Distribution of antigen types and epitopes in the seen-epitope independent test set of CDR3β+others data. l, Number of epitopes that correspond to different TCR numbers in the seen-epitope independent test set of CDR3β+others data. m, Distribution of antigen types and epitopes in the unseen-epitope independent test set of CDR3β+others data. n, Number of epitopes that correspond to different TCR numbers in the unseen-epitope independent test set of CDR3β+others data. Heatmaps (b, d, f, i, k, m) show the log10-transformed number of TCRs corresponding to each epitope, with x-axis representing epitopes and y-axis representing antigens.
Extended Data Fig. 5 Performance of retrained CDR3β-only and CDR3β+others models on seen- and unseen-epitope predictions in terms of multiple metrics.
a-c, Performance of retrained CDR3β-only models in seen-epitope test (a), independent test (b) and unseen-epitope independent test (c) using AS negatives based on CDR3β-only data. d-e, Performance of retrained CDR3β+others models (d) and retrained CDR3β-only models (e) in seen-epitope test using AS negatives based on CDR3β+others data. f-g, Performance of retrained CDR3β+others models (f) and retrained CDR3β-only models (g) in seen-epitope independent test using AS negatives based on CDR3β+others data. h-i, Performance of retrained CDR3β+others models (h) and retrained CDR3β-only models (i) in unseen-epitope independent test using AS negatives based on CDR3β+others data. All heatmaps show results of multiple metrics: AUPRC, Precision, Specificity, Recall and F1.
Extended Data Fig. 6 Impact of key factors on model performance: sequence similarity and source effects of negative data.
a-c, AUPRC comparison between models retrained with CDR3β-only features in predicting seen-epitope test data (a), seen-epitope independent test data (b), and unseen-epitope independent test data (c) with and without removing similar TCR sequences using AS/PS/HS negatives. Dots represent individual model AUPRC, and lines connect the same models across evaluation settings. P-values were from two-sided Wilcoxon signed-rank test (n = 24 for seen-epitope predictions and n = 21 for unseen-epitope predictions) with Benjamini-Hochberg correction. d, AUPRC performance of PS- and HS-based retrained models on AS-based test, seen-epitope independent test and unseen-epitope independent test data with CDR3β-only features. e, AUPRC performance of AS-based retrained models on PS- and HS-based seen-epitope test, seen-epitope independent test and unseen-epitope independent test data with CDR3β-only features.
Extended Data Fig. 7 Impact of key factors on model performance: cross-reactive TCRs and refined AS-based reshuffling methods.
a, Distribution of cross-reactive and non-cross-reactive TCRs in our datasets after preprocessing. b-c, AUPRC comparison between models retrained with and without cross-reactive TCRs under the refined AS-based negative sample generation approach when testing with data comprising both cross-reactive and non-cross-reactive entries. d, AUPRC comparison between models retrained with and without cross-reactive TCRs under the random AS-based negative sample generation. e-f, AUPRC comparison of models retrained with cross-reactive TCRs under two negative data reshuffling strategies: the refined AS-based and the traditional random AS-based reshuffling approach, when testing with data comprising both cross-reactive and non-cross-reactive entries. Dots (b, d, e) represent individual model AUPRC, and lines connect the same models across evaluation settings. P-values were from two-sided Wilcoxon signed-rank test (n = 24 for seen-epitope predictions and n = 21 for unseen-epitope predictions) with Benjamini-Hochberg correction. All metrics (c, f) were rounded to three decimals to enable clearer comparison of subtle performance differences across models.
Extended Data Fig. 8 Performance of the retrained CDR3β-only models on low prevalence of true TCR-epitope pairs.
a-c, Performance of CDR3β-only models using AS negatives in predicting seen-epitope test data (a), independent test (b) and unseen-epitope independent test (c) data with different prevalences (0.1%, 1%, 10%, and 50%) of positive samples in terms of Precision, F1, Recall and Specificity. In consideration of the relatively small magnitude of many metric values, all metrics were rounded to three decimals to enable clearer comparison of subtle performance differences across models.
Extended Data Fig. 9 Additional results of testing the effects of TCR counts on model performance and correlation between the heterogeneity of TCRs and model performance.
a, Performance saturation analysis for TEIM, TCR-BERT, ERGO-AE, VitTCR, NetTCR, PiTE and ATM-TCR, using five epitopes with most TCR counts, showing per-epitope AUPRC and mean performance (red line). b, AUPRC comparison of average AUPRC of models obtained by five epitopes across different TCR numbers. c, Growth trend of AUPRC across TCR count intervals. The x-axis denotes three intervals of TCR counts employed in model training. The heatmap shows the slopes, calculated as AUPRC change divided by the TCR count range within each interval. d, Correlation between TCR sequence heterogeneity and AUPRC for models: epiTCR, TCRGP, TEPCAM, VitTCR, TEIM, TCR-BERT, PiTE, NetTCR, ATM-TCR, and ERGO-AE; dots represent epitopes, colored by antigen group. The heterogeneity between TCR sequences was measured by average Levenshtein distance per epitope. Spearman correlation was used, and P-values were from two-sided t-test (n = 389). e, Differences in the strength of the negative correlation between intra-epitope TCR heterogeneity and model AUPRC across different models based on the results from d. P-values of Fisher’s r-to-z transformation were from two-sided z-test with Benjamini-Hochberg correction (n = 389).
Extended Data Fig. 10 Time and memory usage of models in training and testing under different data sizes.
a-d, Training time (a), memory usage during training (b), testing time (c), and memory usage during testing (d) for various data sizes; CDR3β+others models are highlighted in red.
Supplementary information
Supplementary Information (download PDF )
Supplementary Notes 1–9 and Figs. 1–3.
Supplementary Table 1 (download XLSX )
Details of TCR–epitope binding datasets utilized in this study.
Supplementary Table 2 (download XLSX )
Basic information about the collected TCR–epitope prediction models.
Supplementary Table 3 (download XLSX )
Details of training datasets used by original models.
Supplementary Table 4 (download XLSX )
Detailed performance results across multiple metrics for Fig. 2.
Supplementary Table 5 (download XLSX )
Number of TCRs and epitopes in positive samples for training different original models.
Supplementary Table 6 (download XLSX )
Number of TCRs corresponding to the epitopes in positive samples for training original models.
Supplementary Table 7 (download XLSX )
Detailed performance results across multiple metrics for Fig. 3.
Supplementary Table 8 (download XLSX )
Detailed performance results across multiple metrics for Fig. 4.
Supplementary Table 9 (download XLSX )
Detailed performance results across multiple metrics for Fig. 5.
Supplementary Table 10 (download XLSX )
Detailed performance results across multiple metrics for Fig. 6.
Supplementary Data 1 (download XLSX )
Source data for Supplementary Fig. 1.
Supplementary Data 2 (download XLSX )
Source data for Supplementary Fig. 2.
Supplementary Data 3 (download XLSX )
Source data for Supplementary Fig. 3.
Source data
Source Data Fig. 2 (download XLSX )
Source data of data distribution, model performance and statistical results.
Source Data Fig. 3 (download XLSX )
Source data of data distribution, model performance and statistical results.
Source Data Fig. 4 (download XLSX )
Source data of model performance and statistical results.
Source Data Fig. 5 (download XLSX )
Source data of model performance and statistical results.
Source Data Fig. 6 (download XLSX )
Source data of model performance and statistical results.
Source Data Extended Data Fig. 1 (download XLSX )
Source data of data distribution.
Source Data Extended Data Fig. 2 (download XLSX )
Source data of data distribution, model performance and statistical results.
Source Data Extended Data Fig. 3 (download XLSX )
Source data of model performance and statistical results.
Source Data Extended Data Fig. 4 (download XLSX )
Source data of data distribution.
Source Data Extended Data Fig. 5 (download XLSX )
Source data of model performance.
Source Data Extended Data Fig. 6 (download XLSX )
Source data of model performance and statistical results.
Source Data Extended Data Fig. 7 (download XLSX )
Source data of data distribution, model performance and statistical results.
Source Data Extended Data Fig. 8 (download XLSX )
Source data of model performance.
Source Data Extended Data Fig. 9 (download XLSX )
Source data of model performance and statistical results.
Source Data Extended Data Fig. 10 (download XLSX )
Source data of computational time and memory usage of models.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, Y., Wang, Y., Xu, M. et al. Assessment of computational methods in predicting TCR–epitope binding recognition. Nat Methods 23, 248–259 (2026). https://doi.org/10.1038/s41592-025-02910-0
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41592-025-02910-0
This article is cited by
-
Decoding adaptive immunity: advanced strategies in T and B cell repertoire analysis
Journal of Translational Medicine (2026)
