
Abstract
Microglia and neuroinflammation are involved in amyotrophic lateral sclerosis (ALS), but the precise underlying molecular mechanisms remain elusive. We generated single-nuclei transcriptomes from the spinal cord and motor cortex of patients with sporadic ALS (sALS) and C9orf72 ALS (C9-ALS). Here we confirmed that C9orf72 is highly expressed in microglia and observed that the hexanucleotide repeat expansion (HRE) results in haploinsufficiency. Whereas sALS microglia transitioned toward disease-associated cell states, C9orf72 HRE microglia exhibited a diminished response, with alterations in endolysosomal pathways. We confirmed these observations using a human microglia xenograft model, in which C9orf72 mutations led to a reduced activation. We also confirmed the endolysosomal alterations in C9orf72 HRE and C9orf72-deficient induced pluripotent stem cell (iPSC)-derived microglia. We also found a diminished response of C9orf72 HRE astrocytes and provided a map of dysregulated ligand−receptor pairs in microglia and astrocytes. Our data highlight variations in the cellular substrate of sporadic and inherited forms of ALS, which have implications for patient stratification and selection of appropriate treatments.
Similar content being viewed by others


Main
ALS is a neurodegenerative disease characterized by progressive loss of motor neurons in the motor cortex, brainstem and spinal cord1. Loss of motor neurons is accompanied by neuroinflammation and alterations in both microglia and astrocytes2. ALS typically presents with focal muscle weakness, which spreads to neighboring body regions. Other non-motor manifestations, such as cognitive and behavioral impairment, occur in a subset of patients1. Effective therapies are scarce, and the median survival after symptom onset is limited to 3 years1. The widespread nature of this disease, with contributions from distant central nervous system (CNS) regions, represents a challenge when trying to elucidate the underlying cellular mechanisms. To achieve a deep understanding of ALS pathophysiology, it is essential to investigate cell-specific disease mechanisms acting in both the spinal cord and the motor cortex.
ALS has a strong genetic component, where the most common monogenic cause is the GGGGCC repeat expansions in the C9orf72 (chromosome 9 open reading frame 72) gene3, with a repeat length ranging from 38 to approximately 1,600 repeats4,5. C9orf72 has multiple transcript variants that are differentially enriched across cell types in the brain. Both loss-of-function (LOF) and toxic gain-of-function (GOF) mechanisms in neuronal and non-neuronal cells have been implicated in the disease pathogenesis6. C9orf72 is predominantly expressed in myeloid cells, including microglia7. Although the exact mechanisms by which the C9orf72 mutations alter microglia function are not fully understood8, experimental studies have shown that its deletion leads to altered immune responses, exacerbated myeloid proliferation and lysosomal dysfunction9,10.
In this study, we investigated the impact of the C9orf72 pathogenic repeat expansion (HRE) in microglia and astrocytes. By single-nuclei RNA sequencing (snRNA-seq) of C9-ALS, sALS and healthy control motor cortex and spinal cord samples, we demonstrate that the pathogenic expansion in C9orf72 results in haploinsufficiency in microglia and leads to an impaired phenotypic transition of microglia toward reactive cell states when compared to sALS. We confirmed these findings using a xenografted model where both C9orf72−/− and C9orf72 HRE iPSC-derived microglia were transplanted in the mouse brain. We observed an alteration in endolysosomal transcriptional pathways, which was confirmed using both LOF and patient iPSC-derived microglia in vitro systems. Finally, we found that astrocytes are also transcriptionally altered in C9orf72 carriers when compared to sALS, and we suggest that this might be due to defective microglia−astrocyte communication. Our work indicates that, whereas sALS and C9-ALS are clinically indistinguishable, the cellular and molecular substrates underlying both diseases might be different, thus posing additional challenges when aiming at developing new therapeutic strategies.
Results
The HRE reduces C9orf72 expression in microglia
We performed snRNA-seq on matching motor cortex and spinal cord samples from sALS, C9-ALS and healthy controls (n = 5 per group). We analyzed a total of 156,252 nuclei after batch correction and removal of low-quality nuclei and doublets (Extended Data Fig. 1a). Using publicly available data from recent mouse and human spinal cord and brain atlases11,12,13,14,15,16,17, we defined the different major cell types across the spinal cord and motor cortex. In the motor cortex, we identified six cell types, including astrocytes (SLC1A3, SLC1A2, RYP3), endothelial cells (COBLL1, IGFBP7, SYNE2), neurons (SYT1, GABRB2, RBFOX), microglia (MEF2A, DOCK8, ST6GAl1), oligodendrocytes (MBP, PRP4K2A, RLP1) and oligodendrocyte precursor cells (DSCAM, VCAN, LHFPL3) (Fig. 1a,c). By contrast, in the spinal cord, we identified seven major transcriptionally distinct cell types that we annotated into astrocytes (GFAP, CTNND2, NRG3), endothelial cells (SPAG17, DNAH7, DCDC1), microglia (DOCK4, MEF2C, LRMDA), natural killer cells (PARP8, MBNL1, CBLB, B2M), neurons (SYT1, SNAP25, CALY), oligodendrocytes (MBP, PLP1, PEX5L) and oligodendrocyte precursor cells (DSCAM, VCAN, NRCAM) (Fig. 1b,c). We did not observe any major changes in the proportion of the different cell types across the different experimental conditions (Extended Data Fig. 1c,d).

a, UMAP plot visualizing 93,176 nuclei isolated from postmortem cortical samples of patients with ALS (C9-ALS and sALS) and healthy controls. Cells are annotated and colored according to their assigned cell identity into astrocytes, endothelial cells, neurons, microglia, oligodendrocytes and oligodendrocyte precursor cells. b, UMAP plot visualizing 63,076 nuclei isolated from postmortem spinal cord samples of patients with ALS (C9-ALS and sALS) and healthy controls. Cells are annotated and colored according to their assigned cell identity into astrocytes, endothelial cells, microglia, natural killer cells, neurons, oligodendrocytes and oligodendrocyte precursor cells. c, Heatmaps showing the top 10 most expressed genes per cluster. d, Heatmap depicting the z-scored average expression of C9orf72 across different cell types in both motor cortex and spinal cord. NK, natural killer.
Haploinsufficiency of C9orf72 is a key proposed disease mechanism, along with toxicity derived from HRE-containing expanded RNAs and aggregating dipeptide repeat proteins (DPRs)6,18. Thus, we assessed the expression of C9orf72 in all the different cell types identified in both the spinal cord and motor cortex. We confirmed that microglia show the highest expression of C9orf72 in the CNS (Fig. 1d) and found that the presence of the C9orf72 HRE results in a reduced expression that is limited to these cells and not observed in other cell types (Fig. 1d). This is consistent with previous findings in blood samples and iPSC-derived microglia from C9orf72 HRE carriers19,20,21.
The C9orf72 HRE diminishes microglial activation
Next, we sought to determine the impact of the C9orf72 HRE-derived haploinsufficiency in the transcriptome of microglia. We subset, integrated, quality controlled and reclustered the microglial nuclei from the spinal cord (n = 12,547) and motor cortex (n = 4,216) (Supplementary Table 3). We identified six distinct microglia transcriptional states across the spinal cord and motor cortex (Fig. 2a and Extended Data Fig. 2). To increase the sample size and enhance the accuracy, we incorporated recent data from Gerrits et al.22 into our analysis, which consists of 132,628 microglial nuclei across three brain regions (frontal, temporal and occipital cortex) from both FTD-TDP (n = 88,237) and healthy controls (n = 44,391) (Fig. 2b and Supplementary Table 5). After integration of both datasets (total of 149,172 nuclei), we retained the exact same six microglial subclusters (Fig. 2b and Extended Data Fig. 2c,d). Using previously described microglial signatures23, we annotated these clusters as homeostatic microglia (HM) (MG1; MERTK, MRC1, NHSL1), transitioning microglia (TM) (MG2; lower expression of CX3CR1, P2RY12 and a background of HSP1A1, HSSPA1B, KCNPI1), ribosomal microglia (RM) (MG3; FTH1, RP transcripts), disease-associated microglia (DAM) (MG4; GLDN, SPP1), hypoxia-associated microglia (MG5; NAMPT, HIF1A) and cytokine response microglia (CRM) (MG6; CCL3, CCL4, IL1B, NFKBIZ) (Fig. 2c). The distribution of these clusters among all individuals and experimental groups is shown in Extended Data Fig. 2e.

a, UMAP plot visualizing 16,763 microglia nuclei from ALS (C9 and sALS) and healthy postmortem samples. Microglia subsets are colored based on their cluster into MG1, MG2, MG3, MG4, MG5 and MG6. b, UMAP plot displaying 149,172 nuclei after integration with the dataset by Gerrits et al.22. The correlation between the clusters in a and b is shown in Extended Data Fig. 2. c, Heatmap showing the top 20 most expressed genes per cluster from b. d, Graphical representation of the neighborhood analysis using Milo. e, Relative enrichment of the neighborhood groups (left) in brain and spinal cord samples. The pie charts (right) depict the contribution of cells in patient group and sampling locations in the neighborhood groups. f, Overlap of markers from MG5 and differential abundance (DA) group 6. g, Volcano plot depicting the pairwise comparisons of C9-ALS versus sALS for the combined microglial dataset (DESeq2 negative binomial distribution; P values adjusted with Bonferroni correction based on the total number of genes in the dataset). Vertical lines correspond to the log2FC threshold of 0.2, and the horizontal line corresponds to the adjusted P value threshold at <0.05. Colors indicate marker genes of microglia clusters in b. Positive log2FC values are enriched in C9-ALS; negative log2FC values are enriched in sALS. NA, not assigned to any cluster. h, Seurat module scores of microglia markers as described by Mancuso et al.26 visualized in the single-nuclei dataset, displayed per patient group and location. Black dots indicate the mean, and lines indicate the mean of healthy controls per location. i,j, Scatter plot depicting two pairwise comparisons of microglia from C9-ALS and healthy controls in the spinal cord versus GRN-FTD across cortical regions (R = 0.15, P < 2.2 × 10−16, DESeq2 negative binomial distribution) (i) and sALS and healthy controls in the spinal cord versus GRN-FTD and healthy controls across all cortical regions (R = −0.47, P < 2.2 ×10−16) (j). Lines represent the log2FC threshold of ±0.2. Positive log2FC values display enrichments in disease states; negative log2FC values display enrichments in healthy controls. P values are included in Supplementary Tables 7, 8 and 16. FC, fold change.
Given the widespread anatomical distribution of the samples included in our analysis, we wondered whether microglia displayed any regional transcriptomic differences. To explore this, we quantified shifts in abundances of cell states across brain and spinal cord using Milo24. We grouped transcriptionally similar microglia into neighborhoods of 50–200 cells, and, for each neighborhood, we determined the proportion of cells from each patient group as well as each anatomical region. Then, we assessed the differential abundance of each CNS region in each neighborhood and categorized them into seven distinct groups (Fig. 2d). Although most of the groups did not show any regional enrichment, we found that group 6 was specifically enriched with microglia from the spinal cord (Fig. 2e). Additionally, we observed that the marker genes from group 6 overlap mostly with the transcriptomic profile of MG5 (hypoxia) microglia (Fig. 3f). Nevertheless, we observed no correlation between the gene expression of each neighborhood group with the differentially expressed analysis between either C9-ALS or sALS versus controls (Extended Data Fig. 2g,h). This suggests that, whereas microglia display certain transcriptional heterogeneity across major regions of the CNS, there is no evidence that links this diversity to the pathological changes observed in ALS.

a, Schematic overview of the experimental design. MPs, microglial precursors; hMG, human microglia. Created with BioRender.com. b, UMAP plot visualizing microglia cell states colored based on their assigned cluster: HM, RM, tCRM, CRM1, CRM2, HLA, DAM and IRM. c, Heatmap showing the most expressed genes per microglial cluster. Volcano plots comparing C9-HRE versus control microglia (d) and C9KO versus control microglia (e). Top differentially downregulated and upregulated genes are reported and color coded based on the corresponding microglial cluster. NA, not assigned to any cluster. Wilcoxon test; P values adjusted with Bonferroni correction based on the total number of genes in the dataset (adjusted P < 0.05). P values can be found in Supplementary Tables 19 and 20. Number of mice per experimental group: C9-ISO n = 4, C9-HRE n = 2, CTRL n = 7, C9KO n = 10. Number of cells analyzed per group: C9-ISO = 1,184, C9-HRE = 554, CTRL = 11,104, C9KO = 10,386. Timepoint analyzed: C9-HRE and C9-ISO 3 months, CTRL and C9KO 6 months. FC, fold change.
Next, we performed a differential expression analysis between C9-ALS versus healthy controls and sALS versus healthy controls to determine the transcriptional impact of the C9orf72 haploinsufficiency in microglia (Fig. 2g, Extended Data Fig. 2f and Supplementary Tables 7–15). We observed strong differences between groups, with C9-ALS microglia generally displaying a much lower general transcriptional response to pathology in the motor cortex and especially the spinal cord (Extended Data Fig. 2e). Comparison between C9-ALS and sALS microglia showed that spinal sALS microglia display transcriptomic changes consistent with a transition toward the reactive RM, DAM and cytokine response microglia (CRM) states, with an increased expression of marker genes of clusters MG3, MG4 and MG6, such as FTH1, CD14, FTL, FCGR1A, HLA-DPB1 and HLA-E, and a downregulation of homeostatic markers such as P2RY12. On the contrary, spinal C9-ALS microglia did not show any clear upregulation of genes associated with reactive states and, in turn, expressed higher levels of P2RY12, which is classically defined as an HM marker25 (Fig. 2g and Extended Data Fig. 2f). This indicates that, whereas sALS microglia are able to transit toward responsive states, C9-ALS microglia are less able to engage in this phenotypic switch. Next, we mapped the atlas of transcriptomic signatures of human microglia that we previously obtained using a human brain cellular xenograft (HBCX) model onto our single-nuclei dataset26. We confirmed that, contrary to sALS, spinal C9-ALS microglia displayed a reduced enrichment in both DAM and antigen-presenting HLA gene sets (Fig. 2h). This is consistent with our differential expression analysis as well as with previous findings in C9orf72 knockout mice that show an impaired phenotypic transition toward DAM27 and, therefore, resembles the LOF scenario described in several Alzheimer’s disease genetic mutations, such as TREM2 or APOE26,28. This suggests that there might be strong similarities in the impact of disease-causing or risk-modifying mutations in microglia across different neurodegenerative disorders23.
Given that mutation in both C9orf72 and GRN results in frontotemporal dementia (FTD) with TDP-43 pathology, and that both genes are highly expressed in microglia29,30,31, we wondered whether C9-ALS microglia shared any similarities with GRN-deficient cells. We performed a comparison of the differentially expressed genes in patients with C9-ALS, sALS and GRN-FTD22. Interestingly, we observed that, whereas C9 and GRN-deficient microglia gene expression were positively correlated, sALS and GRN-deficient microglial responses were negatively correlated (Fig. 2i,j and Supplementary Table 16). This could indicate that C9orf72 HRE and GRN deficiency may result in similar downstream functional alterations in microglia.
C9orf72 HRE results in altered endolysosomal pathways
To link the transcriptional alterations that we observed with relevant microglial functional pathways, we performed an unbiased weighted gene co-expression network analysis (WGCNA). We focused on the spinal cord, where we found the most pronounced changes in our differential expression analysis. We identified seven distinct gene co-expression clusters, many of them with differential significant enrichment across sALS, C9-ALS and healthy controls (Extended Data Fig. 3 and Supplementary Table 17). The blue cluster appeared particularly relevant, as it contained a high number of genes (Extended Data Fig. 3b) and was significantly enriched in sALS versus both C9-ALS and healthy controls (Extended Data Fig. 3c). Genes in this cluster were associated with functional pathways related to neurodegeneration, immune system, phagocytosis, autophagy−lysosomal function, ubiquitin−proteasome system, oxidative stress and apoptosis, confirming a strong immune component in the pathogenesis of sALS (Extended Data Fig. 3d). C9-ALS microglia showed no enrichment in these pathways, which is consistent with the C9orf72 HRES leading to a defective engagement in reactive DAM and HLA states (Fig. 3c). In turn, C9-ALS microglia showed an enrichment of clusters turquoise and yellow, which encompassed general biological pathways such as cell proliferation, migration, general metabolic processes, protein biogenesis and degradation (Extended Data Fig. 3c,d). To confirm these observations, we performed a targeted gene set enrichment analysis (GSEA) focusing on lysosomal, phagocytic, inflammatory and metabolic transcriptional pathways. Whereas sALS microglia showed increased transcription of genes involved in lysosomal and phagocytic pathways, both C9-ALS and sALS displayed an enrichment in genes related to oxidative phosphorylation (Extended Data Fig. 4). This is consistent with our differential expression analysis and is in line with previous findings32,33,34.
Our data indicate that the C9orf72 HRE prevents the transition of microglia toward reactive states as opposed to sALS, therefore indicating that the pathogenic contribution of microglia could be different in sporadic and inherited forms of ALS.
The C9orf72 HRE affects engrafted microglial transcriptome
To further validate our postmortem findings and determine whether the transcriptomic changes that we observed are at least partially mediated by a direct effect of the C9orf72 haploinsufficiency in microglia, we used an HBCX model (Fig. 3a)26,35,36. We transplanted iPSC-derived microglial precursors from a C9orf72 patient line (C9-HRE) carrying the repeat expansion and the corresponding isogenic control in which the mutation was corrected via CRISPR−Cas9 genome editing (C9-ISO)37. We also transplanted microglial precursors generated from C9orf72 knockout (C9KO) and isogenic control iPSC lines, which we previously characterized38, to evaluate the impact of C9orf72 LOF in human engrafted microglia without the potentially confounding factor of other C9orf72-associated phenotypes. Microglial precursors were transplanted into the brain of wild-type immunodeficient mice (Rag2−/−Il2g−/−hCSF1KI)26,35,36.
After 3 months or 6 months upon transplantation, we isolated the human microglia by fluorescence-activated cell sorting (FACS) labeled with CD11b and hCD45 (refs. 26,35,36) (Extended Data Fig. 5a) and performed single-cell RNA sequencing (scRNA-seq). In total, we analyzed 21,826 cells, after removal of low-quality cells and doublets and correction for batch effects (Extended Data Fig. 5b–d). Using a combination of unbiased clustering, differential expression and microglial gene signatures scores that we previously described in xenograft mouse models26, we identified eight distinct microglial clusters: HM (P2RY12, CX3CR1), transitioning CRM (tCRM: HSPA1A, JUN, RHOB), cytokine response microglia 1 and 2 (CRM1 and CRM2: CCL2, CCL3, CCL4), RM (RPL3, RPL13A, RPL29), antigen-presenting response (HLA: HLA-DPA1, HLA-DRB5), DAM (CD9, LGALS3, GPNMB) and interferon response microglia (IRM: CXCL10, IFIT3, IFITM1) (Fig. 3b,c and Supplementary Table 18).
In line with our postmortem data, we observed reduced C9orf72 expression in the C9-HRE microglia compared to the isogenic control, again indicating an HRE LOF scenario (Fig. 3d). We also found that C9-HRE microglia showed a downregulation of HLA genes (for example, HLA-DPA1, XACT) and SPP1, a key regulator of DAM and one of the top downregulated genes in C9orf72 postmortem microglia. Moreover, we observed an upregulation of several homeostatic genes (for example, NAV3, AFF3, DISP1). Overall, these data suggest that the C9-HRE in microglia is sufficient to impair the transition of these cells into reactive cell states. Next, we sought to confirm that these observations were due to the loss of C9orf72 expression. Transplanted C9KO microglia partially phenocopied C9-HRE cells (Fig. 3e) and displayed a significantly lower shift into the HLA-reactive transcriptional state when compared to their isogenic controls (Extended Data Fig. 5e,f and Supplementary Tables 19 and 20). We also observed downregulation of several of HLA-related genes, overlapping with what we found altered in postmortem C9-HRE microglia (Fig. 3e).
These data indicate that, although we cannot exclude the contribution of other C9orf72 HRE downstream molecular mechanisms in microglia, the defective transition to reactive states and the downregulation of HLA genes are likely directly driven by a loss of C9orf72 function.
C9orf72 loss alters endolysosomal fitness in iPSC microglia
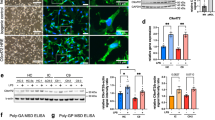
Our data in C9orf72 postmortem brain and transplanted microglia point toward a C9orf72-induced defect in the endolysosomal system. To validate this observation further, we assessed the expression levels of cathepsin D (CTSD), a known lysosomal protease, in the spinal cord of an independent experimental cohort of patients (2 healthy controls, 5 C9-ALS and 5 sALS). Consistent with our WGCNA analysis, patients with C9orf72 HRE showed reduced CTSD levels compared to sALS, especially in the spinal cord (Fig. 4a–c). Additionally, we differentiated a C9-HRE iPSC line into microglia cells35,36 (Extended Data Fig. 6a,b) and observed an increased percentage of cells containing enlarged CTSD-positive lysosomes in vitro (Fig. 4d and Extended Data Fig. 6d), further confirming the role of the C9orf72 HRE in altering the microglial endolysosomal compartment.

a, CTSD protein levels in cortex (top) and spinal cord (bottom) of C9-ALS and sALS. Western blot quantification of cortex (b) and spinal cord (c). Data were normalized to GAPDH. ANOVA with Tukeyʼs post hoc multiple correction testing (n = 2 healthy controls, n = 5 C9-ALS and n = 5 sALS). Western blot was repeated two independent times, and the technical replicates were averaged. d, Quantification showing increased abundance of lysosomal (CTSD) clusters in C9-HRE iPSC microglia versus C9-ISO. Unpaired t-test (C9-ISO n = 4 independent replicates; C9-HRE n = 5 independent replicates). e, High-resolution expansion microscopy images and quantification showing increased abundance of lysosomal clusters in C9KO microglia. Scale bar, corrected for a 4× expansion of the sample, 2.5 µm. f, Quantification showing increased abundance of CTSD clusters in C9KO microglia versus controls. Unpaired t-test (CTRL n = 5 independent replicates; C9KO n = 5 independent replicates). g,h, TEM images and quantification showing increased abundance of late endosomes/lysosomes in C9KO compared to control microglia. Unpaired t-test (CTRL n = 5 independent replicates; C9KO n = 5 independent replicates). Scale bar, upper images, 10 µm; lower images, 20 µm. i, Microscopy images showing RAB7-positive and LAMP1-positive structures (that is, late endosomes) in C9KO and control iPSC microglia. Scale bar, 5 µm. Box plots in b−d, f and h show the median, interquartile range and full data range. Live-cell imaging experiment showing the kinetics of pHrodo-conjugated E. coli particles accumulation in C9KO versus control microglia over 15 hours (j). Data points represent log10-transformed median pHrodo intensities, aggregated via a two-step median process (Methods). LOESS curves with 95% confidence intervals (gray area) were plotted to illustrate temporal trends (n = 6 independent replicates per genotype). k, Quantification of AUC and pHrodo median intensity at 30 minutes (l) and 900 minutes (m) as a proxy for uptake and cargo degradation processes, respectively. Ratio paired t-test (n = 6 independent replicates per group).
Next, we explored the impact of a full C9orf72 loss in iPSC-derived C9KO and isogenic controls (Extended Data Fig. 6a–c). Using resolution-enhancing expansion microscopy, we observed an enlargement and increased clustering of CTSD-positive lysosomes in C9KO microglia versus controls (Fig. 4e). Quantification showed an elevated proportion of cells containing CTSD puncta in the C9KO iPSC-derived microglia compared to controls (Fig. 4f and Extended Data Fig. 6d), consistent with what we observed in C9-HRE microglia (Fig. 4d). Using transmission electron microscopy (TEM), we found an increased density of electron-lucent vesicles in the C9KO microglia (Fig. 4g,h and Extended Data Fig. 6e,f). Immunofluorescence experiments revealed that these structures were LAMP1 and RAB7 positive (a marker of late endosomes) (Fig. 4i) but not EEA1 positive (a marker of early endosomes) (Extended Data Fig. 6g), indicating that the loss of C9orf72 leads to alterations of lysosomal homeostasis with an accumulation of late endosomes/lysosomes. Next, we performed phagocytosis assays to further validate the functional consequences of C9orf72 loss in microglia. We treated the cells with Escherichia coli pHrodo BioParticles that, once internalized, emit fluorescence within the acidic environments of endosomes and lysosomes (Fig. 4j). We monitored both the uptake and the degradation of the pHrodo particles over a timecourse of 15 hours using live-cell imaging. We found a significant increase in the overall phagocytosis in the C9KO microglia versus control as measured by the area under the curve (AUC) (Fig. 4k). To explore differences between uptake and degradation, we assessed the levels of fluorescence intensity in the microglia at 30 minutes and 900 minutes, respectively. We observed no differences in the initial uptake of pHrodo (Fig. 4l), which we also confirmed independently via flow cytometry (Extended Data Fig. 6h–k). However, we found significant differences at late stages of the live imaging, indicating a reduced ability of the C9KO microglia to degrade the phagocytic material compared to controls (Fig. 4m). Overall, our data further confirm the impact of C9 LOF on the microglial endolysosomal pathway, potentially leading to a defective functioning of late phases of the degradative pathway.
Brain and spinal astrocytes are transcriptionally distinct
To determine whether C9orf72 mutations also result in alterations in astrocytes, we first subset, integrated and reclustered both spinal (n = 12,978) and cortical (n = 9,657) astrocytes to define their transcriptomic landscape (Fig. 5a,b, Extended Data Fig. 7a–c and Supplementary Table 4). Unbiased clustering analysis showed eight independent transcriptional states (Fig. 5a). However, after integration with the data from Gerrits et al.22, we refined our analysis to six transcriptional states in 216,574 nuclei (Fig. 5b, Extended Data Fig. 7c and Supplementary Table 6), which parallels previously described astrocyte heterogeneity39,40,41,42. The correlation between the clustering analysis before and after integration can be found in Extended Data Fig. 7c. These six transcriptional states were the following: cluster AS1 was linked to axon guidance and neuronal development (NRP1, GFRAI1, KCNQ3, UNC5C)43; AS2 was related to axon guidance, calcium signaling and heparan sulphate production (CABLES1, HS3ST5, NRP1, SLC1A2); AS3 showed markers indicative of a type of reactive astrocytes (CHI3L1, RASGEF1B, SERPINA3)43,44; AS4 expressed genes associated with cell migration, cell morphogenesis and axonogenesis (SYNPO2, ROBO2, DCLK1, NFASC); AS5 was linked to glutamate receptor and TGFβ signaling (SEMA5A, GRIA1, ID3, DPP10); and AS6 was transcriptionally related to neuron development and modulation of chemical synaptic transmission (RASGRF1, CCK, GPRIN3). AS1 and AS2 were similar to the recently reported astroHO, SLC1A2+ and NRNX1+ subtypes. These clusters were not significantly enriched in any of the experimental groups and were present in all individuals (Extended Data Fig. 7d,e).

a, UMAP plot visualizing 22,635 astrocytes from postmortem samples of patients with ALS (C9-ALS and sALS) and healthy controls. Astrocyte subsets are colored based on assigned cluster: AS1, AS2, AS3, AS4, AS5, AS6, AS7, AS8. b, UMAP plot displaying 216,574 nuclei after integration with the dataset from Gerrits et al.22. New astrocyte subsets are colored based on their assigned cluster: AS1, AS2, AS3, AS4, AS5, AS6. The correlation between clusters in a and b is shown in Extended Data Fig. 5. c, Top 10 most expressed genes per cluster in b. Graphical representation of Milo’s differential abundance (DA) testing (d) and neighborhood group (e). f, Pie charts displaying the ratio of cells sampled in each neighborhood from patient groups and sampling locations. g, Changes in the distribution of neighborhood groups across brain and spinal cord (left) and average expression of differentially expressed genes (right) from the C9-ALS and sALS versus controls. Note that neighborhoods 5 and 6 are enriched in genes differentially expressed in C9-ALS and sALS astrocytes, respectively. Pathway analysis of the neighborhood markers of group 5 (h) and group 6 (i), analyzed using one-sided hypergeometric test. j, Volcano plot depicting pairwise comparisons between C9-ALS and healthy controls and sALS and healthy controls for the combined astrocytes dataset (DESeq2 negative binomial distribution; P values adjusted with Bonferroni correction on the total number of genes in the dataset). Vertical lines correspond to a log2FC = 0.2 threshold, and horizontal line corresponds to an adjusted P < 0.05 threshold. Positive log2FC values are enriched in C9-ALS (purple) or sALS (yellow); negative log2FC values are enriched in healthy controls (yellow and purple). k, Volcano plot showing a pairwise comparison of C9-ALS versus sALS for the combined astrocyte dataset (DESeq2 negative binomial distribution; P values adjusted with Bonferroni correction on the total number of genes in the dataset). Vertical lines depict a log2FC = 0.2 threshold, and horizontal line depicts the adjusted P < 0.05 threshold. Genes are colored based on the clusters in b. Positive log2FC values are enriched in C9-ALS, and negative log2FC values are enriched in sALS. P values are presented in Supplementary Tables 22 and 23. FC, fold change.
Astrocytes are heterogeneous both morphologically45 and transcriptionally46 across different CNS regions. Therefore, we wondered whether regional diversity could be underlying some of our transcriptomic observations. We performed differential abundance testing using Milo to evaluate whether certain transcriptomic profiles had a relative enrichment in the brain or spinal cord. Astrocyte transcriptional neighborhoods were categorized into seven groups (Fig. 5d,e and Supplementary Tables 30–32). The neighborhoods in groups 3, 5 and 6 showed an enrichment in the spinal cord despite not showing any association with any patient group, thus pointing to baseline transcriptomic differences in astrocytes across these two CNS regions (Fig. 5f,g). Overlaying the expression patterns of these neighborhoods with the differential expression performed across C9-ALS, sALS and controls revealed that neighborhoods in group 5 were linked to genes that were specifically upregulated in C9-ALS and involved in pathways such as ribosome, Rap 1 signaling and oxytocin signaling (Fig. 5h). On the contrary, neighborhood 6 was linked to genes specifically upregulated in patients with sALS and grouped in pathways such as MAPK, FoxO and calcium signaling (Fig. 5i). Collectively, the presence of subpopulations of astrocytes specific to the spinal cord that are transcriptionally linked to C9-ALS-associated and sALS-associated genes hints to a role of astrocyte heterogeneity in the selective vulnerability of the spinal cord in ALS, which will require further investigation.
Astrocytes are known to have a reactive profile in ALS47. We first assessed for alterations in the response of astrocytes by performing a differential expression analysis between C9-ALS versus healthy controls and sALS versus healthy controls (Supplementary Tables 21–29). Similar to what we observed in microglia, we found an overall higher extent of transcriptomic changes in spinal astrocytes from sALS compared to C9-ALS (Fig. 5f and Extended Data Fig. 7e). When diving into the nature of these changes, we observed that spinal sALS astrocytes exhibited an upregulation of genes associated with the AS3 reactive state (SERPINA3, RASGEF1B, CHI3L1, F3). On the contrary, compared to sALS, spinal C9-ALS astrocytes showed higher levels of genes characterizing AS1 and AS5 clusters and linked to axonal guidance and development as well as glutamate receptor and TGFβ signaling (AQP1, DLC1, SLC35F1) (Fig. 5g,h and Extended Data Fig. 7f). These data indicate that the C9orf72 HRE also results in a defective transition toward reactive astrocytes.
C9orf72 HRE alters microglia−astrocyte communication
Microglia−astrocyte interactions have been implicated in multiple neurodegenerative disorders and inflammatory conditions48,49,50. It is well accepted that microglia can interact and modulate the response of astrocytes, for example through secretion and physical contact51,52,53,54,55; however, the molecular mechanisms underlying this interplay remain unclear. Given that C9orf72 is primarily expressed in microglia56, we wondered whether the impact on astrocytes could be indicative of a defective microglia-to-astrocyte communication. We used CellChat to infer the enrichment of different ligand−receptor pair interactions in the spinal cord of C9 and sALS57. This analysis returned 84 significant predicted ligand−receptor interaction patterns across the three groups (Fig. 6 and Supplementary Table 32), 35 of which were differentially enriched across the experimental groups (Fig. 6). We constructed a map of cellular interactions, stratified into each of these ligand−receptor pairs into four categories based on the sender and receiver, including microglia to microglia, microglia to astrocyte, astrocyte to microglia and astrocyte to astrocyte (Fig. 6, Extended Data Fig. 8 and Supplementary Table 32). We computed the communication probability and clustered the interaction pairs based on whether they showed an increased communication in C9-ALS, sALS or controls. Additionally, we calculated the levels of expression of each ligand and receptor in every microglia and astrocyte cell state to provide a link to specific cellular phenotypes. Some of the interactions that we report here have already been shown in other diseases—for example, the SEMA4−PLEXIN tandem mediates a detrimental astrocyte−microglia connectivity in experimental autoimmune encephalomyelitis and multiple sclerosis55. Another example is the GAS6-AXL/MERTK axis, activated in microglia−microglia interactions, which have been previously characterized and are expressed in the human brain58 (Fig. 6). It was previously shown that GAS6 can negatively regulate microglial activation59, and GAS6 overexpression worsened cognitive decline in APP/PS1 mice60. On the other hand, MERTK/AXL knockout resulted in a reduced plaque load in APP/PS1 Alzheimerʼs disease mice61. This could indicate that the increased GAS6-AXL/MERTK interaction may partially explain the lack of microglial activation and could contribute to C9orf72 HRE-induced pathology. Interestingly, we found multiple putative interactions that have not been described before within the context of microglia−astrocyte communication in neurodegeneration. For example, the microglia-to-astrocyte SPP1/CD44 axis was engaged in sALS (Fig. 6). SPP1 has been shown to be increased in the cerebrospinal fuid of patients with ALS62, and CD44 expression correlates with disease progression in SOD1G93A mice63.

Here we present a unique resource compiling patterns of microglia−astrocyte communication in ALS, including ligand−receptor pairs and their expression across the different cellular states that arise in response to disease. These data identify candidate signals between microglia and astrocytes that may contribute to the pathological changes observed in ALS and serve as a resource for future functional testing of glial communication in neurodegeneration.
Discussion
Neuroinflammation is a key pathological hallmark for ALS8,64,65. Experimental and genetic evidence66,67,68 indicates that microglia and neuroinflammation are active players, rather than simple bystanders, in the pathogenic events that lead to motor neuron degeneration in ALS. Our study confirms that C9orf72 is highly expressed in microglia in the human CNS7 and that a C9orf72 HRE leads to reduced microglial C9orf72 expression. We report that C9orf72 HRE impairs microglial cell state transition toward DAM and HLA cell states when compared to sporadic cases and induces alterations in transcriptomic pathways related to phagocytosis and lysosomal function. We validated this using an HBCX model, in which the engrafted C9KO and C9orf72 HRE human microglia displayed a downregulation of HLA-related genes. In addition, we identified alterations in the endolysosomal pathways, which were validated using both LOF and patient-derived iPSC microglia in vitro. We also showed that astrocytes from C9orf72 HRE carriers display a generally lower extent of transcriptomic change compared to sporadic ALS, with a defective transition toward reactive states particularly evident in spinal cord samples. Linked to this observation, we found a specific transcriptional subtype of astrocyte enriched in the spinal cord, which could point to a contribution of these cells in the selective vulnerability of this CNS region in ALS. Finally, we provide a resource of altered microglia and astrocyte signaling pathways in sALS and C9-ALS that may serve as foundation for future studies delving into alterations in cellular networks underlying ALS.
Several proposed mechanisms explain the toxic role of the C9orf72 HRE in ALS, including LOF due to reduced C9orf72 mRNA and protein levels69. We show that C9orf72 is highly expressed in microglia compared to other cell types of the brain, as also observed in vitro in two recent studies21,70, suggesting that these cells might be particularly vulnerable to reduced C9orf72 levels. Several studies reported widespread alterations in microglia (and other myeloid cells) after conditional71,72,73,74 or complete C9orf72 deletion9,75,76, including transcriptional alterations, exacerbated production of inflammatory cytokines and lysosomal dysfunction7,56,77,78,79. In line with a predominant impact of C9orf72 LOF in microglia, we found that diminished levels of C9orf72 results in an impaired transition into DAM and HLA microglial responses. In mouse studies, upregulation of a type I interferon signature in C9orf72 knockout microglia and myeloid cells was reported27,80. Although this evidence is interesting and might suggest different effects of the C9orf72 HRE on different microglial subsets, we did not observe upregulation of type I interferon genes in C9orf72 postmortem or xenografted microglia, which can be due to a poor representation of the IRM cell state in our postmortem dataset and/or a mouse-specific microglial response to C9orf72 depletion.
A recent study by Wang et al.81 performed snRNA-seq of the prefrontal cortex from C9orf72 ALS/FTD stratified donors based on phosphorylated TDP-43 (pTDP-43) abundance. Although the extent of transcriptomic changes was limited, a reduced anti-inflammatory transcriptional program was associated with patients with high levels of pTDP-43 (ref. 81), indicating that the response of microglia could be different depending on the stage of the disease (that is, presence or absence of pTDP-43); thus, investigating microglial activation throughout the disease progression might shed light on additional microglial pathogenic mechanisms. Another recent single-cell study examining prefrontal healthy cortex samples of C9-ALS and FTD cases reported minor gene expression changes in C9-ALS versus healthy control microglia82. All these data collectively indicate a minimal microglial response to disease in the context of C9orf72 HRE. This limited response may stem from various factors, including potential variations in disease stages, as proposed by Wang et al.81, or baseline differences in the biological function of microglia. Our findings revealed more pronounced differences in spinal cord compared to regions such as the motor cortex, underscoring the importance of stratifying patients based on the disease-affected regions. It is also important to consider the parameters of comparison in these studies. In our study, although C9-ALS cases show a modest increase toward activated states when compared to controls, their lack of response to disease becomes more apparent when compared to sporadic cases. Specifically, our findings indicate that, although sporadic cases robustly respond to disease, C9-ALS microglia fail to transition to reactive states. Given these observations, additional studies will be crucial to address the following aspects: (1) stratify patients based on TDP-43 pathology and disease stages with larger sample sizes; (2) conduct a comprehensive comparison of spinal versus cortical material in C9-ALS and C9-FTD cases; and (3) evaluate responses in comparison to other patients with ALS, such as sporadic or other familial cases, to elucidate the distinct and shared cellular responses in disease. The hampered microglial transition to an activated state, as proposed in this study, is not unique to C9-ALS; similar observations exist for other neurodegeneration-linked genetic variants, such as TREM2 and APOE26,28. These results underscore distinct inflammatory substrates and microglial roles in C9orf72 and sALS pathology, and further insights are essential to define tailored therapeutic strategies for groups of patients with different pathogenic contributors.
C9orf72 localizes within lysosomes and phagosomes, and its functional alteration has been associated with disruptions in endosomal–lysosomal and autophagic pathways83. Although most of these findings were obtained from non-immune cells, recent work revealed a negative enrichment in lysosome-associated pathways and alterations in the autophagic process in C9orf72 mutant and deficient human iPSC-derived microglia21,70. In line with this, we observed enlarged CTSD-positive lysosomes in microglia carrying the repeat expansion and upon C9orf72 depletion, mirroring observations in C9orf72 knockout mice and in the motor cortex and spinal cord of patients with C9orf72 ALS56,84,85. In addition, we showed that C9-ALS microglia, compared to sALS, fail to engage in lysosomal and phagocytic pathways, which is accompanied by a defective transition into a phagocytic DAM-like state. At present, the molecular mechanisms through which C9orf72 HRE influences lysosomal and phagocytic pathways in human microglia, as well as the impact on microglial responses, require further investigation. Our in vitro data indicate that C9orf72 LOF alone is sufficient to perturb lysosomal homeostasis in human microglia, although we cannot exclude the potential contribution of GOF mechanisms70,86. Recent studies suggested a synergistic effect of C9orf72 LOF and GOF mechanisms18,84,85,87,88. In vivo studies showed that C9orf72 deficiency exacerbates motor phenotypes and premature death in a C9orf72 GOF mouse model18,84,85. It was proposed that reduced C9orf72 levels might further enhance the pathological accumulation and toxicity of DPRs in neurons18,84,87,88. Moreover, recent studies using C9orf72 GOF mouse models showed that neuronal stress induced by the expression of poly(GR) or poly(GA) activates the microglial NLRP3 inflammasome pathway89,90. As these studies were performed in mice with normal C9orf72 levels, it remains to be elucidated how neuronal stress would act on microglia in which there is a reduced expression of C9orf72.
Several co-culture studies demonstrated deleterious effects of C9orf72 astrocytes on neuronal survival91,92,93,94,95, further supporting the non-cell autonomous nature of the diseases. Despite these findings, the precise mechanisms by which C9orf72 HRE leads to astrocyte-mediated neurotoxicity remain unclear. We found no differences in C9orf72 expression levels between astrocytes in C9orf72 carriers versus healthy controls, in line with previous in vitro observations91. Intriguingly, we found distinct transcriptional profiles in astrocytes derived from sALS and C9-ALS cases. Although sALS astrocytes exhibited an upregulation of genes associated with a reactive state, C9-ALS astrocytes manifested increased levels of homeostatic genes linked to axonal guidance, glutamate receptor function and TGFβ signaling. Supporting our observations, a recent transcriptomic study on sALS postmortem tissue showed a multifaceted astrocyte landscape, with a wide variety of transcriptional states39. However, a recent single-cell study by Li et al.82 did reveal alterations in astrocytes from C9orf72 carriers with FTD and ALS, demonstrating increased activation and structural remodeling. Specifically, increased astrocyte reactivity was found in the prefrontal cortex of patients with C9-ALS/FTD. Moreover, a distinct pattern of transcriptome dysregulation was found in C9-ALS versus C9-FTD, suggesting a possible disease-specific effect of C9orf72 HRE on astrocytes82.
Given that the molecular mechanisms governing the transcriptional changes in both microglia and astrocytes are yet unclear, we sought to construct a ligand−receptor pair interaction map for both C9-ALS and sALS glia. We found several interaction partners that are predicted to be altered, possibly reflecting and altered microglia−astrocyte cellular crosstalk that could potentially contribute to disease. Targeting molecular processes in glial cells was recently successful in SOD1G93A mice, demonstrating the potential of investigating such pathways for new therapeutic opportunities62. Interestingly, beyond the reactive changes that we observed in astrocytes, additional layers of complexity must be considered, as these cells are highly heterogeneous across the CNS. Here we report transcriptomic differences in astrocytes in the spinal cord versus the cortex, with specific subsets of spinal astrocytes showing a higher baseline gene expression of C9-ALS-associated as well as sALS-associated markers. This indicates that astrocyte regional diversity may partially contribute to the increased susceptibility of the spinal cord in ALS and poses new interesting questions as to whether glial regional heterogeneity may serve as a susceptibility factor in neurodegeneration.
In summary, our study indicates that C9orf72 HRE results in reduced C9orf72 expression levels in the microglia and a subsequent reduction in their response to disease, consistent with deficits in phagocytic and lysosomal pathways. We hypothesize that this leads to a defective general glial response that extends to astrocytes, hindering a coordinated response that increases the risk for developing ALS.
Methods
Ethics statement
This study was conducted in accordance with all relevant ethical regulations and guidelines. The postmortem study protocol was reviewed and approved by the University Hospitals Leuven (UZ Leuven), Netherlands Brain Bank and the Hospital Clinic de Barcelona−d’Investgiacions Biomédiques August Pi I Sunyer (HCB-IDIBAPS) under approval numbers S65097, S59292 and S60803. All human material was collected from donors from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes was obtained by UZ Leuven. All animal experimental procedures were carried out in accordance with protocols approved by the Ethical Committee for Laboratory Animals at the University of Antwerp (ethical file 2021-13), in compliance with national and European Union regulations.
Human donor tissue
The brain and spinal cord samples were obtained from the Department of Pathology at UZ Leuven, the Netherlands Brain Bank, the Netherlands Institute for Neuroscience, the HCB and the IDIBAPS. Healthy controls are described as individuals without neurological diseases, excluding hematological and oncological conditions. Samples were obtained prior to the COVID-19 pandemic. Information on demographic data can be found in Supplementary Tables 1 and 2.
RNA isolation and RNA integrity of tissues
One milliliter of TriPure per 50−100 mg of postmortem tissue was added into a vial containing beads and postmortem tissue. The sample was homogenized in a tissue homogenizer (Magnalyser) for 3 × 30 seconds at 3,970g. The homogenate was transferred to a new RNase-free tube. Then, 200 µl of chloroform (=1/5 volume of TriPure) was added. The sample was mixed by inversion for 15 seconds and incubated at room temperature for 2−10 minutes. The samples were centrifuged for 15 minutes at 13,528g and 4 °C. Three phases became visible: an aqueous phase (colorless) containing RNA, which was transferred to a new RNase-free tube; an interphase (white color) containing DNA; and an organic phase (red color) containing protein. Only in case of low expected yields of RNA, 1 µl of glycogen was added and precipitated with 500 µl of isopropanol (=1/2 volume of TriPure). The sample was mixed by inversion for 15 seconds, incubated at room temperature for 5−10 minutes and centrifuged for 10 minutes at 13,528g and 4 °C. The supernatant was discarded, and the pellet was washed with 1 ml of 75% EtOH and centrifuged for 5 minutes at 13,528g and 4 °C. The supernatant was again discarded, and, after drying of the pellet, RNase-free water was added. Per sample, 1 µg of total RNA was used as input. RNA concentration and purity were determined spectrophotometrically using a NanoDrop ND-1000 (NanoDrop Technologies), and RNA integrity was assessed using a Bioanalyzer 2100 (Agilent).
Nuclei isolation of postmortem human brain and spinal cord tissues
To optimize nuclei isolation96, starting from 50 mg of tissue, we tested the standard nuclei isolation protocol by Habib et al.97, Tween with salts and Tris (TST)96 and Soma-seq98. We observed marginal advantages using the Soma-seq protocol (Extended Data Fig. 1b) and proceeded with it for the following experiments.
Standard nuclei isolation protocol
The tissue was transferred immediately into a homogenizer containing 1 ml of ice-cold homogenization buffer (320 mM sucrose, 5 mM CaCl2, 3 mM Mg(OAc)2, 10 mM Tris 7.8, 0.1 mM EDTA, 0.1% IGEPAL CA-360, 0.1 mM phenylmethylsulfonyl fluoride, 1 mM β-mercaptoethanol with 5 µl of RNasin Plus). Tissues were homogenized with 10 manual gentle strokes (pestle A) plus 10 manual gentle strokes (pestle B). The homogenate was filtered through 70-µm cell mesh and strainer and washed with 1.65 ml of homogenization buffer to a final volume of 2.65 ml. The nuclei suspension was supplemented with 2.65 ml of gradient medium (5 mM CaCl2, 50% OptiPrep, 3 mM Mg(OAc)2, 10 mM Tris 7.8, 0.1 mM phenylmethylsulfonyl fluoride, 1 mM β-mercaptoethanol). A 29% cushion was prepared by diluting OptiPrep with OptiPrep Diluent Medium (150 mM KCl, 30 mM MgCl2, 60 mM Tris (pH 8.0), 250 mM sucrose). The nuclei suspension in the homogenization buffer + gradient medium mix was then layered over the 29% cushion and ultracentrifuged in an SW 41 Ti Swinging-Bucket Rotor (Beckman Coulter) at 5,574g and 4 °C for 30 minutes. The upper supernatant and lower supernatant were removed, respectively, with a Pasteur pipette and a P200 micropipette. Nuclei were resuspended in 50 µl of resuspension buffer (PBS, 1% BSA) and transferred to a new tube. Resuspended nuclei were counted using a LUNA dual fluorescence cell counter (Logos Biosystems).
TST nuclei isolation protocol
The tissue was transferred immediately into a homogenizer containing 1 ml of ice-cold homogenization buffer (146 mM NaCl, 10 mM Tris 7.5, 1 mM CaCl2, 21 mM MgCl2, 0.03% Tween 20, 1% BSA, 25 mM KCl, 250 mM sucrose, 1 mM β-mercaptoethanol, 0.5× cOmplete Protease Inhibitor (Roche), 0.2 U µl−1 RNasin Plus Ribonuclease Inhibitor (Promega)). The tissue was homogenized with 15 gentle strokes with a pestle and douncer. The resulting nuclei suspension was filtered using a 40-µm cell strainer. The filtrate was transferred into a low binding tube and centrifuged at 500g for 5 minutes. The supernatant was discarded, and the pellet was resuspended thoroughly in 1 ml of homogenization buffer without Tween (146 mM NaCl, 10 mM Tris 7.5, 1 mM CaCl2, 21 mM MgCl2, 1% BSA, 25 mM KCl, 250 mM sucrose, 1 mM β-mercaptoethanol, 0.5× cOmplete Protease Inhibitor (Roche), 0.2 U µl−1 RNasin Plus Ribonuclease Inhibitor (Promega)) and transferred to a 15-ml Falcon. An additional 1.65 ml of homogenization buffer without Tween buffer was added to a final volume of 2.65 ml. The rest of the protocol for density centrifugation and nuclei enrichment was performed as in the standard protocol.
Soma-seq nuclei isolation protocol
The frozen tissue was immediately transferred into a homogenizer containing of 1 ml of ice-cold homogenization buffer (10 mM Tris 8.0, 5 mM MgCl2, 25 mM KCl, 250 mM sucrose, 1 mM β-mercaptoethanol, 0.5× cOmpleteProtease Inhibitor (Roche), 0.2 U µl−1 RNasin Plus Ribonuclease Inhibitor (Promega)). The tissue was homogenized with 15 gentle strokes with a pestle and douncer. The nuclei suspension was filtered using a 70 -µm cell strainer. The filtrate was transferred into a low binding tube and centrifuged at 400g for 5 minutes. The supernatant was discarded, and the pellet was resuspended thoroughly in 1 ml of homogenization buffer and transferred to a 15-ml Falcon. An additional 1.65 ml of homogenization buffer was added to a final volume of 2.65 ml. The density centrifugation and nuclei enrichment were performed as in the standard nuclei isolation protocol.
Optimization of single-nuclei isolation and sequencing
We performed snRNA-seq of spinal cord and motor cortex from healthy controls and from patients with C9-ALS and sALS and compared the three methods for number of reads, genes and unique molecular identifiers (UMIs). Both the TST and Soma-seq protocols performed well (Extended Data Fig. 1b). All following experiments followed the Soma-seq method.
Library preparation
cDNA libraries for the scRNA-seq were prepared using 10x Genomics Chromium Single Cell 3’ Kit version 3.1 NextGEM chemistry (10x Genomics). The cell count and the viability of the samples were assessed using a LUNA dual fluorescence cell counter (Logos Biosystems) and loaded to the Chromium at a targeted nuclei recovery of 6,000 nuclei per sample. Samples (up to three) were pooled in equal numbers and loaded to a single 10x reaction. scRNA-seq libraries were prepared using the manufacturerʼs recommendations (Single Cell 3’ Reagent Kits version 3.1 user guide; CG000204 Rev D), and, at the different checkpoints, the library quality was accessed using a Qubit (Thermo Fisher Scientific) and a Bioanalyzer (Agilent). cDNA libraries were sequenced with an expected coverage of 50,000 reads per cell on Illumina’s NovaSeq 6000 platform using paired-end sequencing workflow and with recommended 10x version 3.1 read parameters (28-8-0-91 cycles). Library preparation took place in the Fall of 2020.
Data analysis
Data preprocessing
Raw sequencing data were demultiplexed using the Cell Ranger (5.0.1) mkfastq command; FASTQ files were processed using Cell Ranger count with the –include-introns enabled and using the GRCh38-2020-A index provided by 10x Genomics. Cells passing the default Cell Ranger filter were kept for subsequent analyses. Samples that were multiplexed for droplet generation were demultiplexed into patient-specific matrices. First, a library of single-nucleotide polymorphisms (SNPs) present in the general population identified by the 1000 Genomes Project was obtained from http://ftp.1000genomes.ebi.ac.uk specifically; files ALL.chr(1-22,X,Y)_GRCh38_sites.20170504.vcf.gz were obtained and merged into a single vcf file. Chromosomes were renamed to match the scheme used in the index provided by 10x Genomics using BCFTools annotate. SNPs were filtered to remove rare variants and to only include SNPs in the regions of the genome included in the Cell Ranger index using BCFTools filter with the parameters –regions-file (with a BED file containing the abovementioned regions) and -i ‘INFO/AF[0] < 0.90 & & INFO/AF[0] > 0.10’. This final vcf file was used to run freemuxlet from the popscle package (https://github.com/statgen/popscle) with default parameters other than nsample = 2 and using the tools from 7.0 to speed up computation. We used verifyBamID (http://griffan.github.io/VerifyBamID/) to compare the freemuxlet genotype to the bulk RNA-seq data of patient; the highest-scoring match was used to assign patient labels to freemuxlet clusters.
Data processing and cell type annotation
Libraries were individually processed using Scanpy 1.7.2 (ref. 99). First, patient labels and associated metadata were added to the cells, any doublets identified by freemuxlet being removed. Next, scrublet was used to remove remaining identifiable doublets (threshold = 0.25), and samples were combined into two datasets based on their tissue of origin: motor cortex and spinal cord. Nuclei with fewer than 250 genes or more than 5% of total counts associated with mitochondrial genes were removed. The resulting matrices were normalized (to 10,000 counts) and log transformed, and highly variable genes were identified using default parameters. Number of UMIs and percentage of mitochondrial reads were regressed out, and the data were scaled to a maximum of 10. A principal component analysis (PCA) was used to reduce the dimensions of the data; Harmony100 was used to integrate the PCA components based on the ‘Patient’ batch key; and the pcacv workflow from vsn-pipelines (https://doi.org/10.5281/zenodo.3703108) was then used to determine the number of principal components to continue with (76 for motor cortex and 82 for spinal cord). Neighborhood embedding, uniform manifold approximation and projection (UMAP) and t-distributed stochastic neighbor embedding (tSNE) were calculated using the number of principal components listed above, and clusters were identified at various resolutions (0.4, 0.8, 1.0, 1.2, 1.6, 2.0, 4.0, 8.0) using the Leiden algorithm. The resulting data were exported to a loom file and uploaded to SCope; cell type annotations were provided via SCope by experts based on marker gene expression. All plots were generated with ggplot2 (version 3.3.5) in R version 4.1.2.
Processing of the microglia and astrocytes
To investigate microglia subsets, we isolated microglia nuclei from both the motor cortex and spinal cord and merged them using Seurat (version 4.1.0) under R version 4.1.2 (ref. 101). The data were log normalized, and the highest variable genes were identified, followed by scaling of the data and PCA. The data were batch corrected with Harmony (version 0.1.0) based on patient identifiers and location. Cells were clustered using the first 20 principal components, and UMAP plots were generated with standard methods. Five other cell subsets were identified (peripheral macrophages n = 898, synaptosomes n = 821, oligodendrocytes n = 200 and proliferating cells n = 143). The clusters contained low expression of microglia markers CXCR3 and P2RY12 with subset-specific markers for peripheral macrophages (CD163, MRC1, F13A1), oligodendrocytes (MBP, SHROOM4, PLP1, PCDH9, EDIL3) and synaptosomes/astrocytes (SNAP25, RORA, DPP10, ROBO2, LSAMP). The oligodendrocytes were most likely doublets of microglia and oligodendrocytes, the same for the synaptosomes. Proliferating cells (TOP2A, MIK67, MELK, KIF11) were excluded from final analysis. Next, we extracted the astrocytes (n = 23,483) from the spinal cord and motor cortex and re-identified the astrocyte population. Apart from astrocytes, two other clusters were identified: one of oligodendrocytes n = 500 cells (ELMO1, IL1RAPL1, SLC24A2, SHROOM4) and one with doublet/low-quality n = 348 cells (PCLO, WARS, RUNX1), the latter being a cluster with markers of multiple cell types that could not be confidently annotated. All plots were generated with ggplot2 (version 3.3.5) in R version 4.1.2.
Processing of microglia and astrocytes from Gerrits et al
Individual libraries were extracted from GSE163122, and nuclei with at least 250 features and features in at least three cells were retained. Scrublet was used to remove doublets as described previously. Nuclei with a number of features higher or lower than mean ±2s.d. and more than 5% mitochondrial transcripts were excluded. All libraries were integrated, resulting in a total of 402,569 nuclei. Microglia and astrocytes were extracted individually and integrated with the microglia and astrocytes from the ALS cohort using Seurat r-PCA in R version 4.1.2.
Differential expression
Differential expression testing was performed using DESeq2 in Seurat FindMarkers. Genes with adjusted P < 0.05 and log2 fold change ± 0.20 were considered as differentially expressed. Heatmaps were generated with ComplexHeatmap_2.10.0.
Gene enrichment analysis
Specific microglia functions were assessed by extracting human-specific genes (taxon: 9606), three Gene Ontology terms (GO:0005764, GO:0006954 and GO:006906) and one pathway in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (HSA00190).
WGCNA
Matrices for microglia and astrocytes were extracted from each tissue and were used to perform WGCNA102 to identify modules of co-expressed genes using the hdWGCNA package (version 0.1.1.9005)103. Genes were filtered to include only those expressed within at least 5% of the cells within a dataset using the SetupForWGCNA function, and MetacellsByGroups was used to create metacells by the combination of ALS status, patient and library of origin using values of 25 for the number of cells to be combined and a maximum number of shared cells of 10. Finally, metacells were normalized using NormalizeMetacells. Next, the groups of interest (healthy, sALS and C9-ALS) were selected to build the co-expression network using SetDatExpr, and TestSoftPowers was used per dataset to select a soft threshold (Spinal Cord – Astrocytes: 5, Spinal Cord – Microglia: 7, Motor Cortex – Astrocytes: 5, Motor Cortex – Microglia: 4). ConstructNetwork was used with default parameters to generate the co-expression networks and modules. Finally, module eigengenes were corrected for between-patient batch effects using Harmony.
WGCNA modules−trait relationships
To identify WGCNA genes modules that may be correlated with ALS status, a one-way ANOVA (via the scipy Python package version 1.7.3) was performed using the module eigengene values for each module across all datasets. P values across all tests were corrected using Benjamini−Hochberg correction (via the statsmodels Python package version 0.12.2). A Tukey honestly significant difference test was then performed (also using statsmodels) on modules discovered in the ANOVA as having at least one significantly different mean, with the goal of identifying the specific statuses that are different.
Gene Ontology enrichment analysis
Gene Ontology enrichment was performed using the g:OSt functionality of gProfiler104 via the gprofiler-official Python package (version 1.0.0).
Differential abundance testing and correlations with MiloR
Differential abundance testing was performed using MiloR (2.0.0) in R version 4.4.1 (ref. 24). Microglia and astrocytes were randomly downsampled to equal numbers of nuclei per each location/patient. Milo was run with a proportion of 0.2 as input sample and k = 15 and d = 15 as well as a false discovery rate set of 0.1. The differences in regional abundance in neighborhoods were tested as a comparison of the brain versus spinal cord.
Single-cell analysis of xenografted human microglia
The FASTQ files were aligned by Cell Ranger (version 7.1.0) against the human genome database (GRCh38, Ensembl 98, refdata-gex-GRCh38-2020-A). Filtered count matrices were imported in R (version 4.4.0) for data analysis. The datasets were analyzed using the Seurat R package (version 5.1.0). Cells were demultiplexed with HTODemux with positive.quantile = 0.99. Subsequently, low-quality cells (thresholds defined by is.Outlier in scuttle 1.14.0, with high and/or low library size; a high percentage of reads mapping to mitochondrial genes; or cells with a low number of detected genes) were removed from each individual library. For one of the libraries, the high nCount threshold was determined to be 33,306; this threshold was adjusted to 26,439, aligning it with the maximum value observed in the other libraries. Individual libraries were processed with SCTransform with the parameters layer = ‘counts’ and vst.flavor = ‘v2’. PCA was calculated on the ‘SCT’ assay. Layers were integrated with rPCA on the SCTransformed data. Standard clustering analysis was performed in Seurat on the integrated dataset—that is, FindNeighbors() and FindClusters with resolution parameter set to 0.1 and, finally, RunUMAP(). Microglia (n = 21,826), proliferating microglia (n = 425), macrophages (n = 2,591) and doublets (n = 522) were assigned. Subsequent analysis was performed on microglia alone. Cells were clustered using the first 26 principal components, and UMAP plots were generated with Seurat standard methods. Differentially expressed genes were calculated with Seurat’s FindMarkers using default settings. Seurat version 4 fold changes were calculated according to Seurat version 4. Plots were generated in R version 4.4.2 with ggplot2 version 3.5.1.
Microglia−astrocyte communication with CellChat
Microglia−astrocyte communication was assessed with CellChat (version 1.5.0)57 in R version 4.1.2. Microglia and astrocytes transcriptomes were extracted from the spinal cord and combined to generate a CellChat object per patient group using CellchatDB.human. Overexpressed genes and interactions were detected with default settings before computing and filtering the communication probabilities and calculating centrality scores. CellChat objects of the different patient groups were merged to assess differences in communication patterns with mergeCellChat. For the identification of pairwise changes, computeNetSimilarityPairwise was used. Average expression of the ligands and receptors was extracted from Seurat (version 5.1.0) with AverageExpression. Plots were generated in R version 4.4.2 with ggplot2 version 3.5.1.
Western blot
Quantification of CTSD levels was performed on brain lysates from healthy controls, C9-ALS cases and sALS cases. Protein extraction was carried out by transferring 50 mg of tissue into 1.5-ml RNase-free microfuge tubes (Thermo Fisher Scientific, AM12400). Next, 0.5 ml of 2% sodium dodecyl sulphate (SDS) containing TBS and protease inhibitor (cOmplete, EDTA-free protease inhibitor cocktail; Sigma-Aldrich, 1836170001) was added. Each sample was mechanically homogenized by using pellet pestles (Sigma-Aldrich, Z359971) under a laminar flow without putting the samples on ice. The samples were centrifuged at 20,000g for 30 minutes at room temperature. The supernatant was transferred to a new 1.5-ml RNase-free microfuge tube, and the pellet was discarded. Protein quantification was performed on the supernatant using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific, 23235) according to the manufacturer’s instructions. Subsequently, samples were supplemented with SDS containing reducing sample buffer (Thermo Fisher Scientific, 39000), boiled for 10 minutes at 95 °C and loaded into 4–20% precast polyacrylamide gels (Bio-Rad, 4568094). After electrophoresis (100 V, 1.5 hours), the gel was transferred onto a 0.2-µm PVDF membrane (trans-blot turbo transfer pack; Bio-Rad, 1704156) for 10 minutes at 1.5 Å. After transfer, the membrane was blocked with 5% non-fat dried milk for 1 hour at room temperature (Sigma-Aldrich, M7409-1BTL) diluted in TBS (Sigma-Aldrich, T5912-1L) with Tween (Sigma-Aldrich, P1379-500ML) (TBST). We incubated the membrane with anti-CTSD (1/1,000; Abcam, ab75852) and GAPDH (1/1,000; Cell Signaling Technology, 2118) and imaged them with an ImageQuant LAS 4000 Biomolecular Imager (GE Healthcare). Protein bands were quantified with ImageQuant TL version 7.0 software (Cytiva). Samples (n = 2 healthy controls, n = 5 patients with C9-ALS and n = 5 patients with sALS) were run in parallel across two independent membranes.
iPSC lines
The C9orf72 HRE iPSC lines used (C9-1 and C9-2 and corresponding isogenic controls used for in vivo and in vitro experiments, respectively) were previously generated and characterized37. The C9KO line was generated using a SYNTHEGO multi-guide system targeting exon 2 of the C9orf72 gene, as described in ref. 38. IPSC lines were cultured on 0.5 mg ml−1 growth-factor-reduced Matrigel (Corning) with mTeSR 1 basal medium, supplemented with mTeSR 1 5× supplement (STEMCELL Technologies), 100 U ml−1 penicillin−streptomycin (Thermo Fisher Scientific) and 100 U ml−1 RevitaCell (Thermo Fisher Scientific). The experiments were performed under ethical approval 2021-1705 by the Antwerp University Hospital.
Microglial differentiation
Microglia progenitors were produced as previously described in Fattorelli et al.35. At day 25 or day 32 of the microglial differentiation protocols, cells were plated in TIC medium and allowed to mature into microglial-like cells for 10 days. Medium change was performed every 2 days. TIC medium consisted of DMEM/F12 (Thermo Fisher Scientific), supplemented with 2 mM L-glutamine (Thermo Fisher Scientific), 5 µg ml−1 N-acetyl-L-cysteine (Merck), insulin (1:2,000) (Merck), 100 µg ml−1 apo-transferrin (Merck), 100 ng ml−1 sodium selenite (Merck), 1.5 µg ml−1 cholesterol (Merck), 1 µg ml−1 heparan sulphate (Merck), 50 ng ml−1 M-CSF, 50 ng ml−1 IL-34 (STEMCELL Technologies), 25 ng ml−1 TGFβ (STEMCELL Technologies) and 10 ng ml−1 CX3CL1 (STEMCELL Technologies). For in vitro experiments, no statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications21,70. When sample size allowed, data were assessed for normality. Data were normally distributed unless otherwise stated.
Bulk RNA-seq
Cells were harvested and RNA was extracted using an RNeasy Micro Kit (Qiagen, 74004), and sequencing was performed by BGI Genomics. The analysis was performed in-house with a standardized pipeline integrated in GenomeComb105. The pipeline used fastq-mcf for adapter clipping. Reads were aligned to the hg38 genome reference106 using STAR107 in two-pass mode, and the resulting SAM file was converted to CRAM using SAMtools108. BAM files were sorted109,110, and, at the initial stage, positions with a coverage <8 or a genotype quality score <25 were considered unsequenced. Gene counts were obtained using RNA-SeQC111. The matrix with gene counts was read into R. Genes with zero count in all samples were removed. Raw gene counts were normalized using size factors estimated via the median ratio method described by Anders and Huber112 and subsequently transformed using variance stabilizing transformation (VST) from the DESeq2 package (version 1.44.0) with blind = TRUE to stabilize variance across the mean113. Plots were generated in R version 4.4.2 with ComplexHeatmap version 2.22.0.
Xenotransplantation of microglia precursors into mouse brain
We xenotransplanted microglial progenitors from C9KO, CTRL, C9-HRE and C9-ISO into the brain of Rag2−/−Il2g−/−hCSF1KI immunodeficient mice (male and female), as described in Fattorelli et al.35. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications26. We transplanted approximately 500,000 cells per mouse. We used Total-Seq A hashing antibodies (BioLegend) to demultiplex individual mouse replicates. Mice were provided with unrestricted access to food and water and kept in groups of 2−5 under a 14-hour light/10-hour dark cycle at a temperature of 21 °C and a relative humidity of 32%.
Human microglia isolation for scRNA-seq
At 3 months (C9-HRE and C9-ISO) or 6 months (CTRL and C9KO) of age, xenotransplanted mice were euthanized with an overdose of sodium pentobarbital and immediately perfused with ice-cold 1× DPBS (Gibco, 14190-144) supplemented with 5 U of heparin (LEO). After perfusion, one brain hemisphere per mouse was placed in FACS buffer (1× DPBS, 2% FCS, 2 mM EDTA) + 5 µM actinomycin D (ActD; Sigma-Aldrich, A1410-5MG) for transcriptomics. Brains were dissociated using Miltenyi Neural Tissue Dissociation Kit P (Miltenyi Biotec, 130-092-628) supplemented with 5 µM ActD. Samples were then passed through a 70-µm strainer (BD2 Falcon), washed in 10 ml of ice-cold FACS buffer with 5 µM ActD and spun at 300g for 15 minutes at 4 °C. ActD was included during tissue collection and enzymatic dissociation to stop transcriptional activity, and it was stopped after the myelin removal step to minimize toxicity from prolonged exposure. After dissociation, myelin was removed using 30% isotonic Percoll (GE Healthcare, 17-5445-02) solution. Cells resuspended in Percoll were centrifuged at 300g for 15 minutes at 4 °C. After centrifugation, layers of myelin and cellular debris were discarded, and FC receptors were blocked in FcR blocking solution (1:10; Miltenyi Biotec, 130-092-575) in cold FACS buffer for 10 minutes at 4 °C. Cells were then washed in 5 ml of FACS buffer, pelleted and incubated with the following antibodies: PE-Pan-CD11b (1:100; Miltenyi Biotec, 130-113-806), BV421-mCD45 (1:100; BD Biosciences, 563890), APC-hCD45 (1:50; BD Biosciences, 555485), Total-Seq A cell hashing antibodies (1:500; BioLegend) and viability dye (1:2,000; eFluor 780; Thermo Fisher Scientific, 65-0865-14), in cold FACS buffer for 30 minutes at 4 °C. Cells were then washed, pelleted down and resuspended in 500 µl of FACS buffer. Cells were passed through a 35-µm strainer before sorting and loaded into the input chamber of a MACSQuant Tyto Cartridge. Human cells were sorted based on CD11b and hCD45 expression at 4 °C (Extended Data Fig. 5a).
Single-cell library preparation and sequencing
For scRNA-seq, 15,000–20,000 human microglia (CD11b+hCD45+) from each mouse were sorted on a MACSQuant Tyto and diluted to a final concentration of 1,000 cells per microliter. In total, 2,000 human microglia per mouse were pooled and loaded onto a Chromium Next GEM Chip G (PN no. 2000120). The DNA libraries were generated following the manufacturer’s instructions (CG000315 Chromium Next GEM Single Cell 3′ Reagent Kits version 3.1 Dual Index). Hashtag oligo libraries were prepared according to the manufacturer’s instructions (BioLegend, Total-Seq A Antibodies and Cell Hashing with 10x Single Cell 3′ Reagent Kit version 3 3.1 Protocol) using 16 cycles for the index polymerase chain reaction (PCR). A total of five libraries were sequenced, targeting a 90% mRNA and 10% hashtag oligo library (50,000 reads per cell), on a DNBSEQ-G400 platform following the 10x Genomics workflow. Libraries were sequenced by BGI Genomics.
Immunocytochemistry
Cells were fixed using 4% paraformaldehyde (Thermo Fisher Scientific) for 15−20 minutes and washed three times, 5 minutes, with PBS. Permeabilization was performed for 20 minutes at room temperature, using permeabilization solution (0.1% Triton X-100 (Thermo Fisher Scientific) in PBS). Samples were incubated for 30 minutes at room temperature in blocking solution containing 5% normal donkey serum (NDS) (Bio-Connect) and 0.1% Triton X-100 in PBS. Next, the cells were incubated with the primary antibodies diluted in PBS containing 0.1% Triton X-100 and 2% NDS at 4 °C overnight. The cells were rinsed three times, 5 minutes, with PBS containing 0.1% Triton X-100 and incubated with the secondary antibodies for 1 hour at room temperature. The cells were washed three times, 5 minutes, with PBS containing 0.1% Triton X-100 and stained for 20 minutes at room temperature with Hoechst (Thermo Fisher Scientific). Coverslips were rinsed three times with PBS and mounted on Superfrost Plus Adhesion Microscope Slides (Epredia) using Dako Fluorescence Mounting Medium (Agilent). The cells were visualized under an LSM 700 confocal laser scanning microscope or with a Zeiss LSM 900 with Airyscan 2, using ZEN software (Zeiss). Images were analyzed, formatted and quantified with ImageJ software (Cell Counter plugin). Anti-Iba1 (Abcam, ab5076, 1:500, goat), anti-CTSD (Abcam, ab75852, 1:500, rabbit), Lamp1 (DSHB, ab2296838, 1:200, mouse), EEA1 (SYSY, 237002, rabbit, 1:100) and Rab7 (Thermo Fisher Scientific, PA5-52369, rabbit, 1:100) primary antibodies were used. As secondary antibodies, donkey anti-goat (Thermo Fisher Scientific, A32758, 1:1,000), donkey anti-rabbit (Thermo Fisher Scientific, A31573, 1:1,000, and Biotium, 20802, 1:500) and donkey anti-mouse (Biotium, 20015, 1:500) were used.
Expansion microscopy
For expansion microscopy, a protocol based on Chozinski et al.112 was used. Microglia progenitor cells were plated in a 24-well plate on 12-mm glass coverslips (Thermo Fisher Scientific, 11708701). The cells were differentiated for 10 days and fixed for 10 minutes at room temperature with a solution containing 3.2% paraformaldehyde and 0.1% glutaraldehyde, followed by a wash with PBS. Next, the cells were reduced for 5 minutes at room temperature using 10 mM NaBH4 (Merck). Cells were washed three times for 5 minutes with PBS. After fixation, cells were blocked with normal serum (1:500) and 0.5% Triton X-100 in PBS-T (PBS with 0.02% Triton X-100 and 0.5% BSA) for 1 hour at room temperature or overnight at 4 °C. Incubation with primary antibodies was performed in PBS-T for 1 hour at room temperature or overnight at 4 °C. After washing steps (four for 5 minutes and one for 30 minutes), samples were incubated with secondary antibody diluted in PBS-T for 1 hour at room temperature or overnight at 4 °C and washed twice in PBS-T for 5 minutes and three times in PBS for 5 minutes. Nuclear staining with Hoechst was performed for 5 minutes in 1/20,000 dilution, followed by four washes in PBS for 5 minutes. Samples were then treated with an anchoring solution (0.25% glutaraldehyde in PBS) for 10 minutes at room temperature and washed three times in PBS. Next, a solution made of monomer solution (1× PBS, 2 M NaCl, 2.5% acrylamide (Bio-Rad, 1610140), 0.15% bisacrylamide (Bio-Rad, 1610142), 8.625% sodium acrylate (Sigma-Aldrich, 408220), 0.2% ammonium persulfate (APS) solution, 0.2% TEMED (ChemCruz, sc-29111)) was added to the samples for 1 hour at 37 °C. After the polymerization of the gel, a digestion step was performed. Digestion mix was made of 1× TE, 0.5% Triton X-100, 0.8 M guanidinium hydrochlodride and 8 U ml−1 proteinase K. Samples were incubated with digestion mix for 30 minutes at 37 °C. The gel was then expanded using high-purity water, which was replaced every 15−30 minutes as the gel expanded for typically 45−90 minutes. The gel was then cut to fit a WillCo dish (50 mm), and a 2% agar solution (UltraPure LMP Agarose; Invitrogen, 16520-050) was added to fill the dish completely. Once the agar solidified, samples were stored at 4 °C until processing. The cells were visualized under an LSM 700 confocal laser scanning microscope (Zeiss) at a magnification of ×63 using a water immersion C-Apochromat 1.2 W Korr ×63 objective UV-VIS-IR (Zeiss) and imaged with ZEN software (Zeiss).
TEM
For TEM experiments, microglia progenitor cells were plated in a 24-well plate on glass coverslips. Cells were differentiated for 10 days and then fixed using a solution containing a 1:1 dilution of medium and fixative (4% formaldehyde (ProSciTech) and 2.5% glutaraldehyde (Merck)) dissolved in 0.1 M cacodylate buffer (Merck) for 10 minutes, followed by 1-hour incubation with fixative only. Both incubations were performed at room temperature on a shaking plate. After fixation, the cells were washed three times for 30 minutes with 0.1 M cacodylate buffer. Cells were post-fixed in 1% OsO4 (Aurion, 19150) at 4 °C overnight on a shaking plate. After washing in 0.1 M cacodylate buffer, cells were left for 2 hours in 1% uranyl acetate at 4 °C on a shaking plate. Cells were then dehydrated through a graded ethanol series (7%, 15%, 30%, 50%, 70%, 95%, 100% ethanol). Dehydration was followed by embedding in Spurr’s resin for 8 hours. The polymerization was performed at 70 °C overnight. The cells were visualized under a Jeol JEM-1400plus TEM microscope using TEM Center software (Jeol). The captured images were analyzed using Ilastik (Anna Kreshuk’s laboratory at the European Molecular Biology Laboratory) by building a machine learning classifier (pixel classification workflow) that segments (1) background (no cells), (2) late-endosomal/lysosomal and (3) cytoplasmic/nuclear regions. The resulting binary masks were used to extract the fraction of late-endosomal/lysosomal pixels to all non-background pixels per image (Fig. 4g). In Extended Data Fig. 6f, images were quantified with ImageJ software (Cell Counter plugin).
Live-cell imaging
For the live-cell imaging, the microglia progenitor cells harvested at days 25 and 32 were plated in an eight-well glass chamber slide (ibidi) in TIC medium (50,000 cells per well) for 10 days. At day 10, pHrodo Green E. coli BioParticles (Thermo Fisher Scientific, P35366, 20×) were added to the iPSC microglia. pHrodo particles are designed to have minimal fluorescence under neutral pH and become increasingly fluorescent in an acidic environment. The plate was loaded into a Zeiss Axiovert imaging system, and the cells were maintained at 37 °C and 5% CO2 to monitor the real-time uptake and degradation of pHrodo BioParticles. The cells were imaged using a ×20 objective at an interval of 15 minutes for 15 hours (900 minutes). To determine the phagocytic and degradation index of each genotype (C9KO and CTRL), images were analyzed using ImageJ. Raw.zvi files containing three-slice z stacks (acquired during imaging) were first processed to generate a maximum intensity projection. In each projection, individual cells were segmented to delineate the regions of interest (ROIs) corresponding to cell areas. Next, the green fluorescence emitted by pHrodo-labeled particles was quantified for each ROI. Background intensity was subtracted from the integrated density measurements and ROI areas. Finally, the summed background-corrected integrated green fluorescence values for each well were calculated and further plotted using RStudio. The median of the pHrodo background-subtracted intensity was first calculated to summarize intra-well measurements (n = 3 images per well) per each timepoint. These values were then further aggregated by computing the median across wells (n = 3 wells for each independent replicate). For visualization, the processed data from the different individual replicates (n = 6 per genotype) were plotted using ggplot2, with geom_smooth() applied to depict temporal trends. This function fits a locally smoothed curve (LOESS) to the data (Fig. 4j).
Phagocytosis assay by flow cytometry
At days 25 and 32, microglia progenitors were plated in a 12-well plate on glass coverslips in TIC medium (500,000 cells per well). After 10 days, cells were incubated either for 1 hour at 37 °C with pHrodo Green E. coli BioParticles (Thermo Fisher Scientific, P35366, 20×) or for 3 hours at 37 °C with β-amyloid (1−42, HiLyte Fluor 488-labeled, AS-60479-01, Anaspec, 2 µM). After three washes with DPBS (1×, Thermo Fisher Scientific), cells were incubated for 15 minutes at 37 °C with 500 µl of viability dye (e780; Thermo Fisher Scientific, 65-0865-14) in 2000 µl of Accutase. After 15 minutes, the cells were collected in 3.5 ml of MACS buffer (0.5% BSA (Miltenyi Biotec) in 1× DPBS) and centrifuged for 5 minutes at 300g. After centrifugation, the supernatant was removed, and the pellet was resuspended in 100 µl of MACS buffer and transferred to an Eppendorf tube and placed on ice. The phagocytic activity of the cells was measured as median fluorescence intensity with a MACSQuant10 analyzer (Miltenyi Biotec) using MACSQuantify software (Miltenyi Biotec). The obtained data were analyzed by FlowJo software (BD Biosciences).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data of human postmortem samples (https://scope.aertslab.org/https://scope.aertslab.org/Raw) have been deposited in the European Genome-phenome Archive with data accession no. EGAS00001006711 (to access the data itself under restricted access). Requests for accessing raw sequencing data will be reviewed by the VIB Data Access Committee (dac@vib.be). Any data shared will be released via a data transfer agreement that will include the necessary conditions to guarantee protection of personal data (according to European General Data Protection Regulation law). Further information and requests for resources and reagents should be directed to, and will be fulfilled by, P.M. (pegah.masrori@kuleuven.be) and P.V.D. (philip.vandamme@kuleuven.be). An online platform for user-friendly access and visualization of the data shown in this study is available and can be accessed through https://mancusolab.bioinf.be/C9orf72/c9orf72-als-microglia.html. The sequencing data of iPSC-derived human microglia in vitro and in vivo experiments are accessible at the Gene Expression Omnibus database under GSE302320. The datasets from Gerrits et al. can be found at GSE16312. Source data are provided with this paper and available at https://scope.aertslab.org/#/Masrori_Nat_Neuro.
Code availability
The code used for the transcriptomic data analysis is available upon reasonable request.
References
Masrori, P. & Van Damme, P. Amyotrophic lateral sclerosis: a clinical review. Eur. J. Neurol. 27, 1918−1929 (2020).
Ilieva, H., Polymenidou, M. & Cleveland, D. W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 (2009).
Mizielinska, S. et al. Amyotrophic lateral sclerosis caused by hexanucleotide repeat expansions in C9orf72: from genetics to therapeutics. Lancet Neurol. 24, 261–274 (2025).
Dedeene, L. et al. Dipeptide repeat protein and TDP-43 pathology along the hypothalamic−pituitary axis in C9orf72 and non-C9orf72 ALS and FTLD-TDP cases. Acta Neuropathol. 140, 777–781 (2020).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Gendron, T. F. & Petrucelli, L. Disease Mechanisms of C9ORF72 repeat expansions. Cold Spring Harb. Perspect. Med. 8, a024224 (2018).
Rizzu, P. et al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 4, 37 (2016).
Masrori, P., Beckers, J., Gossye, H. & Van Damme, P. The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol. Neurodegenr. 17, 22 (2022).
Atanasio, A. et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 6, 23204 (2016).
Amick, J. & Ferguson, S. M. C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Traffic 18, 267–276 (2017).
Blum, J. A. et al. Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons. Nat. Neurosci. 24, 572–583 (2021).
Wang, F. et al. Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level. Nat. Commun. 13, 3620 (2022).
Hardwick, S. A. et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 40, 1082–1092 (2022).
Zhong, S. et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 555, 524–528 (2018).
Gautier, O. et al. Challenges of profiling motor neuron transcriptomes from human spinal cord. Neuron 111, 3739–3741 (2023).
Netskar, H. et al. Pan-cancer profiling of tumor-infiltrating natural killer cells through transcriptional reference mapping. Nat. Immunol. 25, 1445–1459 (2024).
Yang, C. et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat. Commun. 10, 3931 (2019).
Shi, Y. et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325 (2018).
Belzil, V. V. et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905 (2013).
Jackson, J. L. et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol. Neurodegener. 15, 7 (2020).
Banerjee, P. et al. Cell-autonomous immune dysfunction driven by disrupted autophagy in C9orf72-ALS iPSC-derived microglia contributes to neurodegeneration. Sci. Adv. 9, eabq0651 (2023).
Gerrits, E. et al. Neurovascular dysfunction in GRN-associated frontotemporal dementia identified by single-nucleus RNA sequencing of human cerebral cortex. Nat. Neurosci. 25, 1034–1048 (2022).
Fumagalli, L. et al. Microglia heterogeneity, modeling and cell-state annotation in development and neurodegeneration. Nat. Neurosci. 28, 1381–1392 (2025).
Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. & Marioni, J. C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 40, 245–253 (2022).
Dumas, A. A., Borst, K. & Prinz, M. Current tools to interrogate microglial biology. Neuron 109, 2805–2819 (2021).
Mancuso, R. et al. Xenografted human microglia display diverse transcriptomic states in response to Alzheimer’s disease-related amyloid-β pathology. Nat. Neurosci. 27, 886–900 (2024).
Lall, D. et al. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 109, 2275–2291 (2021).
Yin, Z. et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat. Immunol. 24, 1839–1853 (2023).
Petkau, T. L. et al. Progranulin expression in the developing and adult murine brain. J. Comp. Neurol. 518, 3931–3947 (2010).
Philips, T. et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J. Neuropathol. Exp. Neurol. 69, 1191–1200 (2010).
Natarajan, K. et al. Single-cell multimodal analysis in a case with reduced penetrance of progranulin-frontotemporal dementia. Acta Neuropathol. Commun. 9, 132 (2021).
March-Diaz, R. et al. Hypoxia compromises the mitochondrial metabolism of Alzheimer’s disease microglia via HIF1. Nat. Aging 1, 385–399 (2021).
Mehta, A. R. et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 141, 257–279 (2021).
Song, S. et al. Elevated microglial oxidative phosphorylation and phagocytosis stimulate post-stroke brain remodeling and cognitive function recovery in mice. Commun. Biol. 5, 35 (2022).
Fattorelli, N. et al. Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat. Protoc. 16, 1013–1033 (2021).
Mancuso, R. et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 22, 2111–2116 (2019).
Selvaraj, B. T. et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9, 347 (2018).
Beckers, J. et al. A toxic gain-of-function mechanism in C9orf72 ALS impairs the autophagy-lysosome pathway in neurons. Acta Neuropathol. Commun. 11, 151 (2023).
O’Neill, K. et al. ALS molecular subtypes are a combination of cellular and pathological features learned by deep multiomics classifiers. Cell Rep. 44, 115402 (2025).
Sadick, J. S. et al. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 110, 1788–1805 (2022).
Serrano-Pozo, A. et al. Astrocyte transcriptomic changes along the spatiotemporal progression of Alzheimer’s disease. Nat. Neurosci. 27, 2384–2400 (2024).
Lucas, T. A. et al. Integrated cross-disease atlas of human and mouse astrocytes reveals heterogeneity and conservation of astrocyte subtypes in neurodegeneration. Preprint at bioRxiv https://doi.org/10.1101/2025.02.12.637903 (2025).
Escartin, C. et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 24, 312–325 (2021).
Habib, N. et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 23, 701–706 (2020).
Matias, I., Morgado, J. & Gomes, F. C. A. Astrocyte heterogeneity: impact to brain aging and disease. Front. Aging Neurosci. 11, 59 (2019).
Batiuk, M. Y. et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 11, 1220 (2020).
Blackburn, D., Sargsyan, S., Monk, P. N. & Shaw, P. J. Astrocyte function and role in motor neuron disease: a future therapeutic target?. Glia 57, 1251–1264 (2009).
Jha, M. K., Jo, M., Kim, J. H. & Suk, K. Microglia−astrocyte crosstalk: an intimate molecular conversation. Neuroscientist 25, 227–240 (2019).
Vainchtein, I. D. & Molofsky, A. V. Astrocytes and microglia: in sickness and in health. Trends Neurosci. 43, 144–154 (2020).
Greenhalgh, A. D., David, S. & Bennett, F. C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 21, 139–152 (2020).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A. & Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl Acad. Sci. USA 109, E197–E205 (2012).
Rothhammer, V. et al. Microglial control of astrocytes in response to microbial metabolites. Nature 557, 724–728 (2018).
Bezzi, P. et al. CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 4, 702–710 (2001).
Clark, I. C. et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science 372, eabf1230 (2021).
O’Rourke, J. G. et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329 (2016).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Tondo, G., Perani, D. & Comi, C. TAM receptor pathways at the crossroads of neuroinflammation and neurodegeneration. Dis. Markers 2019, 2387614 (2019).
Gilchrist, S. E., Goudarzi, S. & Hafizi, S. Gas6 inhibits toll-like receptor-mediated inflammatory pathways in mouse microglia via Axl and Mer. Front. Cell Neurosci. 14, 576650 (2020).
Owlett, L. D. et al. Gas6 induces inflammation and reduces plaque burden but worsens behavior in a sex-dependent manner in the APP/PS1 model of Alzheimer’s disease. J. Neuroinflammation 19, 38 (2022).
Huang, Y. et al. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 22, 586–594 (2021).
Zelic, M. et al. Single-cell transcriptomic and functional studies identify glial state changes and a role for inflammatory RIPK1 signaling in ALS pathogenesis. Immunity 58, 961–979 (2025).
Matsumoto, T. et al. CD44 expression in astrocytes and microglia is associated with ALS progression in a mouse model. Neurosci. Lett. 520, 115–120 (2012).
Lorenzini, I. et al. Moderate intrinsic phenotypic alterations in C9orf72 ALS/FTD iPSC-microglia despite the presence of C9orf72 pathological features. Front. Cell. Neurosci. 17, 1179796 (2023).
Wilson, D. M. 3rd. et al. Hallmarks of neurodegenerative diseases. Cell 186, 693–714 (2023).
Beers, D. R. & Appel, S. H. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 18, 211–220 (2019).
Henkel, J. S., Beers, D. R., Zhao, W. & Appel, S. H. Microglia in ALS: the good, the bad, and the resting. J. Neuroimmune Pharmacol.4, 389–398 (2009).
Jun, K. Y. et al. Epidemiology of ALS in Korea using nationwide big data. J. Neurol. Neurosurg. Psychiatry 90, 395–403 (2019).
Balendra, R. & Isaacs, A. M. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558 (2018).
Vahsen, B. F. et al. C9orf72-ALS human iPSC microglia are pro-inflammatory and toxic to co-cultured motor neurons via MMP9. Nat. Commun. 14, 5898 (2023).
Zhou, Q. et al. Active poly-GA vaccination prevents microglia activation and motor deficits in a C9orf72 mouse model. EMBO Mol. Med. 12, e10919 (2020).
Hao, Z. et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun. 10, 2906 (2019).
Schludi, M. H. et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 134, 241–254 (2017).
Chew, J. et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol. Neurodegener. 14, 9 (2019).
Sudria-Lopez, E. et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 132, 145–147 (2016).
Burberry, A. et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8, 347ra93 (2016).
Farg, M. A. et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 23, 3579–3595 (2014).
Prudencio, M. et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 18, 1175–1182 (2015).
Nataf, S. & Pays, L. Gene co-expression analysis unravels a link between C9orf72 and RNA metabolism in myeloid cells. Acta Neuropathol. Commun. 3, 64 (2015).
McCauley, M. E. et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 585, 96–101 (2020).
Wang, H. V. et al. Single nucleus multiome analysis of the prefrontal cortex from C9orf72 ALS/FTD patients illuminates pathways affected during disease progression. Preprint at bioRxiv https://doi.org/10.1101/2023.01.12.523820 (2023).
Li, J. et al. Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat. Commun. 14, 5714 (2023).
Beckers, J., Tharkeshwar, A. K., Van Damme, P. C9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 17, 3306−3322 (2021).
Zhu, Q. et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624 (2020).
Shao, Q. et al. C9orf72 deficiency promotes motor deficits of a C9ALS/FTD mouse model in a dose-dependent manner. Acta Neuropathol. Commun. 7, 32 (2019).
Lorenzini, I. et al. Moderate intrinsic phenotypic alterations in C9orf72 ALS/FTD iPSC-microglia despite the presence of C9orf72 pathological features. Front. Cell Neurosci. 17, 1179796 (2023).
Maharjan, N., Künzli, C., Buthey, K. & Saxena, S. C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Mol. Neurobiol. 54, 3062–3077 (2017).
Boivin, M. et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39, e100574 (2020).
Trageser, K. J. et al. Inflammasome-mediated neuronal-microglial crosstalk: a therapeutic substrate for the familial C9orf72 variant of frontotemporal dementia/amyotrophic lateral sclerosis. Mol. Neurobiol. 60, 4004–4016 (2023).
Shu, X. et al. Negative regulation of TREM2-mediated C9orf72 poly-GA clearance by the NLRP3 inflammasome. Cell Rep. 42, 112133 (2023).
Zhao, C. et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 68, 1046–1064 (2020).
Allen, S. P. et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 142, 586–605 (2019).
Meyer, K. et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl Acad. Sci. USA 111, 829–832 (2014).
Varcianna, A. et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40, 626–635 (2019).
Allen, S. P. et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 142, 3771–3790 (2019).
Slyper, M. et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 26, 792–802 (2020).
Habib, N. et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017).
Otero-Garcia, M. et. al. Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease. Neuron 110, 2929–2948 (2022).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Morabito, S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimerʼs disease. Nat. Genet. 53, 1143–1155 (2021).
Raudvere, U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Reumers, J. et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 30, 61–68 (2011).
Schneider, V. A. et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Graubert, A., Aguet, F., Ravi, A., Ardlie, K. G. & Getz, G. RNA-SeQC 2: efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics 37, 3048–3050 (2021).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Acknowledgements
We are thankful to all participants and their relatives for their invaluable contributions to this research. We are indebted to the Department of Pathology at KU Leuven, the Netherlands Brain Bank and the HCB-IDIBAPS for sample and data procurement. We especially thank M. Kooreman, technical coordinator of the Netherlands Brain Bank, for helping us with selecting and transferring the samples. We thank N. Lamaire, ALS clinical trial assistant, for helping with submission of this study protocol to the Ethics Committee Research at UZ Leuven. We thank C. Verfaillie, Stem Cell Institute KU Leuven, for providing the iSOX9 iPSC line. We thank the VIB Nucleomics Core (https://nucleomicscore.sites.vib.be/en), especially L. De Swert, K. Coeck, J. Vandewinkel and S. Derveaux, for their help and expertise. We thank F. Baeke and R. De Rycke, VIB BioImaging Core and VIB-Ugent Center for Inflammation Research, for performing electron microscopy experiments. We thank the VIB Protein Core for the generation of growth factors for microglia differentiation. We thank M. Fiers for his support in bioinformatics. P.M. has been awarded a Postdoctoral Mandate from KU Leuven Internal Funds, is a recipient of the Early-Stage ALS Clinicians Grant from Target ALS and has also received the Innovative Collaborative Projects with Biomarker Consortia Grant from Target ALS. B.B. is recipient of a research fellowship from Research Foundation–Flanders (FWO) (11PD824N). L.F. is recipient of a scholarship from the University of Antwerp (BOF Seal of Excellence, ADP0144), SAO-FRA (20240019) and the BrightFocus Foundation (A2025008F). W.H.D.V., R.G. and J.V.D.D. receive funding from FWO (1274822N, I003420N and I000321N) and SAO (2019/0035). D.R.T. receives additional funds from FWO (G0F8516N and G065721N) and SAO-FRA (2020/017). R.M. receives funding from the European Research Council (XenoMicrogliaAD), FWO (G0C9219N, G056022N, G0K9422N and G067625N) and Alzheimer’s Association USA (2018-AARF-591110, 2018-AARF-591110-RAPID, ABA-22-968700 and ADSF-25-1460010-C, with support from the AD Strategic Fund and the WoodNext Foundation, administered by the Greater Houston Community Foundation). He also receives funding from the BrightFocus Foundation (A2021034S), SAO-FRA (2021/0021 and 20230019) and the University of Antwerp (BOF-TOP 2022-2025). L.V.D.B. is supported by the Generet Award for Rare Diseases. P.V.D. holds a fundamental clinical investigatorship of KU Leuven and is supported by the ALS Liga België and the KU Leuven funds ‘Een Hart voor ALS’ and ‘Laeversfonds voor ALS Onderzoek’. This work was supported by C1 grants from KU Leuven (C14/17/107 and C14/22/132 to L.V.D.B., D.R.T. and P.V.D.), FWO (G073222N and T003519N), the KU Leuven fund ‘Opening the Future’ and in part by the Intramural Research Programs of the National Institutes of Health, National Institute on Aging (Z01-AG000949-02) and the National Institute of Neurological Disorders and Stroke. Computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by FWO and the Flemish Government.
Author information
Authors and Affiliations
Contributions
P.M. conceived and authored the study protocol; obtained ethical approval from KU Leuven; systematically excluded cases with co-occurring FTD, Alzheimerʼs disease or other neurodegenerative disorders based on neuropathological assessment; thoroughly reviewed clinical symptoms at both disease onset and during progression; coordinated the acquisition of institutional biobank samples; prepared and managed regulatory documentation, including ethics amendments and inter-institutional material transfer agreements; assisted with the single-nuclei postmortem analysis; and led the project. B.B. designed and performed the analysis of single-nuclei microglia, single-cell microglia and single-nuclei astrocyte data and performed downstream snRNA-seq analysis and led the project. L.F. designed, performed and analyzed in vivo xenograft and in vitro iPSC-derived microglia experiments and led the project. R.M. and P.V.D. contributed to designing this study, provided supervision and led the project. P.M. and D.R.T. collected the postmortem human samples. D.R.T. performed neuropathological analysis and dissected brain and spinal cord samples for analysis. P.M. performed extraction of nuclei from postmortem tissues. K.D. processed snRNA-seq data and performed quality control and snRNA-seq analysis of motor cortex and spinal cord atlas. S.K.P. performed the 10x Genomics, with the assistance of P.M., A.S. and N.H. T.M. processed scRNA-seq data, performed quality control and analyzed single-cell microglia and bulk RNA-seq data of in vitro microglia. P.M., B.B., L.F., K.D., L.V.D.B., R.M. and P.V.D. interpreted the snRNA-seq data. B.B., L.F. and R.M. interpreted scRNA-seq data of xenografted microglia. R.F. performed and analyzed western blot on human postmortem samples. D.v.d.B. and H.D. provided technical support with iPSC culture and differentiation. D.v.d.B., P.P.M. and B.A. assisted with the in vitro experiments and analysis. B.A. and S.M. performed expansion microscopy experiments. R.G, J.V.D.D. and W.H.D.V. contributed to the cell-to-cell communication experiments. S.C. provided C9-HRE and isogenic control lines. P.M., B.B., L.F., R.M. and P.V.D. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.R.T. received speaker honoraria from Biogen and travel reimbursement from UCB and collaborated with Novartis Pharma AG, Probiodrug, GE Healthcare and Janssen Pharmaceutical Companies. P.V.D. has served on advisory boards for Biogen, CSL Behring, Alexion Pharmaceuticals, Ferrer, QurAlis, Cytokinetics, Argenx, UCB, Muna Therapeutics, Alector, Augustine Therapeutics, VectorY, Zambon, Amylyx, Novartis, Prilenia, Verge Genomics, Sapreme Technologies, Trace Neuroscience and NRG Therapeutics (paid to institution). R.M. has scientific collaborations with Alector, Nodthera, Alchemab and Roche and is a consultant for Alector and Muna Therapeutics. The other authors declare that they have no conflicts of interest. L.V.D.B. is head of the Scientific Advisory Board of Augustine Therapeutics and is part of the Investment Advisory Board of Droia Ventures.
Peer review
Peer review information
Nature Neuroscience thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overview of the full dataset and quality control.
(a) UMAP plot visualization of the single nuclei from motor cortex and spinal cord prior and after batch correction. (b) Quality control of the three isolation protocols (Habib, Soma-seq, TST): percentage of mitochondrial RNA, number of molecules detected per cell, number of genes detected per cell. (c) Contribution of each patient (y-axis) to each cluster (x-axis) of the motor cortex and spinal cord. (d) Contribution of each patient (y-axis) to each annotated cell type of the motor cortex and spinal cord.
Extended Data Fig. 2 Extended clustering and quality control in the microglial subset.
(a) UMAP plot visualizing 4,845 nuclei from the motor cortex and 13,980 nuclei from the spinal cord of the ALS dataset. Cell populations are coloured based on their assigned into the following clusters: microglia, peripheral macrophages, doublets (neurons), doublets (oligodendrocytes) and cycling cells. (b) Heatmap showing the top 20 most expressed genes per cluster. (c, d) Enrichment of transcriptomic signatures of different human microglial cell states from Mancuso et al.26. in (c) the clusters from the ALS dataset and (d) the final clusters after integration with Gerrits et al. (e) Fraction of cells (y-axis) from each cluster in each subject, divided by anatomical regions (x-axis). (f) Heatmaps showing differentially expressed genes (log2FC, p-value adjusted <0.05) in both C9-ALS or sALS patients compared to healthy controls in the spinal cord, the motor cortex, or both locations combined. (g) Log fold change of the neighbourhood groups distribution between brain and spinal cord and (h) expression values of differentially expressed genes in C9-ALS and sALS, vs. healthy controls.
Extended Data Fig. 3 WGCNA reveals dysregulated gene modules in C9 dWGCNA reveals dysregulated gene modules in C9.
(a) Dendrogram showing the result of unsupervised hierarchical clustering of modules identified by WGCNA. (b) Bar plots showing the number of genes per module. (c) Boxplots visualizing the module eigengenes (y-axis) per genotype (x-axis) for the blue, turquoise, yellow, black, brown, red and green cluster in the spinal cord. (d) Pathway enrichment analysis of the WGCNA clusters blue, turquoise and yellow, with the associated GO term (y-axis) and –log10 p-value (colour heatmap), coloured per direction of expression changes across the patient groups.
Extended Data Fig. 4 Microglia from C9orf72 HRE patients show transcriptional alterations in functional pathways.
Heatmaps depicting differentially expressed genes (log2FC < +/-0,20, p-value adjusted <0,05) from selected gene sets associated with lysosomal, inflammatory, phagocytic and oxidative phosphorylation functional pathways.
Extended Data Fig. 5 Extended data on the HBDX model.
(a) Gating strategy for the isolation of human microglia from the HBDX model (CD11b + hCD45 + ). (b) Scatter plot showing the number of unique genes (nFeature_RNA) and total RNA counts (nCount_RNA) per cell, across five single-cell RNA sequencing libraries (LIB14, LIB17, LIB28, LIB59, and LIB60). Each dot represents an individual cell, coloured according to the percentage of mitochondrial gene expression. Marginal density plots display the distribution of nFeature_RNA (top) and nCount_RNA (right) for each library (c) UMAP plot showing cell populations coloured based on their assignment into the following clusters: microglia, macrophages, doublets (neurons), cycling cells, and doublets. (d) Heatmap showing the top 15 most expressed genes per cluster. (e) Fraction of cells per genotype in each cluster. (f) Percentage of cells in HLA cluster, showing reduced percentage of cells in C9KO versus controls. Mann-Whitney test (CTRL n = 7, C9KO n = 10 mice). Box plots show the median, interquartile range, and full data range.
Extended Data Fig. 6 Extended data related to Fig.4.
(a) Schematic representation of the microglial differentiation protocol and plating for in vitro experiments, based on Fattorelli et al.35. Created in BioRender. Polanco, P. (2025) https://BioRender.com/83a97ah (b) Representative images of IBA1 immunostaining for control and C9KO iPSC-derived microglia. Scale bar, 20 µm35. (c) Heatmap showing enrichment of microglial-related genes versus macrophages after iPSC differentiation in vitro (n = 7 independent replicates). (d) Example of low-magnification confocal images used for quantification of CTSD clusters in Fig. 4d and f. Upper panel: C9-HRE and C9-ISO; lower panel: C9KO and control microglia. Cells containing at least one CTSD puncta were considered for quantification in ImageJ. The percentage of cells containing CTSD positive puncta was calculated on total DAPI positive cells. (e) Representative low-magnification TEM images for C9KO and control iPSC-derived microglia that were used for quantification of endolysosomes/lysosomes clusters in Fig. 4h. Scale bar, 20 µm. (f) Quantification of percentage of cells containing enlarged endolysosomes/lysosomes. Unpaired T-test (CTRL n = 5 independent replicates, C9KO n = 5 independent replicates). Box plots show the median, interquartile range, and full data range. (g) Microscopy images showing EEA1 (that is, early endosomes) and Lamp1 positive structures in control and C9KO microglial cells. Scale bar, 5 µm. Flow cytometry analysis of uptake of (h, i) amyloid beta and (j, k) pHrodo E. coli particles, showing no difference in the median fluorescence intensity (MFI) between C9KO and controls, at 3 h and 1 h upon treatment, respectively. Ratio paired T-test (amyloid beta, CTRL n = 3 independent replicates, C9KO n = 3 independent replicates; pHrodo E. coli CTRL n = 4 independent replicates, C9KO n = 4 independent replicates).
Extended Data Fig. 7 Extended clustering and quality control in the astrocytic subset.
(a) UMAP plot visualizing 10,119 nuclei from the motor cortex and 13,364 nuclei from the spinal cord. Cell populations are coloured based on their assigned cluster into astrocytes, oligodendrocytes, doublets/low quality. (b) Heatmap showing the top 20 most expressed genes per cluster. (c) Heatmap showing average expression of the top 100 markers from the original 8 astrocyte clusters (Fig. 5a) onto the novel clustering after integration with Gerrits et al.22. (Fig. 5b). (d) Proportion of cells per cluster (y-axis) for each individual, across all the different anatomical regions. (e) Heatmaps showing differentially expressed genes (log2FC, p-value adjusted <0.05) between C9-ALS or sALS and healthy controls, and displayed for the spinal cord, motor cortex or both locations combined.
Extended Data Fig. 8 Extended CellChat analysis to astrocyte-astrocyte communication pathways.
Dot Plots depicting (a) scaled communication probabilities of differential pathways in healthy, C9-ALS and sALS patients’ spinal cord, and (b) scaled average expression of the source and target in the astrocyte cell states from Fig. 5b.
Supplementary information
Supplementary Tables 1–32 (download XLSX )
Supplementary Table 1. Demographic data of participants. Supplementary Table 2. Demographic ranges of experimental groups. Supplementary Table 3. Cluster markers of ALS microglia. Supplementary Table 4. Cluster markers of ALS astrocytes. Supplementary Table 5. Cluster markers of ALS and FTD microglia. Supplementary Table 6. Cluster markers of ALS and FTD astrocytes. Supplementary Table 7. Differential expression between microglia from healthy and C9 ALS in the spinal cord and motor cortex. Supplementary Table 8. Differential expression between microglia from healthy and sALS in the spinal cord and motor cortex. Supplementary Table 9. Differential expression between microglia from C9ALS and sALS in the spinal cord and motor cortex. Supplementary Table 10. Differential expression between microglia from healthy and C9 ALS in the motor cortex. Supplementary Table 11. Differential expression between microglia from healthy and sALS in the motor cortex. Supplementary Table 12. Differential expression between microglia from C9ALS and sALS in the motor cortex. Supplementary Table 13. Differential expression between microglia from healthy and C9 ALS in the spinal cord. Supplementary Table 14. Differential expression between microglia from healthy and sALS in the spinal cord. Supplementary Table 15. Differential expression between microglia from C9ALS and sALS in the spinal cord. Supplementary Table 16. Differential expression of patients with FTD versus controls in all sampling locations. Supplementary Table 17. Overview of the WGCNA results of microglia in the spinal cord. Supplementary Table 18. Cluster markers of xenografted microglia. Supplementary Table 19. Differential expression of C9KO xenografted microglia versus controls. Supplementary Table 20. Differential expression of C9HRE xenografted microglia versus isogenic controls. Supplementary Table 21. Differential expression between astrocytes from healthy and C9 ALS in the spinal cord and motor cortex. Supplementary Table 22. Differential expression between astrocytes from healthy and sALS in the spinal cord and motor cortex. Supplementary Table 23. Differential expression between astrocytes from C9ALS and sALS in the spinal cord and motor cortex. Supplementary Table 24. Differential expression between astrocytes from healthy and C9 ALS in the motor cortex. Supplementary Table 25. Differential expression between astrocytes from healthy and sALS in the motor cortex. Supplementary Table 26. Differential expression between astrocytes from C9ALS and sALS in the motor cortex. Supplementary Table 27. Differential expression between astrocytes from healthy and C9 ALS in the spinal cord. Supplementary Table 28. Differential expression between astrocytes from healthy and sALS in the spinal cord. Supplementary Table 29. Differential expression between astrocytes from C9ALS and sALS in the spinal cord. Supplementary Table 30. nHoodGroup marker genes of astrocytes. Supplementary Table 31. KEGG pathway analysis of nHood marker genes. Supplementary Table 32. Microglia and astrocyte communication pathways.
Source data
Source Data Fig. 4 (download JPG )
Uncropped membranes from western blots shown in Fig. 4a, displaying CTSD (mature, approximately 28 kDa) and GAPDH (approximately 37 kDa) protein levels in control (n = 2), C9orf72 (n = 5) and sporadic ALS (n = 5) patients in motor cortex (a) and spinal cord (b).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Masrori, P., Bijnens, B., Fumagalli, L. et al. C9orf72 hexanucleotide repeat expansions impair microglial response in ALS. Nat Neurosci 28, 2217–2230 (2025). https://doi.org/10.1038/s41593-025-02075-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41593-025-02075-1
This article is cited by
-
Microglia response altered by ALS mutation
Nature Reviews Neurology (2025)

{kind=link}