
Abstract
Motor neuron hyperexcitability is a broadly observed yet poorly understood feature of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Nuclear depletion and cytoplasmic aggregation of the RNA splicing protein TAR DNA-binding protein 43 (TDP-43) are observed in most ALS and FTD patients. Here we show that TDP-43 dysfunction causes mis-splicing of KCNQ2, which encodes a voltage-gated potassium channel (Kv7.2) that regulates neuronal excitability. Using iPSC-derived neurons and postmortem ALS/FTD brain and spinal cord tissue we find widespread, disease-specific and TDP-43-specific skipping of an exon encoding the KCNQ2 pore domain. The mis-spliced mRNA escapes degradation and is translated into a nonfunctional protein with severely reduced ion conductance that aggregates in the endoplasmic reticulum and causes intrinsic hyperexcitability in ALS neuronal models. This event, which correlates with higher phosphorylated TDP-43 levels and earlier age of disease onset in patients, can be rescued by splice-modulating antisense oligonucleotides that dampen hyperexcitability in induced pluripotent stem cell cortical neurons and spinal motor neurons with TDP-43 depletion. Our work reveals that nuclear TDP-43 maintains the fidelity of KCNQ2 expression and function and provides a mechanistic link between established excitability disruption in ALS/FTD patients and TDP-43 dysfunction.
Similar content being viewed by others

Main
ALS is a fatal neurodegenerative disorder that eliminates motor neurons (MNs) responsible for skeletal muscle contraction and locomotion1. Although the disease is inherited in approximately 10% of cases, most patients are diagnosed with sporadic disease2. Consequently, there remain significant challenges in identifying molecular biomarkers for early diagnosis and therapeutic targets for precision medicine. Nuclear depletion and cytoplasmic aggregation of TDP-43 represents a shared histopathological feature that is found in more than 95% of all ALS cases3. Cytoplasmic aggregation of TDP-43 also occurs in approximately half of patients with frontotemporal dementia (FTD-TDP), a disease that shares genetic and clinical features with ALS1,3. TDP-43 pathology is also seen in a considerable proportion (~57%) of patients with Alzheimer’s disease4,5. TDP-43 is a nuclear RNA-binding protein with conserved roles across the RNA lifecycle, from transcriptional regulation to RNA splicing modulation, RNA transport and stability6. The direct link between TDP-43 pathology and neurodegeneration is additionally underscored by the identification of rare mutations in TARDBP, the gene that encodes TDP-43, in ALS and FTD families7,8. However, the cellular mechanisms that relate TDP-43 to neuronal dysfunction and neurotoxicity remain unclear. The recent interrogation of human induced pluripotent stem cell (iPS cell)-derived neurons with loss of TDP-43 function and postmortem tissue from patients with ALS has revealed hundreds of target mRNAs that undergo mis-splicing9,10,11,12,13. For example, in neurons depleted of TDP-43, the pre-mRNA encoding stathmin 2 (STMN2), a modulator of microtubule stability that impacts axonal outgrowth, yields a truncated variant through activation of a cryptic premature last exon11,12,13. Similarly, TDP-43 depletion leads to the inclusion of a cryptic exon in UNC13A9,10, which encodes a protein with critical functions in synaptic vesicle release14, resulting in nonsense-mediated decay and downregulation of the protein. Intriguingly, polymorphisms in UNC13A that overlap with TDP-43 binding sites are strongly associated with increased ALS and FTD risk15. These findings have shifted the focus from gain-of-function effects associated with aggregated TDP-43 to loss-of-function effects driven by its nuclear depletion, as they underscore direct relationships between TDP-43 pathology, neuronal function and disease risk. However, a link between the role of TDP-43 in splicing and commonly observed clinical features of ALS/FTD has yet to be established.
Neuronal hyperexcitability is a broadly observed clinical feature in familial and sporadic ALS16,17,18,19 and FTD20,21,22. MN excitability, measured by transcranial magnetic stimulation and threshold tracking nerve conduction studies, is an early indicator of MN disease16,17,18,19, and greater excitability is prognostic of faster disease progression and worse survival23,24. Excitability disturbances have been linked to a loss of cortical inhibition25,26, noradrenaline reduction27 and changes in the expression and activity of key ion channels that serve to regulate intrinsic membrane potential28,29,30. Hyperexcitability has been observed in murine TDP-43 models31,32 and iPS cell-based ALS models and can be modulated by the anti-seizure medication retigabine29,33,34 (also known as ezogabine), an activator of Kv7 channels, which are known to be widely expressed in the nervous system and have a central role in regulating excitability35,36. Treatment of ALS iPS cell MNs with ezogabine reduced endoplasmic reticulum (ER) stress and improved survival, suggesting a mechanistic interaction between excitability and proteotoxic degeneration33,34. Activation of Kv7 channels has also been shown to improve neuromuscular function and survival of MNs in ALS mouse models37. Notably, retigabine was proven to be effective at decreasing both cortical and spinal MN excitability in individuals with ALS in a phase 2 clinical trial38. Despite its wide prevalence in the patient population and its therapeutic potential, the mechanisms that drive neuronal hyperexcitability in ALS are poorly understood.
Here, we sought to examine a potential association between TDP-43 dysfunction and neurophysiology. By integrating gene expression datasets from human iPS cell-derived neurons depleted of TDP-43 and postmortem ALS/FTD tissue, we identify spurious skipping of exon 5 of the voltage-gated potassium channel gene KCNQ2 (KCNQ2∆E5). KCNQ2 encoded protein (Kv7.2) forms heterotetrameric channels with other Kv7 subunits to conduct M-current and regulate repetitive firing and excitability in neurons39. We show that KCNQ2 splicing is sensitive to TDP-43 levels, and aberrant pre-mRNA processing yields a non-functional protein that disrupts neuronal excitability, accumulates within the ER of iPS cell-derived neurons and forms ubiquitin-positive accumulations in postmortem spinal cord tissue from patients with ALS. Critically, we demonstrate that splice-modulating antisense oligonucleotides (ASOs) that restore KCNQ2 splicing can rescue hyperexcitability in iPS cell cortical and spinal neurons with TDP-43 depletion. Collectively, our findings highlight KCNQ2∆E5 as a reliable molecular biomarker of TDP-43 dysfunction and provide a direct connection between TDP-43 pathology and intrinsic neuronal hyperexcitability in ALS/FTD.
Results
Spinal iPS cell MNs exhibit several TDP-43-dependent splicing events
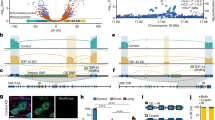
We previously reported gene expression differences in purified Hb9–GFP positive human MNs derived from iPS cells depleted of TDP-43 (ref. 11). Here, we analyzed these RNA sequencing (RNA-seq) datasets using the MAJIQ pipeline40 and identified a stringent set of 125 differential splicing variants, statistically supported by differential splicing of at least 10% (Fig. 1a and Supplementary Table 1). Classification of variants revealed that most TDP-43 sensitive events include cassette exons; that is, splicing events involving exon inclusion or skipping (Extended Data Fig. 1a,b). We discovered 11 unique and statistically significant de novo processing events that are neither detected in control-treated neurons nor defined within current gene annotations, including previously reported premature termination events in STMN2 and ARHGAP32, cryptic exon inclusions in KALRN and others9,10,11,12 (Fig. 1a). Among these, we identified a spurious exon skipping event in the voltage-gated potassium channel KCNQ2 (Fig. 1a). We asked whether the splice junctions we identified in purified spinal MNs were prevalent across other datasets of TDP-43 dysfunction reported in iPS cell-derived cortical neurons9 and neuronal nuclei from postmortem brains of patients with ALS/FTD41. Despite using different criteria for defining differential splicing, we observed significant overlap, with 87 out of 125 of MN events also perturbed in at least one of the other datasets (Extended Data Fig. 1c). This was restricted to not only annotated alternative splicing events, but also de novo or derepressed cryptic splicing events (Extended Data Fig. 1c). These analyses add further clarity to TDP-43-regulated processes in neurons and provide a framework to investigate the mechanistic interaction between TDP-43 loss-of-function and disease processes.

a, Differential splicing analysis of stem cell-derived MNs treated with TDP-43 (n = 6) or scrambled (n = 6) siRNAs reported previously11. Annotated alternative splicing events are shown in green, de novo events are shown in orange and a de novo KCNQ2 event is shown in red. b, Mis-spliced genes after TDP-43 loss-of-function (LOF) involved in neuronal excitability. Left, integration of TDP-43 mis-spliced genes derived from purified MN11, i3Neurons9 and TDP-43-low neuronal nuclei41 yields 522 genes; 47 are involved in the regulation of ‘membrane potential’ or ‘synaptic signaling’. Right, two genes (KCNQ2 and CACNA1E) are mis-spliced in all three datasets. c, Sashimi plot depicting HISAT2-mapped sequencing coverage along exons 4–6 of KCNQ2. The gene model indicates the locations of exons and introns. Junction spanning reads mapping to the canonical junctions (exons 4–5 and 5–6) are displayed with solid lines, and de novo junction (exons 4–6) reads are displayed with a dashed line. Read counts and percent spliced index (PSI) scores for each splice junction are denoted. KCNQ2 exon skipping is only detected under TDP-43 depletion. siTDP-43, siRNA against TDP-43; siSCR, scrambled control siRNA. d, RT–PCR assay shows KCNQ2 mis-splicing in cortical neurons depleted of TDP-43 by siRNA. e,f, RT–qPCR of mis-spliced KCNQ2∆E5 that increases (e) and total KCNQ2 transcript that remains unaltered (f) after TDP-43 depletion. For e and f, circles represent n = 3 biological replicates, values represent mean expression; error bars, s.e.m. g–i, TDP-43 directly binds to KCNQ2 pre-mRNA. g, iCLIP sequencing coverage9 in the KCNQ2 locus suggests that TDP-43 binds KCNQ2. TDP-43 consensus motifs (UGNNUG) and (UG)n simple repeat are shown below the gene model. RIP amplicon used in h and i is indicated. h, Cross-linking immunoprecipitation followed by RT–PCR analysis shows enrichment of KCNQ2 in the TDP-43-bound fraction compared to IgG immunoprecipitates (IP) using the same quantity of lysate for IPs. i, Quantification of results present in h. Circles represent n = 3 biological replicates; values represent mean relative enrichment; error bars, s.e.m. For e, f and i, P values are the result of an unpaired two-sided t-test.
TDP-43 is required for the inclusion of a constitutive exon in KCNQ2
The identification of KCNQ2 as a differentially spliced gene in TDP-43-depleted MNs (Fig. 1a) suggested a potential connection between TDP-43 dysfunction and neuronal excitability. To investigate this connection further, we integrated splicing changes from three datasets examining TDP-43-depletion (iPS cell-derived MNs11, iPS cell cortical neurons9 and ALS/FTD neuronal nuclei41) to arrive at 522 significantly differentially spliced genes (Fig. 1b, left and Extended Data Fig. 1d, left). Consistent with our hypothesis, this stringent set was enriched for genes with connections to human phenotypes such as ‘motor seizures’, ‘abnormal motor function’ and related terms (Supplementary Table 1). Intersecting differentially spliced genes with Gene Ontology terms such as ‘regulation of membrane potential’ and ‘synaptic signaling’ highlighted 47 target genes (Fig. 1b, right and Extended Data Fig. 1d, right), only two of which were commonly affected across all three datasets: KCNQ2 and the voltage-gated calcium channel CACNA1E (Fig. 1b, right). Critically, a similar analysis integrating gene expression rather than splicing alterations did not yield common targets dysregulated in all three datasets (Extended Data Fig. 1e,f). Although the TDP-43-dependent modulation of CACNA1E represents the preferential splicing of a known annotated isoform (Extended Data Fig. 1g), loss of TDP-43 results in the skipping of exon 5 in the KCNQ2 transcript (Fig. 1c). Notably, this is not an annotated cassette exon, as we were unable to detect regulated alternative splicing at this locus in diverse human central nervous system (CNS) tissue. The unexpectedly skipped exon codes for a deeply conserved segment of the ion channel (Extended Data Fig. 1h), including the entire S5 transmembrane helix and a portion of the pore loop that conducts K+ ions (Extended Data Fig. 1i). Therefore, we categorize this alteration as an aberrant splicing event and refer to the resulting exon 5 skipped transcript as KCNQ2∆E5.
We next used PCR with reverse transcription (RT–PCR) coupled with Sanger sequencing to validate the presence of KCNQ2∆E5 in iPS cell-derived cortical neurons42 (Fig. 1d and Extended Data Fig. 1j) and spinal MNs43 (Extended Data Fig. 2a) that were depleted of TDP-43. The product corresponding to exon 5 skipping was substantially upregulated in neurons (~60-fold) treated with short interfering RNA (siRNA) against TDP-43 but not in neurons treated with a scrambled control siRNA sequence (Fig. 1e). KCNQ2∆E5 was also identifiable in published TDP-43 loss-of-function RNA-seq datasets generated using SH-SY5Y and SK-N-BE(2) neuroblastoma lines9, as well as i3 cortical neurons9, suggesting that this target is reliably sensitive to TDP-43 levels across neurons and neuron-like cells (Extended Data Fig. 2b). Moreover, using SH-SY5Y cells expressing the aggregation-prone version of TDP-43 in which the Q/N-rich domain is repeated 12 times, we also observed robust exclusion of KCNQ2 exon 5, further corroborating that KCNQ2 is highly responsive to TDP-43 dysfunction (Extended Data Fig. 2c–e). Other recently discovered human neuronal TDP-43 targets, such as STMN2 and UNC13A, undergo cryptic exon inclusion that results in a decrease of the host gene either through premature transcription termination or nonsense-mediated decay9,10,11,12,13. However, skipping of exon 5 in KCNQ2 does not alter the reading frame or induce a premature termination codon, and analysis by RT–quantitative PCR (qPCR) showed that the overall expression levels of KCNQ2 in TDP-43-depleted neurons remain unchanged despite the dramatic shift to KCNQ2∆E5 (Fig. 1f).
TDP-43 is known to bind UG repeats within long introns44. To explore whether TDP-43 may directly associate with KCNQ2 mRNA, we first examined sequence content surrounding the skipped exon 5. Although there are many UG dinucleotides within the 5 kb locus containing exon 5 and flanking introns, we observed an annotated ~1 kb-long simple repeat 6 bp upstream of exon 5 (Fig. 1g). This region consists of UG repeats, suggesting the potential for direct TDP-43 recruitment. Mapping of published TDP-43 individual nucleotide resolution UV cross-linking and immunoprecipitation (iCLIP) datasets in human neuroblastoma SH-SY5Y cells9 to this locus showed broad TDP-43 binding within KCNQ2, including upstream of exon 5 (Fig. 1g, top). To validate this CLIP enrichment, we UV-crosslinked SH-SY5Y cells and used a protocol to immunoprecipitate TDP-43 and associated RNA targets. We detected specific and TDP-43-immunoprecipitate-dependent KCNQ2 pre-mRNA enrichment (Fig. 1h,i). Notably, although the human and mouse KCNQ2 exons are highly conserved, the intronic regions around exon 5 show very little homology (Extended Data Fig. 2f). Accordingly, skipping of Kcnq2 exon 5 is not detected in published TDP-43 mouse models45 (Extended Data Fig. 2g,h), suggesting that this splicing event is not similarly regulated in mice, like other TDP-43 targets such as STMN2 and UNC13A9,10,11,12. To directly test this hypothesis, we differentiated MNs from mouse embryonic stem cells and knocked down Tardbp using siRNA and observed no evidence for mis-splicing of Kcnq2 exon 5 (Extended Data Fig. 2i). To exclude the possibility that mouse Kcnq2 is erroneously spliced past exon 5, we examined published RNA-seq datasets from mouse brains depleted of Tdp-43 (ref. 45) and again found no evidence for Tdp-43 dependent mis-splicing (Extended Data Fig. 2j). Our findings demonstrate that TDP-43 has a critical role in ensuring the integrity of KCNQ2 processing in human neurons that is not conserved in mice.
TDP-43 processing of KCNQ2 is altered in postmortem ALS/FTD CNS
The identification of KCNQ2 mis-splicing in iPS cell-derived neurons depleted of TDP-43 prompted us to investigate the extent of this event in ALS postmortem tissue. We first examined RNA-seq datasets generated by the Target ALS/New York Genome Center, consisting of 1,124 libraries of diverse tissues from patients with ALS (216), non-neurological disease controls (28) and individuals with other neurological diseases (8) (Fig. 2a). To evaluate the state of KCNQ2 processing, we counted sequencing reads that stringently aligned to the KCNQ2 exon 4/6 boundary and thus correspond to the aberrant KCNQ2∆E5 mRNA. (Fig. 2b). This analysis revealed that KCNQ2 misprocessing was detectable and highly specific to the ALS cohort (Fig. 2c). Evidence for KCNQ2∆E5 was found in most CNS tissues, including different spinal cord regions, as well as in temporal, frontal and motor cortex within the brain (Fig. 2d). Notably, as much as 29.5% of motor cortex and 17.3% of lumbar spinal cord samples exhibited KCNQ2∆E5, regions that are notably involved in the pathophysiology of ALS/FTD (Fig. 2d). This contrasted with only 4.1% and 4.3% of cerebellum and occipital cortex samples, respectively, which are two regions not typically affected in patients (Fig. 2d and Extended Data Fig. 3a). These tissue-specific differences in the detection of KCNQ2∆E5 probably reflect differences in neuronal cell type composition, disease burden, TDP-43 pathology and KCNQ2 expression level within the CNS (Extended Data Fig. 3b,c). Comparison of aberrant KCNQ2 levels with other known TDP-43 targets, like STMN2 and UNC13A, further revealed differential detection of aberrantly spliced products, with STMN2 misprocessing prominent within the spinal cord and motor cortex, and UNC13A misprocessing within the motor cortex (Extended Data Fig. 3d). For these genes as well, differential levels of mis-splicing across tissues and neuronal subtypes are not strictly driven by their relative abundance (Extended Data Fig. 3b–e).

a, Target ALS/New York Genome Center RNA-seq datasets, including 118 non-neurological controls, 982 ALS and 24 other neurological disease samples from distinct CNS regions. b, Strategy to detect KCNQ2∆E5 reads. c, Violin plots of CNS samples with KCNQ2∆E5 reads in RNA-seq datasets stratified by disease status reveal specificity to ALS. d, Percentage of ALS samples across CNS regions with KCNQ2∆E5 transcripts. Red shade intensity is relative to the proportion of samples with detectable KCNQ2∆E5. e, Violin plots of ALS samples with KCNQ2∆E5 reads in RNA-seq datasets stratified by genetics reveal specificity to TDP-43 pathologies. In c and e, each point represents a unique sample, and the y axis indicates the number of unique KCNQ2∆E5 reads. P values are the result of an unpaired one-sided Wilcoxon rank sum test. f–i, RT–qPCR analysis in brain samples from patients with ALS/FTD (n = 89) or controls (n = 27) for KCNQ2∆E5 (f) and wild type KCNQ2 (g). Boxplots represent interquartile range (IQR), whiskers indicate IQR limits ±1.5× IQR. h,i, KCNQ2∆E5 abundance increases with pTDP-43 levels (h), and KCNQ2∆E5 abundance is higher in individuals with early disease onset (i). For additional statistics, see Supplementary Table 2. For f–i, points indicate data obtained from unique postmortem samples. P values are the result of linear regression models adjusted for age at death, sex and RIN. j,k, Abundance of KCNQ2∆E5 in TDP-43-high and TDP-43-low neuronal nuclei from patients with ALS/FTD with C9orf72 hyperexpansions41. j, Top, schematic of neuronal nuclei sorted by flow cytometry into fractions with high and low TDP-43 levels. Bottom, quantification of KCNQ2 shows 100% wild type KCNQ2 transcripts in TDP-43-high nuclei and ~50% reduction in TDP-43-low nuclei and a concomitant ~50% increase in KCNQ2∆E5 transcripts. Violin plots of KCNQ2 levels; each point represents a unique sample. k, Sashimi plot depicting HISAT2-mapped sequencing coverage along exons 4–6 of KCNQ2 in purified neuronal nuclei from individual patients. Canonical junctions (exons 4–5 and 5–6) are displayed with solid lines; KCNQ2 exon 5 skipping reads are displayed with red dashed lines; read counts are denoted. KCNQ2 exon skipping is only detected in TDP-43-low neurons.
As KCNQ2 splicing is dependent on TDP-43 functional activity in iPS cell-derived neurons, we next evaluated this correlation in postmortem patient tissue. Cytoplasmic TDP-43 aggregation is observed in an overwhelming majority of patients with ALS (>95%) and is typically accompanied by nuclear loss-of-function3. Therefore, to interrogate whether detection of KCNQ2∆E5 was related to TDP-43 status, we grouped ALS datasets into those known to have typical cytoplasmic TDP-43 neuropathology (C9orf72 and sporadic cases) and those without (patients with SOD1 mutations; non-TDP-43). We found that KCNQ2∆E5 was only observed in patient samples that typically exhibit TDP-43 aggregation (Fig. 2e). To provide more direct evidence for this relationship, we analyzed RNA from a cohort of 89 frontal cortex brains from patients diagnosed with ALS/FTD and 27 controls from the Mayo Clinic Brain Bank (Supplementary Table 2). Along with documented clinical history, these samples were characterized for TDP-43 pathology by immunohistochemistry and a phosphorylated TDP-43 (pTDP-43) immunoassay. Using RT–qPCR primers that distinguish wild type (WT) and KCNQ2∆E5, we found a highly significant difference, with KCNQ2∆E5 detected exclusively in patient samples (P < 0.0001 adjusting for age, sex and RNA integrity number (RIN); Supplementary Table 2), while the levels of WT KCNQ2 transcript were not different (P = 0.9947 adjusting for age, sex and RIN; Supplementary Table 2) (Fig. 2f,g). Although the RT–qPCR assay can detect unique KCNQ2∆E5 transcripts, it is probably not sensitive enough to detect a potential reduction in WT KCNQ2 levels, as these RNA samples were produced from bulk cortical tissue, and TDP-43 pathology is typically seen only in a small proportion of neurons41. Critically, the level of misprocessed KCNQ2∆E5 was directly and significantly associated with the level of pTDP-43 (higher KCNQ2∆E5 = higher pTPD-43; P < 0.0001, adjusting for age, sex, RIN and gene mutation status; Fig. 2h and Supplementary Table 2). Additionally, there was an inverse relationship between the abundance of KCNQ2∆E5 and the age of disease onset (higher KCNQ2∆E5 = lower age of onset; P = 0.0053, adjusting for sex, RIN and gene mutation status; Fig. 2i and Supplementary Table 2), suggesting a potential contribution of KCNQ2 dysfunction to disease pathophysiology. Although there was also an indication that the higher abundance of KCNQ2∆E5 was associated with shorter disease duration, this was not statistically significant after correcting for confounding variables (P = 0.0322, adjusting for age at onset, sex, RIN and gene mutation status; Extended Data Fig. 3f and Supplementary Table 2).
Lastly, to assess the extent of KCNQ2 mis-splicing within purified patient neurons with TDP-43 dysfunction, we analyzed a previously published RNA-seq dataset41 in which neuronal nuclei from postmortem brains of individuals with ALS/FTD with C9orf72 expansions were purified by fluorescence-activated cell sorting (FACS) based on TDP-43 abundance41. We specifically compared the transcriptomes of neuronal nuclei with high and low TDP-43 levels. Strikingly, for six out of six individuals, we observed reads indicative of KCNQ2∆E5 exclusively in neuronal nuclei with low TDP-43 levels (Fig. 2j,k). The level of disruption in splicing amounted to ~50% of total KCNQ2 gene expression, suggesting that if KCNQ2∆E5 encodes a dysfunctional protein, it could reduce half of the total KCNQ2 available to neurons (Fig. 2j,k). Collectively, these analyses demonstrate that KCNQ2∆E5 is a molecular biomarker of TDP-43 ALS/FTD pathology.
KCNQ2∆E5 exhibits dominant negative loss-of-function activity
Having established that the KCNQ2∆E5 variant is induced by TDP-43 loss-of-function in iPS cell-derived neurons and ALS/FTD patient tissue, we next wondered how mis-splicing of this transcript might affect the biology of the encoded protein. KCNQ2 encodes a voltage-gated, slow-activating and non-inactivating K+ channel (KCNQ2 or Kv7.2) subunit that is expressed very early in the developing brain and forms homo-and hetero-tetrameric K+ channels in combination with KCNQ3 (Kv7.3)46,47. The resulting Kv7 channels underlie the repolarizing M-current, which contributes to the resting membrane potential (RMP), action potential (AP) threshold and limits repetitive firing during prolonged depolarizing inputs48,49. Heterozygous mutations in KCNQ2 can cause epilepsy ranging in severity from benign self-limiting familial neonatal epilepsy (SLFNE) to severe developmental and epileptic encephalopathy (DEE), which is characterized by neonatal seizures and a spectrum of developmental deficits50,51,52,53. The drastically different clinical outcomes between SLFNE and DEE cases are associated with the impact of the variants on channel function that can cause haploinsufficiency or dominant negative loss-of-function effects. To interrogate the function of the KCNQ2∆E5-encoded protein, we generated a KCNQ2 expression plasmid lacking the corresponding exon 5. We transfected this plasmid (KCNQ2∆E5) or WT KCNQ2 (KCNQ2WT) in a Chinese hamster ovary (CHO) cell line that stably expresses KCNQ3 (ref. 39) and performed patch-clamp experiments to measure K+ current density across a series of voltage steps (Fig. 3a,b). Untransfected CHO cells expressing KCNQ3 exhibit negligible endogenous outward currents39; therefore, any measurements reflect the activity of the heterologous KCNQ2/KCNQ3 tetramer. To record across a large cohort of cells (n = 24–154), we conducted high-throughput automated patch clamp by using the SynchroPatch system (Fig. 3b, right). As an additional control, we included constructs expressing two recurrent, severe KCNQ2 DEE-associated variants known to exert a dominant negative effect (KCNQ2G279S and KCNQ2T274M)54. We first interrogated channel function in the homozygous state, in which the WT channel or mutant transgenes were introduced into KCNQ3-expressing CHO cells (Fig. 3b, top left). These experiments clearly showed that in stark contrast to the WT, KCNQ2∆E5-expressing cells conducted no outward currents (Fig. 3c–e, Extended Data Fig. 4d,e and Supplementary Table 3). As KCNQ2 functions in tetrameric complexes, we also considered whether the TDP-43-specific KCNQ2∆E5 possesses dominant negative activity. To examine this possibility, we tested channel function in the heterozygous state, in which mutant and WT KCNQ2 constructs were co-expressed at a 1:1 ratio (Fig. 3b, bottom left). KCNQ2∆E5 demonstrated a sharp reduction in current density at all voltage steps relative to WT/WT channels (approximately 44% reduction at +40 mV), an effect identical to DEE-KCNQ2T274M and slightly less severe than DEE-KCNQ2G279S (Fig. 3f–h, Extended Data Fig. 4b,c, Extended Data Fig. 4f,g and Supplementary Table 3). Notably, KCNQ2∆E5 did not impact the activation kinetics of the co-expressed WT KCNQ2 (Extended Data Fig. 3c). A comparison of the impact of KCNQ2∆E5 on conductance with other KCNQ2 variants (n = 74), including ones associated with SLFNE and DEE cases that we previously analyzed using the same assay54, clearly demonstrated that KCNQ2∆E5 acted like the average severe DEE variant typically associated with dominant negative loss-of-function activity (Fig. 3i).

a, Amino acid sequence (220–280) and illustration for KCNQ2WT, KCNQ2∆E5 and DEE variants KCNQ2G279S and KCNQ2T274M. Exon 5 deletion is shown by the dashed line. b, Schematic outlining heterologous expression model and SyncroPatch recordings. Left, KCNQ2 transgenes are transfected into CHO-Kv7.3 cells either alone (homozygous; top left) or with KCNQ2WT (heterozygous; bottom left). Right, cells are plated into 384-well plates for automated voltage-clamp recordings. c, SyncroPatch results for homozygous expression of KCNQ2 transgenes. Summary data (means; error bars, s.e.m.) for peak outward current density as a function of command voltage. P value determined by one-way ANOVA, #P < 0.001 for all steps starting at −50 to +40 mV for KCNQ2∆E5, G279S and T274M relative to WT. n represents cells denoted within the panel. d, Representative KCNQ2 current (XE991-sensitive) traces. No significant current is observed from KCNQ2∆E5, KCNQ2T274M or KCNQ2G279S co-expressed with KCNQ3. Scale bar, 50 pA, 200 ms. e, Peak current density at +40 mV plotted as a percentage of KCNQ2WT current (means; error bars, s.e.m.); n represents cells as in c. P value determined by one-way ANOVA, pairwise multiple comparisons by Holm–Sidak method. Circle represents the average of all cells, which are shown individually in Extended Data Fig. 4d,e. f, SyncroPatch results for heterozygous (×2) expression of KCNQ2 transgenes. Summary data (means; error bars, s.e.m.) for peak outward current density. One-way ANOVA, #P < 0.001 for all steps starting at −40 to +40 mV for KCNQ2∆E5, G279S and T274M relative to WT; n represents cells denoted within the panel. g, Representative traces as in d. Scale bar, 50 pA, 200 ms. h, Peak current density at +40 mV plotted as a percentage of KCNQ2WT (×2) current (means; error bars, s.e.m.); n represents cells as in f. P value determined by one-way ANOVA, pairwise multiple comparisons by Holm–Sidak method. Each circle represents the average of cells recorded from n = 3 independent experiments, as shown in Extended Data Fig. 4f,g. See Supplementary Table 3 for statistics for c and f. i, Top, schematic of correlation between clinical severity of KCNQ2 variants and M-current reduction. Variants are classified as PV (non-pathogenic population variants), SLFNE (mild cases of epilepsy) and DEE (severe developmental and epileptic encephalopathy). Bottom, SyncroPatch data from grouped KCNQ2 variants (heterozygous with KCNQ2WT, previously reported54) in comparison to KCNQ2∆E5. Each circle represents the mean value recorded from all cells of a particular variant. PVs (n = 15) result in 101 ± 8% of WT, SFLNE (n = 15) in 74.1 ± 6% of WT and DEE variants (n = 40) in 56.6 ± 5% of WT current density. KCNQ2∆E5-expressing cells are identical to the average of DEE variants (56.6 ± 5% of WT). No statistics were run; error bars, s.e.m. j, Top, schematic of cell surface biotinylation experiments. Bottom, western blot of input and cell surface fractions from WT/WT (GFP–KCNQ2WT + mCh–KCNQ2WT) and ∆E5/WT (GFP–KCNQ2∆E5 + mCh–KCNQ2WT). k,l, Quantification of surface levels of GFP–KCNQ2 (k) and mCh–KCNQ2 (l) normalized to transferrin receptor (TfR). Relative intensities are normalized to WT/WT in each experiment and plotted as fold change. Each shape represents the result from one of n = 3 independent experiments. P value determined by two-tailed paired t-test; error bars, s.e.m.
Structure prediction suggests that the exon skipping event in KCNQ2 mRNA is translated into an aberrant protein by disrupting the formation of a functional pore domain (Extended Data Fig. 1i and Extended Data Fig. 4a), which may explain decreased ion conductance. However, given the severity of this disruption within a highly conserved transmembrane region of the protein, it is also possible that KCNQ2∆E5 does not traffic properly to the membrane. To address this possibility, we generated a series of constructs with WT or aberrant KCNQ2∆E5 transgenes fused to GFP and introduced these into KCNQ3-expressing CHO cells. We first used manual patch clamp to confirm that the fusion did not impede the functional capacity of the WT channel and that the fused KCNQ2∆E5 protein was still unable to conduct current (Extended Data Fig. 4h). We then performed cell surface biotinylation assays to quantify the amount of WT KCNQ2 and KCNQ2∆E5 protein that localizes to the membrane (Fig. 3j, top). Total cell lysate (input), cell surface and flow-through fractions were analyzed by western blot for the membrane marker transferrin receptor and negative control histone H3 to confirm enrichment of cell surface proteins (Fig. 3j, bottom and Extended Data Fig. 4i,j). Cell surface fractions isolated from CHO cells transfected with GFP-tagged WT KCNQ2 or KCNQ2∆E5 that were co-expressed with mCherry-tagged WT KCNQ2 showed a dramatically lower surface abundance of monomeric mutant KCNQ2 compared to WT (>90% reduction) (Fig. 3k). Notably, we also observed an approximately 25% lower amount of mCherry-tagged WT KCNQ2 localized to the cell surface when co-expressed with mis-spliced KCNQ2 protein, suggesting a dominant negative trafficking effect (Fig. 3l). These results demonstrate that KCNQ2∆E5 protein does not traffic to the membrane efficiently and that 50% of KCNQ2∆E5 is sufficient to disrupt the appropriate trafficking of as much as 25% of WT protein. The dominant negative effect of KCNQ2∆E5 is in line with the fact that tetrameric Kv7 channels are typically assembled within the ER and subsequently transported to the membrane55. Collectively, our characterization reveals that TDP-43 dysfunction causes the formation of an aberrant KCNQ2 protein variant that is unable to conduct current or localize to the cell surface and exerts dominant negative effects.
KCNQ2∆E5 accumulates in the ER and causes neuronal hyperexcitability
We next sought to address the relevance of these findings in the context of human neurons. To interrogate the impact of KCNQ2∆E5 on neuronal homeostasis in the absence of all the additional effects of TDP-43 dysfunction, we used CRISPR–Cas9 genome engineering to delete exon 5 in a healthy control human embryonic stem cell line (Fig. 4a and Extended Data Fig. 5a). We generated a clone with a homozygous deletion of KCNQ2 exon 5 (KCNQ2∆E5/∆E5) and confirmed that the cells had correct on-target zygosity by performing an allele copy number assay (Extended Data Fig. 5b) and a normal karyotype (Extended Data Fig. 5c). We then differentiated the KCNQ2∆E5/∆E5 and parental isogenic WT cell lines into cortical excitatory glutamatergic neurons42,43 (Extended Data Fig. 5d), validated the KCNQ2∆E5/∆E5 variant expression (Fig. 4b) and performed immunocytochemistry with a KCNQ2 antibody to assess the subcellular localization of the endogenous protein after 50–60 days in culture. To mark the axon initial segment (AIS), which is the site where KCNQ2 channels are predominantly localized in mature neurons, we immunostained cells with an antibody against ankyrin-G (ANK3). We found that WT neurons exhibited diffuse KCNQ2 signal in the cell body and, occasionally, a distinct signal within the AIS (Fig. 4c, top). In stark contrast, all KCNQ2∆E5-derived neurons exhibited an irregular KCNQ2 distribution pattern with very little to no signal within the AIS and dramatic intracellular focal accumulations within the cell body (Fig. 4c, bottom and Extended Data Fig. 5e). Quantification revealed that 100% of mutant neurons exhibited KCNQ2 accumulation (Fig. 4d), while analysis of the KCNQ2 signal distribution showed a significant increase in standard deviation (Fig. 4e), mean and maximum intensity (Extended Data Fig. 5f,g) in mutant neurons relative to isogenic controls, suggesting severe mislocalization of the TDP-43-associated KCNQ2∆E5 protein variant. The dramatic focal accumulation of mis-spliced protein prompted us to determine the precise site of localization. Immunostaining with the ER marker calnexin revealed substantial overlap, demonstrating that the majority of KCNQ2∆E5 protein is retained within the ER and does not traffic to the neuronal surface membrane (Fig. 4f), in agreement with the surface localization analysis in heterologous cells (Fig. 3j–l).

a, Schematic of CRISPR–Cas9 editing of embryonic stem cells (ESCs) to generate homozygous KCNQ2∆E5/∆E5 lines and differentiation into cortical excitatory neurons. b, RT–PCR shows that KCNQ2∆E5/∆E5 neurons express KCNQ2∆E5 and KCNQ2WT/WT express KCNQ2WT. c, Representative immunocytochemical images of neurons stained with DAPI, MAP2, KCNQ2 and ANK-G. Arrowheads denote the beginning of AIS. Asterisk (*) denotes KCNQ2 localization in the AIS for WT (top) and accumulated in the soma for KCNQ2∆E5/∆E5 neurons (bottom). Yellow dashed line outlines the cell body. Scale bar, 10 μm. d, Percentage of WT (n = 51, 0%) and KCNQ2∆E5/∆E5 (n = 79, 100%) neurons with somatic accumulation of KCNQ2. e, Quantification of KCNQ2 signal intensity variation. Statistical significance determined by unpaired, two-tailed Student’s t-test. Data are shown as mean ± s.e.m.; each circle corresponds to one neuron. a.u., arbitrary units. f, Representative image of KCNQ2∆E5/∆E5 neuron stained with DAPI, MAP2, calnexin and KCNQ2. Top, maximum Z-projection; middle, 3D views with neuron rotated forward; bottom, 3D views from below. g, Representative raster plot of neuronal activity recorded in a MEA well for control (top) and KCNQ2∆E5/∆E5 (bottom). Rows depict individual electrodes; black lines represent single spikes; blue lines indicate ‘bursts’. h–o, Longitudinal analysis of neuronal MEA recordings for days 9–43 (h–k) or 12–43 (l–o). Data are presented as means from n = 3 independent experiments (n = 59 wells for WT and n = 64 for KCNQ2∆E5/∆E5); circles represent means; shaded areas, s.e.m. Two-way repeated-measures ANOVA was used for h, i and l; mixed-effects model restricted maximum likelihood) for j, k, m, n and o. P values: in black indicate genotype effects and in pink reflect genotype × day interactions. MEA metrics are indicated within each panel. p, Experimental schematic (top) and representative raster plot from MEA wells (bottom) during treatment with the Kv7 agonist ICA-069673 (1 μM). For each metric, pre-ICA-069673 and post-ICA-069673 values are represented as the percent of baseline values (right). Each circle-pair represents the change in activity recorded from a well (total number of wells from two replicate MEA plates were combined for analysis: n = 20 for WT and n = 19 wells for KCNQ2∆E5/∆E5. P value determined by unpaired, two-tailed Student’s t-test.
To determine the neurophysiological implications of the KCNQ2∆E5 variant, we plated equal numbers of mutant and isogenic control neurons on multi-electrode arrays (MEAs) and recorded spontaneous neuronal activity over the course of several weeks in culture (days 10–45; n = 3 independent differentiations; Fig. 4g–o). Neurons started firing after 10–15 days and became progressively more active thereafter (Fig. 4h). Although there were only minor differences in the number of active electrodes (Fig. 4h), KCNQ2∆E5 neurons exhibited a dramatic shift in their pattern of firing activity (Fig. 4g), with a significant increase in the number of spikes (Fig. 4i), weighted mean firing rate (Fig. 4j), increased irregularity as measured by an increase in inter-spike interval coefficient of variation (Fig. 4k) and several bursting metrics relative to isogenic WT controls (Fig. 4l–o). Most of their spikes occurred within bursts, reflected by a significant increase in number of bursts (Fig. 4l), burst percentage (Fig. 4m) and number of spikes per burst (Fig. 4n), while there were only minor effects on the duration of each burst (Fig. 4o). Notably, treatment of KCNQ2∆E5 neurons wih the Kv7 agonist ICA-069673 had no significant effect on firing activity (active electrodes, weighted mean firing rate, number of bursts), consistent with non-functional Kv7 channels, whereas it caused a substantial reduction in most metrics in isogenic WT controls (Fig. 4p). These widespread alterations in neuronal spiking behavior are consistent with the critical role of Kv7 channels in regulating excitability39.
TDP-43-depleted MNs exhibit intrinsic hyperexcitability that can be rescued by splice-modulating KCNQ2 ASOs
Having established that expression of KCNQ2∆E5 alone is sufficient to induce dramatic neuronal excitability alterations, we next sought to dissect the contribution of this mis-spliced protein in the context of TDP-43 loss. We first set up a screening assay to identify splice-modulating ASOs (smASOs) that selectively repress the expression of the KCNQ2∆E5 isoform in iPS cell-derived spinal MNs56 (Extended Data Fig. 6a). We designed a series of ~20 bp ASOs with 2’-O-methoxyethyl sugars and phosphorothioate linkages, spanning the intronic regions upstream and downstream of exon 5, including the simple UG-rich repeat (Fig. 5a, top and Supplementary Table 4). We then differentiated WT MNs, depleted TDP-43 (Extended Data Fig. 6b), delivered the smASOs gymnotically57 and quantified the expression of mis-spliced KCNQ2∆E5 by RT–qPCR (Fig. 5a, bottom). We found several candidate smASOs that significantly repressed the level of KCNQ2∆E5, which targeted both the UG-rich repeat sequence as well as the 3’ flanking intronic region of exon 5 (Fig. 5a).

a, Top, KCNQ2 and smASO design strategy. Bottom, RT–qPCR of KCNQ2∆E5 expression. P value determined by an unpaired, two-tailed t-test of each smASO relative to splice-modulating control ASO (smCTRL). b, Experimental schematic: iPS cell MNs are treated with a control ASO (CTRL) or an ASO to knock down TDP-43 (TDP-KD) and smASOs between days 20 and 30 for analysis; images of neurons analyzed by patch clamp. Three treatment groups: (1) CTRL + smCTRL, (2) TDP-KD + smCTRL and (3) TDP-KD + smKCNQ2. c, RT–qPCR of TARDBP expression. P value determined by a paired, two-tailed t-test; ns: not significant. d, RT–PCR shows KCNQ2 mis-splicing upon TDP-43 KD and partial rescue with smASO treatment. e, RT–qPCR of KCNQ2∆E5. P value determined by a paired, two-tailed t-test. For a, c and e, each circle represents one of n = 4 biological replicates; data are presented as means; error bars, s.e.m. f, Representative traces of spontaneous APs counted while cells were at RMP for all three groups. Asterisks denote individual spontaneous APs; black dashed line denotes 0 mV; colored dashed line denotes RMP. Scale bar, 10 mV, 1 s. g, RMP recorded from all three groups; number of neurons: (1) n = 53, (2) n = 63, (3) n = 30. P value determined by one-way ANOVA (F2,144 = 8.3, P = 0.0004) followed by Tukey’s multiple comparisons test. h, Spontaneous AP frequency recorded from all three groups; number of neurons: (1) n = 54, (2) n = 63, (3) n = 30. P value determined by one-way ANOVA (F2,143 = 16.82, P < 0.0001) followed by Tukey’s multiple comparisons test. i, Representative traces of the post-burst mAHP. Scale bar, 5 mV, 250 ms. j, Post-burst mAHP recorded from all three groups; number of neurons: (1) n = 51, (2) n = 55, (3) n = 28. P value determined by one-way ANOVA (F2,131 = 4.7, P = 0.0105) followed by Tukey’s multiple comparisons test. k, Post-burst sAHP recorded from all three groups; number of neurons: (1) n = 51, (2) n = 55, (3) n = 28. P value determined by one-way ANOVA (F2,131 = 5.01, P = 0.008) followed by Tukey’s multiple comparisons test. For g, h, j and k, each circle represents neurons recorded from n = 4 biological replicates. Data are presented as means; error bars, s.e.m.
We next set up an experimental paradigm to interrogate the intrinsic excitability of mature iPS cell MNs by whole-cell patch-clamp electrophysiological analysis (Fig. 5b). In these experiments, conducted at Northwestern University, we depleted TDP-43 levels by treating MNs with an RNase H-dependent gapmer ASO (Fig. 5c), and repressed mis-spliced KCNQ2∆E5 by treating with a lead candidate KCNQ2 smASO identified in our screen (Extended Data Fig. 6c,d), or a scrambled control ASO of matched chemistry (Fig. 5d,e). The substantial reduction in TDP-43 levels (~85% reduction) (Fig. 5c) caused a substantial increase in the expression level of KCNQ2∆E5 (~37-fold increase), which was significantly, but not completely, repressed by simultaneous treatment with the KCNQ2 smASO (~35% reduction) (Fig. 5d,e). Kv7 encoded M-current contributes to the RMP and AP thresholds that attenuate excitability, as well as to modulate the post-burst afterhyperpolarization (AHP), which limits repetitive AP firing48,49,58. Therefore, we performed current-clamp measurements to analyze RMP and AP properties, spontaneous firing and post-burst AHP (Fig. 5f–k and Extended Data Fig. 6e–n). TDP-43 knockdown (KD) MNs exhibited a significantly depolarized RMP relative to MNs treated with scrambled control ASO (Fig. 5f,g). Consistent with a depolarized state, TDP-43 depletion caused hyperexcitability as MNs were more active than controls, as measured by the number of spontaneous APs recorded at rest (Fig. 5f,h). These changes, which are consistent with dysfunctional Kv7 channels, were restored in MNs treated with the KCNQ2 smASO. The treatment resulted in a significant recovery in RMP and associated reduction in spontaneous AP frequency (Fig. 5f–h). At the same time, although there was a substantial reduction in post-burst medium AHP (mAHP) and slow AHP (sAHP) upon TDP-43 KD, these properties were not restored by KCNQ2 smASO treatment (Fig. 5i–k). Similarly, changes in fast AHP and mAHP measured following a single AP were not restored by KCNQ2 smASO treatment (Extended Data Fig. 6m,n), which may either reflect the contribution of other K+ channels to them or insufficient repression of KCNQ2∆E5 by the smASO. We did not find any substantial effects upon TDP-43 KD on other intrinsic cell or AP properties (Extended Data Fig. 6e–l). These experiments suggest that TDP-43 loss causes intrinsic neuronal depolarization and hyperexcitability that can be rescued by repressing mis-spliced KCNQ2∆E5.
TDP-43-depleted cortical neurons exhibit hyperexcitability that can be rescued by KCNQ2 smASOs
To increase the rigor of our findings and substantiate their relevance across other neuronal subtypes, we performed a series of patch-clamp measurements in iPS cell-derived cortical neurons59 that were independently conducted at University College London. We used a recently developed iPS cell line in which endogenous TARDBP was edited to harbor a HaloTag, which allows for complete degradation of TDP-43 through a proteolysis targeting chimera (PROTAC)60 (Extended Data Fig. 7a,b). Although tagging TDP-43 slightly compromises its function, HaloPROTAC-mediated KD caused a substantial increase in the expression of KCNQ2∆E5 (Extended Data Fig. 7c). We differentiated NGN2-based cortical neurons59 and performed whole-cell patch-clamp analysis 2 weeks after TDP-43 KD (Fig. 6a, top). Voltage-clamp recordings revealed a significant reduction in M-current deactivation kinetics in neurons depleted of TDP-43 relative to controls (Fig. 6c). Critically, acute treatment with the M-current inhibitor XE991 showed a reduction in control neurons but had no effect in TDP-43 KDs, suggesting loss of Kv7 channels (Fig. 6c). To dissect the contribution of KCNQ2 mis-splicing to these TDP-43-dependent effects, we treated TDP-43 KD neurons with the smASO and found a significant, but not complete, repression in the expression of KCNQ2∆E5 (Fig. 6a, bottom and Fig. 6b). The repression of KCNQ2∆E5 expression resulted in a marked recovery of M-current, which was responsive to XE991 treatment (Fig. 6d).

a, Experimental schematic: cortical neurons were differentiated from HaloTag TDP-43 iPS cells and treated with vehicle (CTRL) or PROTAC to knock down TDP-43. Some TDP-43 KD neurons were also treated with control smASO or smKCNQ2 to repress KCNQ2∆E5. b, Left, RT–PCR assay shows KCNQ2 mis-splicing upon TDP-43 KD and partial rescue with smASO treatment. Right, RT–qPCR of KCNQ2∆E5. P value determined by unpaired, two-tailed t-test. Data are means; error bars, s.e.m. Circles are one of n = 3 biological replicates. c, Left, M-current from CTRL (n = 29) and TDP-43 KD (n = 36) neurons. P value determined by unpaired, two-tailed t-test. Right, representative traces and measurements of individual neurons for pre-XE991 and post-XE991 treatment (n = 7). P value determined by paired, two-tailed t-test. d, Left, M-current from TDP-43 KD + smCTRL (n = 27) and TDP-43 KD + smKCNQ2 (n = 28) neurons. P value determined by unpaired, two-tailed t-test. Right, representative traces and measurements of individual neurons for pre-XE991 and post-XE991 treatment (n = 8). P value determined by paired, two-tailed t-test. For c and d, circles represent one cell; data are means; error bars, s.e.m. Scale bar, 100 pA, 500 ms. e,f, Representative traces of spontaneous APs. e, CTRL neurons were not firing at rest (CTRL, −XE991), while XE991 caused increasing spiking (CTRL, +XE991). TDP-43 KDs were active at rest (TDP-43 KD, −XE991), while XE991 did not cause significant firing changes (TDP-43 KD, +XE991). Scale bar, 20 mV, 500 ms. f, TDP-43 KD + smCTRL neurons were active at rest (TDP-43 KD + smCTRL, −XE991) and unresponsive to XE991 (TDP-43 KD + smCTRL, +XE991). TDP-43 KD + smKCNQ2 neurons were quiet at rest (TDP-43 KD + smKCNQ2, −XE991) and responsive to XE991 (TDP-43 KD + smKCNQ2, +XE991). Scale bar, 20 mV, 500 ms. g, Left, RMP from CTRL (n = 29) and TDP-43 KD (n = 35) neurons. Right, RMP from TDP-43 KD + smCTRL (n = 27) and TDP-43 KD + smKCNQ2 (n = 27) neurons. P values determined by unpaired, two-sided t-test; each circle represents one cell. Data are means; error bars, s.e.m. h, Left, spontaneous AP frequency from CTRL (n = 29) and TDP-43 KD (n = 37) neurons. P value determined by unpaired, two-sided t-test. Right, AP measurements of individual CTRL (top) and TDP-43 KD (bottom) neurons for pre-XE991 and post-XE991 treatment. P values determined by paired, two-sided t-test; each circle represents one cell. Data are means; error bars, s.e.m. i, Left, spontaneous AP frequency from TDP-43 KD + smCTRL (n = 27) and TDP-43 KD + smKCNQ2 (n = 27) neurons. P value determined by unpaired, two-sided t-test. Right, AP measurements of individual KD + smCTRL (top, n = 9) and KD + smKCNQ2 (bottom, n = 8) neurons for pre-XE991 and post-XE991 treatment. P values determined by paired, two-sided t-test; each circle represents one cell. Data are means; error bars, s.e.m.
Moreover, as in spinal MNs, we found that loss of TDP-43 in these cortical neurons caused a depolarization of the RMP, which was rescued by the smKCNQ2 ASO (Fig. 6e–g). Similarly, TDP-43 KD neurons showed a substantial increase in the frequency of spontaneous AP firing relative to controls (Fig. 6e and Fig. 6h, left). As expected, treating controls with the Kv7 inhibitor XE991 caused an increase in AP frequency, indicative of functional Kv7.2 channels (Fig. 6e and Fig. 6h, top right), while treating TDP-43 KDs with XE991 had no effect, demonstrating the lack of functional Kv7.2 channels downstream of TDP-43 KD (Fig. 6e and Fig. 6h, bottom). These results suggest that the increase in spontaneous AP in TDP-43 KD neurons is driven by dysfunctional Kv7 channels. Remarkably, the smKCNQ2 ASO repressed this spontaneous hyperexcitability and restored the sensitivity of the TDP-43 KD neurons to XE991 treatment (Fig. 6f,i). We did not find any substantial effects on TDP-43 KD on the capacitance or input resistance of cortical neurons (Extended Data Fig. 7d,e). Lastly, reduction of TDP-43 levels caused a more depolarized mAHP (following a single AP) (Extended Data Fig. 7f), a property that was restored by the smKCNQ2 ASO along with its responsiveness to Kv7 inhibition by XE991 (Extended Data Fig. 7g). Notably, although this metric was similarly affected by TDP-43 KD in spinal MNs, smKCNQ2 treatment did not induce a recovery in those cells (Extended Data Fig. 6n).
Altogether, these results demonstrate that in cortical glutamatergic neurons, TDP-43 loss causes intrinsic hyperexcitability driven by KCNQ2 mis-splicing. The use of the smASO across both spinal and cortical neurons addresses a direct cause-and-effect relationship between mis-spliced KCNQ2 and TDP-43-dependent excitability alterations.
KCNQ2 accumulates focally in postmortem neurons from patients with ALS
To investigate the localization of KCNQ2 protein in ALS, we performed immunohistochemistry on spinal cord sections at two independent sites, using two cohorts of postmortem tissue (Supplementary Table 5). First, at Northwestern University, we used sections from three sporadic ALS cases and three non-neurological disease controls to characterize TDP-43 and KCNQ2 levels. We found that in non-neurological control individuals, large MAP2-positive MNs located in the ventral horn of the spinal cord exhibited a diffuse KCNQ2 pattern of immunoreactivity throughout the soma, whereas TDP-43 had clear nuclear localization (Fig. 7a and Extended Data Fig. 8a). In contrast, MNs from patients with ALS exhibited cytoplasmic focal accumulations of KCNQ2 (Fig. 7a and Extended Data Fig. 8b). This significant difference between ALS cases and controls was quantified by measuring KCNQ2 signal intensity variation across all cases and controls (Fig. 7b). Notably, these KCNQ2 accumulations exhibited substantial overlap with ubiquitin B, suggesting that a significant proportion of KCNQ2 in diseased neurons is likely aggregated (Fig. 7c and Extended Data Fig. 8c,d). To increase the confidence in our findings, we evaluated KCNQ2 in lumbar spinal cord sections of an expanded set of 15 ALS cases (n = 11 patients with sporadic ALS and n = 4 cases of C9orf72 familial ALS) and three non-neurological controls in experiments conducted independently at Columbia University (Supplementary Table 5). Analysis of large neurons found in the ventral horn showed that KCNQ2 was also substantially altered in this cohort of patients with ALS. We specifically found that KCNQ2 intensity was significantly higher in the neurons of patients with ALS than it was in controls (Fig. 7d and Extended Data Fig. 8e–g), and we also observed increased focal accumulations that were independent of increased KCNQ2 expression (Fig. 7d,e and Extended Data Fig. 8e,f) and overlapped with ubiquitin B (Extended Data Fig. 8h), mirroring our original observations at Northwestern University. These alterations were consistent across familial and sporadic cases as well as across genetic subtypes (Fig. 7f). Finally, we note that in ALS MNs, KCNQ2 co-stained with the luminal ER marker GRP78 (Fig. 7g), suggesting that the focal KCNQ2 accumulations in patients with ALS are within the ER, as we observed in in vitro human neurons expressing 100% KCNQ2∆E5 protein. Intriguingly, although there was no clear separation in the levels of KCNQ2 accumulation between neurons with or without pathological pTDP-43 (Fig. 7h,i and Extended Data Fig. 8e,f), in some instances, we observed dramatic co-aggregation of KCNQ2 with pTDP-43 cytoplasmic aggregates (Fig. 7h). These experiments do not confirm that the accumulated KCNQ2 protein in tissue from ALS patients was derived from mis-spliced transcript. However, they demonstrate that at this terminal disease stage, there is substantial focal accumulation and ubiquitination of total KCNQ2 protein, perhaps as a result of the dominant negative effects of KCNQ2∆E5 protein on the trafficking of the WT isoform that we observed in vitro.

a–c, Samples analyzed at Northwestern University (NU cohort). a, Immunohistochemistry of neurons from non-neurological control (n = 3) and sporadic ALS (sALS) (n = 3) spinal cord sections stained for DAPI (blue), MAP2 (white), KCNQ2 (green) and TDP-43 (red). b, KCNQ2 signal intensity variation was significantly higher in sALS cases: controls, (1) n = 25, (2) n = 21, (3) n = 14; sALS, (1) n = 19, (2) n = 20, (3) n = 30. Data are shown as coefficients of variation (mean-normalized standard deviation); each circle represents one neuron. c, Neurons stained for DAPI (blue), MAP2 (white), KCNQ2 (green) and ubiquitin (red). The square box denotes the magnified region shown at the bottom. Scale bar, 10 μm. d–i, Samples analyzed at Columbia University (CU cohort). d, Immunohistochemistry of MNs from non-neurological control and ALS lumbar spinal cord sections stained for DAPI (blue), KCNQ2 (green) and pTDP-43 (red). e, KCNQ2 signal intensity variation was significantly higher in ALS cases: controls, n = 29 neurons from three individuals; ALS, n = 142 neurons from 15 patients. f, KCNQ2 signal intensity variation was significantly higher in C9orf72 repeat expansion and sALS cases: controls, n = 29 neurons from three individuals; C9orf72, n = 40 from four patients; sALS, n = 102 from 11 patients. g, Neurons stained for DAPI (blue), KCNQ2 (green) and GRP78 (orange). h, sALS neurons from spinal cord sections stained for DAPI (blue), KCNQ2 (green) and pTDP-43 (red). Representative cases of localization of KCNQ2 in large globular pTDP-43 aggregates. i, KCNQ2 signal intensity variation was significantly higher in neurons from ALS patients, irrespective of pTDP-43 pathology: control, n = 29; ALS without TDP-43 pathology, n = 86; ALS with TDP-43 pathology, n = 56. P values in b and e determined by unpaired, two-tailed t-test with Welch’s correction; in f and i by Brown–Forsythe and Welch ANOVA test followed by Benjamini and Hochberg correction for multiple comparisons. Scale bars for NU, 10 μm; for CU, 15 μm. Detailed sample information is included in Supplementary Table 5.
Discussion
The functional implications of TDP-43 pathophysiology in human MNs has been intensely investigated since it was identified in ubiquitinated aggregates of postmortem brains from individuals affected with ALS and FTD61. Here, we find that TDP-43 dysfunction yields altered KCNQ2 mRNA that escapes degradation and is translated into a non-functional neoprotein without current conduction capacity that causes inherent hyperexcitability in neurons. This de novo protein variant can accumulate in the ER of neurons, inhibit the trafficking of WT KCNQ2 to the membrane and likely contributes to MN hyperexcitability described in patients with ALS/FTD.
The mis-splicing of KCNQ2 represents a spurious, TDP-43-dependent event. We observed it in transcriptomes of stem cell-derived spinal-like cholinergic MNs and cortical-like glutamatergic neurons in vitro as well as in the spinal cord and motor cortex of tissue from patients with ALS. We detected KCNQ2∆E5 in sporadic and familial ALS with known predisposition for TDP-43 aggregation, whereas it was not detected in neurological controls or typically non-TDP-43 ALS cases, such as those with SOD1 mutations. At present, there is a paucity of reliable biomarkers for the diagnosis of ALS. Our data suggest that KCNQ2∆E5, which is absent in neurotypical individuals and abundant in patients with ALS and FTD-TDP, is a potential molecular biomarker of TDP-43 dysfunction. Indeed, recent work demonstrated that the KCNQ2∆E5 neoprotein can be detected using mass spectrometry in cerebrospinal fluid samples from patients with ALS/FTD spectrum disorders62.
TDP-43 has hundreds of RNA targets in the nervous system45,63, is required for regulated alternative splicing and is known to serve both as a splicing repressor and enhancer. To date, there has been a strong focus on studying splicing events that are derepressed upon TDP-43 loss, particularly non-conserved cryptic exons, which lead to dramatic loss of protein or the expression of neopeptides that can be useful as biomarkers62. KCNQ2 mis-splicing differs from such events, as exon 5 is deeply conserved and constitutively included in mRNA in the CNS and is therefore neither an alternatively spliced locus nor a cryptic exon. Nevertheless, like some other TDP-43-regulated cryptic exons, our analysis demonstrates that KCNQ2∆E5 is not conserved in mice. Murine CNS cells lack the UG repeat element that is possibly critical for TDP-43 recruitment, and do not rely on TDP-43 for correct pre-mRNA processing. Interestingly, mouse models based on the artificial nuclear translocation of TDP-43 have revealed cortical hyperexcitability31,32, and the precise mechanism of these effects has not been well-defined. The lack of conservation in KCNQ2 splicing is similarly observed for other TDP-43 target genes, such as STMN2 (refs. 11,12) and UNC13A9,10, suggesting evolutionarily divergent mechanisms for regulating these genes in neurons. It also underscores a need for the development of humanized mouse models64 to evaluate the significance of TDP-43 pathophysiological mechanisms in the context of an intact nervous system65.
Examination of neuronal nuclei with decreased TDP-43 abundance41 revealed that at least 50% of total KCNQ2 may be mis-spliced in affected cells. This is probably an underestimation, as it is based on bulk RNA-seq and may reflect a range of mis-spliced KCNQ2 within individual neurons (0–100% mis-splicing), depending on the degree of TDP-43 pathology within the neuronal population. The application of single-cell technologies and long-read sequencing will illuminate the precise extent and diversity of misprocessed KCNQ2 transcripts within the CNS. Notably, the dominant negative trafficking effects of KCNQ2∆E5 we identified here, as well as the prominent aggregation of KCNQ2 in patient tissue, suggest that KCNQ2 channel dysfunction may be pronounced, at least at the terminal stages of the disease.
Although TDP-43 is undoubtedly critical for the health of MNs, questions that remain unanswered involve determining which of the many transcripts regulated by TDP-43 control important neuronal pathways that are altered in patients with ALS, which of those are related to the origins of the disease and which hold the most promise as therapeutic targets. KCNQ2 channels mediate the repolarizing M-current, which activates to dampen neuronal excitability and limits repetitive firing following bursts of APs. Mutations in this gene can cause epileptic syndromes46,47,66. Our findings, that misprocessed KCNQ2 results in a non-functional protein—effectively a ‘dead’ channel—and causes intrinsic hyperexcitability and striking alterations in the distribution of spiking in human neurons have strong implications for patients with ALS and FTD-TDP. We recently showed that cortical neurons with a heterozygous loss-of-function mutation in KCNQ2 associated with DEE exhibited a dyshomeostatic adaptation that caused an increased burst firing pattern39. The bursting phenotype we observed in KCNQ2∆E5 neurons is highly similar. Although seizures are not typically seen in patients with ALS, neuronal hyperexcitability is a broadly observed feature in both familial and sporadic disease16,17,18,19. As TDP-43 loss-of-function effects are both gradual and heterogeneous within the CNS, mis-splicing of KCNQ2 would not be expected to cause spontaneous epileptic seizures but could lead to focal disruptions in neuronal firing. Furthermore, TDP-43 mis-splicing of other targets involved in synaptic communication, such as UNC13A60, may also lead to synaptic deficits that prevent network-wide epileptiform activity, despite individual neurons being inherently hyperexcitable.
Our findings provide a direct mechanistic link between TDP-43 dysfunction and decreased neuronal M-currents that are probably contributing to the cortical hyperexcitability seen in patients. Intriguingly, the anticonvulsant ezogabine, which had been evaluated in clinical trials as a treatment for ALS38, augments KCNQ2-5 channel-encoded current amplitudes67,68. Given that the degree of hyperexcitability may be prognostic of ALS disease progression and survival23,24, correction of mis-spliced KCNQ2∆E5 may be an attractive therapeutic target for ALS. Pharmacological strategies aimed at restoring M-current levels may not stem the tide of Kv7.2 activation, given some of the dominant negative trafficking effects of the mis-spliced protein. smASOs like the ones we developed here constitute an alternative therapeutic strategy, as they would selectively increase functional Kv7.2 activity in the most affected neurons with TDP-43 pathology.
Importantly, several of our key findings—including the electrophysiological characterization of TDP-43-depleted neurons, the beneficial effects of the KCNQ2 smASO on excitability and the histopathological characterization of KCNQ2 in ALS postmortem tissue—were verified across two independent laboratories. These experiments—performed using different iPS cell-model systems and patient tissues, operators and reagents—serve as strong proof of the robustness and reproducibility of our observations. At the same time, our work has some limitations, primarily associated with the lack of our ability to study mis-spliced KCNQ2 over the course of disease progression in an appropriate in vivo model, as well as in neurons derived from sporadic and familial ALS iPS cells. Notably, such longitudinal assessment is also difficult in the clinical field, for which interpretation of threshold-tracking transcranial magnetic stimulation (TT-TMS) and short-interval intracortical inhibition measurements of axonal and neuronal excitability are confounded by progressive neuronal loss. Unfortunately, the lack of spontaneous pathological TDP-43 phenotypes such as nuclear depletion or cytoplasmic aggregation in iPS cell-derived neurons69, probably because of their embryonic nature70, constitutes a major bottleneck in the field. Notably, recent findings using iPS cell-derived neurons with ALS-associated TDP-43 mutations highlighted excitability alterations and structural changes in the AIS29, which is the site of the highest density of KCNQ2 channels. Although TDP-43 KD does not emulate potential gain-of-function effects driven by TDP-43 aggregation, it is able to capture key molecular defects that are seen in patients with ALS71,72.
Our observation of the cytosolic accumulation of KCNQ2∆E5 is an unexpected consequence of KCNQ2 misprocessing that underscores the prioritization of examining precise splice variants instead of genetic loss-of-function to study TDP-43 targets. It underscores a direct link between proteotoxic stress and neuronal hyperexcitability acting downstream of TDP-43 dysfunction in patients with ALS and FTD-TDP. Lastly, we note the possibility that the de novo KCNQ2∆E5 variant may trigger autoimmune deregulation, constituting another collateral effect of TDP-43 dysfunction that may be contributing to ALS pathophysiology.
Methods
Transcriptomic datasets
We obtained the following published datasets from the NCBI Gene Expression Omnibus or the European Nucleotide Archive: FACS-purified MNs depleted of TDP-43: Eggan lab11, GSE121569; purified TDP-43-high and TDP-43-low neuronal nuclei datasets: Lee lab41, GSE126543 (we used six out seven datasets with sufficient read coverage for KCNQ2); i3Neurons, SH-SY5Y and SK-N-DZa TDP-43 depleted datasets: Fratta lab9, PRJEB42763; and mouse TDP-43 depletion studies: Cleveland lab45, GSE27394. Raw sequencing data were mapped to the human reference genome sequence (GRCh38.p13) using HISAT2 (ref. 73). The raw reads for TDP-43 iCLIP from SH-SY5Y cells published by the Fratta lab (E-MTAB-10297) were mapped using bowtie with parameters -a -l 10 -m 1 -v 1. We obtained 1,124 RNA-seq datasets from the Target ALS/New York Genome Center ALS Consortium, consisting of de-identified postmortem brain samples from CNS tissues; we mapped fastq files to the human reference genome sequence (GRCh38.p13) using HISAT2 (ref. 73). Differential gene expression analyses were done using the DESeq2 (ref. 74) package in R.
Detection of KCNQ2, STMN2 and UNC13A mis-splicing in postmortem RNA-seq datasets
A custom script was used to count reads that indicate mis-splicing. In brief, we filtered the BAM files to only include unique, high-quality mapped reads using Samtools (v.1.18)75. We mined these files to identify split reads that mapped across junctions of interest with no mismatches and with at least 15 nucleotides mapped across each side of the junction. We used the ‘subject group’ metadata provided by the New York Genome Center to assign sample IDs to control, ALS or other neurological disease. We stratified samples into sporadic, C9orf72 or non-TDP-43 using ‘reported genomic mutations from sites’.
Splicing analysis
Splice junctions were mapped using the MAJIQ algorithm (v.2.0) under default conditions40. Splice graphs and known or novel local splice variants were defined through the use of the MAJIQ builder provided with comprehensive human gene annotations (GENCODE release 36: GRCh38.p13) and uniquely mapped BAM files as input. The MAJIQ Quantifier was used to calculate relative abundances (percent selected index) for all defined junctions from the perspective of sources and targets40. Following this step, the MAJIQ dPSI function was used to compare different conditions, and the Voila methods outputted the results into tabular formats containing local splice variants that underwent significant changes between specified conditions. Custom R scripts were written to process all significant local splice variants to obtain significantly changing splice junctions. We used probability changing > 0.8 and mean_dpsi_per_lsv_junction > 0.1. These criteria provided junctions with a strong statistical likelihood of at least a 10% change between conditions. MAJIQ also defined de novo junctions based on the provided GENCODE annotations. We adopted a more stringent definition in which de novo junctions were required to have a PSI < 0.05 in control conditions. This allowed us to distinguish de novo splicing that was caused by poor annotation of human neurons versus those that are repressed by TDP-43. Sashimi plots were generated using Integrative Genomics Viewer76.
Cell culture
Stem cells at Northwestern University were maintained on plates coated with Matrigel Basement Membrane Matrix (Corning, CLS354234) in mTeSR or mTeSR Plus media (StemCell Technologies, 100-0276) and passaged with Accutase (Gibco, A11105). The homozygous KCNQ2∆E5 engineered cell line was created using the WA01 (H1) human embryonic stem cell line from WiCell. The iPS cell lines 18a77 and KOLF2.1J78 were used to differentiate spinal MNs and cortical NGN2-based neurons at Northwestern University.
Halo-TDP-43 iNeurons59 at University College London were derived from a WTC11 background with a doxycycline-inducible NGN2 construct in the AAVS1 safe-harbor locus and a CRISPRi construct in the CLYBL safe-harbor locus60. These were maintained in E8 Flex Medium (Thermo) on Geltrex (Thermo) coated plates and passaged with TrypLE (Thermo) at 80% confluency.
CHO cells expressing human KCNQ3 (CHO-Q3; Kv7.3, GenBank accession no. NM_004519) were generated using Flp-In-CHO cells54. Cells were grown in F-12 nutrient mixture medium (GIBCO/Invitrogen) supplemented with 10% FBS (ATLANTA Biologicals), Zeocin (100 μg ml−1), hygromycin B (600 μg ml−1), penicillin (50 U ml−1) and streptomycin (50 μg ml−1).
SH-SY5Y cells (ATCC, CRL-2266) were grown in DMEM (Gibco, 11965092) supplemented with 15% FBS (Takara Bio, 631106), GlutaMAX (Gibco, 35050061), penicillin (50 U ml−1) and streptomycin (50 μg ml−1).
Primary glial cell cultures were derived from postnatal day 0–2 CD-1 mice (Charles River). Brain cortices were dissected free of meninges in dissection buffer Hank’s Balanced Salt Solution (Thermo Fisher), then digested with trypsin (Thermo Fisher) and DNaseI (Worthington) for 10 min at 37 °C. The tissue was dissociated in glia medium: DMEM (Corning, 15–013-CV) supplemented with GlutaMAX, D-glucose, 10% normal horse serum (Life Technologies) and penicillin–streptomycin (Thermo Fisher). After centrifugation and resuspension, cells were filtered through a 0.45 µm cell strainer and plated on poly-D-lysine (PDL)-coated plates with glia media for 2 weeks. Glial cultures were tested for mycoplasma, dissociated for expansion and frozen in 10% dimethylsulfoxide (DMSO) and horse serum. All animal experiments were approved and conducted in accordance with the policies and guidelines set forth by the Northwestern University Institutional Animal Care and Use Committee (IACUC47).
All cell cultures were maintained at 37 °C and 5% CO2. All cell lines were regularly tested for the presence of mycoplasma using MycoAlert PLUS Detection Kit (Lonza).
Human stem cell-based neuronal differentiations
Northwestern University
MNs
We used previously described protocols to generate spinal MNs79. In brief, iPS cells were plated in mTESR1 supplemented with 10 μM ROCK inhibitor (DNSK International, Y-27632) at a density of 1.2 million per well of a six-well plate. From the day after plating (day 0) until day 5, the media was replaced daily with 50% DMEM:F-12/50% Neurobasal media, supplemented with non-essential amino acids, GlutaMAX, N2, B27 (Thermo Fischer Scientific) and with 10 μM SB431542 (DNSK International), 100 nM LDN193189 (DNSK International), 1 μM retinoic acid (Sigma-Aldrich). From day 6–13, the media was replaced daily with 50% DMEM:F-12/50% Neurobasal media, supplemented with non-essential amino acids, GlutaMAX, N2, B27 (Thermo Fischer Scientific) and with 1 μM retinoic acid, 1 μM SAG, 5 μM DAPT (DNSK International) and 4 μM SU5402 (DNSK International). Cells were dissociated on day 14 using TrypLE (Thermo Fischer Scientific) with added DNaseI (Worthington) and plated onto plates or cover slips previously coated with Matrigel (BD Biosciences). A half-media change was performed every 2–3 days with Neurobasal media supplemented with non-essential amino acids, GlutaMAX, N2, B27, 1% FBS and the neurotrophic factors BDNF, CNTF, GDNF (10 ng ml−1, R&D Systems) and ascorbic acid (0.2 μg ml−1). ASOs (5 μM) were added directly to the neuronal maturation media (gymnotic delivery) three times, starting on day 20 after differentiation until electrophysiological experiments were performed.
Excitatory neurons
We used previously described protocols to generate cortical-like excitatory neurons42,43. In brief, stem cells were infected with a cocktail of three lentiviral constructs: TetO-Ngn2-Puro, TetO-GFP and rtTA80. Media was replaced after overnight incubation in mTeSR Plus with 10 μM ROCK inhibitor Y-27632. Cells were passaged and differentiated at 70–80% confluency. On days 1–4, cells were grown in induction media consisting of the following minimal components: DMEM/F-12 (Life Technologies, 11320-033), N2 supplement (0.5% v/v; Gibco, 17502048), 1× GlutaMAX (Gibco, 35050061), 0.1 mM MEM non-essential amino acids (Gibco, 11140050) and 0.5% glucose. Media was changed daily, with the addition of the following small molecules: day 1, 10 mM SB431543 (Custom Synthesis), 200 nM LDN193189 (Custom Synthesis), doxycycline hyclate (2 mg ml−1) and 4 mM XAV939 (Stemgent, 04-00046). Days 2–4, puromycin (5 mg ml−1), 10 mM SB431543, 200 nM LDN193189, doxycycline hyclate (2 mg ml−1) and 4 mM XAV939 (Stemgent, 04-00046). Cells were dissociated on day 4 using Accutase (Gibco, A11105), resuspended in NBM media supplemented with ROCK inhibitor, then mixed 1:1 in 80% Hyclone-FBS/20% DMSO and stored in liquid nitrogen until thawed on day 5 for subsequent experiments. NGN2 neurons were initially seeded in neuronally supportive medium described above with the addition of 10 ng ml−1 BDNF (R&D Systems) and supplemented with ROCK inhibitor for the first 24 h. Thereafter, half-media changes were performed every 3 days. For imaging and MEA experiments, neurons were co-cultured with mouse glial cells39.
University College London
Excitatory neurons
Doxycycline-inducible NGN2 overexpression at the AAVS1 safe-harbor locus was used to forward-program human iPS cells into cortical-like neurons. Human iPS cells were passaged using TrypLE into Geltrex-coated plates containing induction media: DMEM/F-12 with GlutaMAX (Thermo), 1× non-essential amino acids (Thermo), 2 μg ml−1 doxycycline hyclate (Sigma-Aldrich), 2 μM XAV939 (Cayman Chemical), 10 μM SB431542 (Cayman Chemical), 100 nM LDN193189 (Cayman Chemical) and 10 μM ROCK inhibitor (Tocris, Y-27632) for the first 24 h. The media was changed daily for 3 days. A total of 500,000 neurons were re-plated onto 18 mm coverslips (Deckglaser) coated with PDL (Gibco) and laminin (Thermo), containing 50,000 primary E18 rat astrocytes81 that were grown in DMEM/F-12 with GlutaMAX, 10% ES-FBS (Thermo) and 1× penicillin–streptomycin (Gibco) for 1 week prior. Neuronal and astrocyte co-cultures were subsequently grown in neuronal maturation media containing BrainPhys (StemCell Technologies), 1× non-essential amino acids, 1× GlutaMAX (Gibco), 2% ES-FBS, 1× N21-MAX (R&D Systems), 1× N-2 MAX (R&D Systems), 20 ng ml−1 BDNF (Peprotech), 20 ng ml−1 GDNF (Peprotech), 20 ng ml−1 NT3, 1 mM dibutyryl cAMP (Sigma-Aldrich), 200 nM L-ascorbic acid (Sigma-Aldrich) and 1 μg ml−1 laminin. For the first 24 h, ROCK inhibitor (Y-27632, 10 μM) and 1× CultureOne (Gibco) were added to the media. Half-media changes were performed twice each week. Halo-iNeurons were cultured for 2 weeks and then treated with 30 nM HaloPROTAC-E to knock down TDP-43. ASOs (1 μM) were added directly to the neuronal maturation media (gymnotic delivery) at the same time to enable free uptake and were included in all subsequent media changes for another 2 weeks until electrophysiological experiments were performed.
Lentivirus production and transduction
TetO-Ngn2-puro (Addgene plasmid, 52047) and TetO-FUW-EGFP (Addgene plasmid, 30130) plasmids were gifts from M. Wernig80,82. FUW-M2rtTA (Addgene plasmid, 20342) was a gift from R. Jaenisch83. Lentiviruses were generated in HEK293T cells using the second-generation packaging vectors psPAX2 and pMD2.G as described previously39,84 by the Northwestern University DNA/RNA Delivery Core or ALSTEM.
CLIP and RIP
SH-SY5Y cells were grown in 6 x 10 cm plates. Media was removed and cells were washed once in PBS. Plates were placed on ice without their lids and irradiated once at 400 mJ cm−2 using a UV 500 Crosslinker. Cells were scraped off the plate and resuspended in 2 ml PBS, 2 ml nuclear isolation buffer consisting of 1.28 M sucrose, 40 mM Tris-HCl pH 7.5, 20 mM MgCl2, 4% Triton X-100 supplemented with protease (Roche, 11836170001) and phosphatase inhibitors (Roche, 4906845001) and 2 ml water, then incubated on ice for 20 min with occasional mixing. Nuclei were pelleted by centrifugation at 2,500g for 15 min. The pellet was resuspended in 500 μl RIPA buffer consisting of 150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM dithiothreitol (DTT), 0.5% IGEPAL CA-630 supplemented with 100 U ml−1 RNasin Plus RNase Inhibitor (Promega, N2611) and protease/phosphatase inhibitors. The nuclei were lysed with 15 –20 strokes of a Dounce homogenizer and centrifuged for 10 min at 16,000g at 4 °C to remove nuclear membrane and debris. Approximately 1 mg of nuclear lysate was used for each RIP assay. To prepare antibody/bead conjugates, Dynabeads Protein A beads (50 μl) (Invitrogen, 10001D) were washed twice in RIP buffer, resuspended in 200 μl of RIP buffer containing 4 μg of TDP-43 or IGG antibody (Proteintech, 10782-2-AP and 30000-0-AP) and incubated for 1 h at 4 °C under rotation. The beads were isolated magnetically and resuspended in 500 μl of immunoprecipitation buffer that contained 1 mg of nuclear lysate and incubated overnight at 4 °C under rotation. The beads were washed four times with RIP buffer and finally in NT2 buffer, which consists of 150 mM NaCl, 50 mM Tris pH 7.4, 1 mM MgCl2 and 0.05% IGEPAL CA-630. One-third of the sample was used for protein extraction, performed with 50 μM glycine pH 2.8 at 23 °C and centrifuged at 200g for 20 min. The remainder was used for RNA collection. RNA was eluted twice with NT2 buffer supplemented with 0.1% SDS and 30 μg of proteinase K. Each round of elution was performed at 55 °C, 200g, for 30 min. Finally, RNA was purified by phenol–chloroform extraction. RNA pellets were resuspended in water and used for cDNA synthesis. We used semi-quantitative RT–PCR to assess the interaction between TDP-43 and KCNQ2, using GAPDH (Hs.PT.39a.22214836, IDT) as a control. KCNQ2 primers were F, GGAGCTGGTCACTGCCTG; R, AATGACCACAACTCACCAGG.
RNA extraction and KCNQ2 isoform analysis
RNA was extracted from iPS cell-neurons plated in 12-well plates coated with PDL and laminin using the TRIzol reagent (Invitrogen). cDNAs were synthesized with approximately 200–800 ng of total RNA using SuperScript IV VILO (Invitrogen) reverse transcriptase. RT–PCR assays were performed for KCNQ2 splicing analysis using primers listed below and the AccuPrime Pfx DNA polymerase (Invitrogen) with 32 cycles. Gene expression was quantified by RT–qPCR using SYBR Green reagent (Bio-Rad) with recommended PCR settings and custom primers and probes from IDT as shown below.
KCNQ2 RT–PCR primers as presented in Figs. 1d and 4b: E4_F, TCATGGTGCTCATCGCCTC; E8_R, CTGTACATGGGCACGGTGAC; as presented in Extended Data Fig. 2a,c and Figs. 5d and 6b: E4_F, AACGTCTTTGCCACATCTGC; E6_R, AGCGCGAAGAAGGAGACAC.
KCNQ2∆E5 RT–qPCR primers as presented in Fig. 1e and Extended Data Fig. 2d,i: qKCNQ2_skip_F, CCACAGCAAGATCACGCTGA; qKCNQ2_skip_R, CCTTCAGGGCAAACCCAGAC; as presented in Figs. 5e and 6b and Extended Data Fig. 7c: qKCNQ2_skip_F, TATGCCCACAGCAAGATCAC; qKCNQ2_skip_R, AGACACCGATGAGGGTGAAG
In Fig. 1f: total KCNQ2 RT–qPCR primers, Hs.PT.58.2443397.g (IDT). In Extended Data Fig. 2i: qTARDBP: PrimeTime qPCR Primers, Hs.PT.58.26912658 (IDT). As control in Fig. 1e,f and Extended Data Fig. 2i: qHPRT: PrimeTime qPCR Primers, Hs.PT.58v.45621572 (IDT).
Mouse embryonic stem cell MN differentiation
Hb9–CD14–IRES–GFP mouse embryonic stem cells were differentiated into spinal MNs as previously described85. On day 6, embryoid bodies were dissociated as follows: embryoid bodies were collected and washed with PBS, then incubated in 0.05% trypsin–EDTA + 1:100 5 mg ml−1 DNaseI for 15 min. Next, one volume of FBS + 1:100 5 mg ml−1 DNaseI and one volume of PBS were added to the embryoid bodies to quench the trypsin. The supernatant was removed after the embryoid bodies settled to the bottom. The embryoid bodies were then triturated in PBS with a 1 ml pipette until dissociated, filtered using a 40 μm strainer and centrifuged through an Ovomucoid protease inhibitor and BSA cushion to yield a single-cell suspension. These cells were plated on 12-well dishes coated with polyethylenimine and laminin. Basal media to culture MNs consisted of Neurobasal media, B27, sodium pyruvate, GlutaMAX, AlbuMAX and penicillin–streptomycin. This basal media was supplemented with 10 ng ml−1 CNTF, GDNF, BDNF and IGF1. MNs were cultured for 1 week. Thereafter, cells were transfected with siRNAs targeting Tardbp (or scrambled) using the RNAiMAX transfection kit. Then, 4 days after transfection, RNA was collected using TRIzol (Invitrogen). A SYBR qPCR assay was used to quantify isoforms with the following primers:
m-qKcnq2_skip_F, ATATCATGGTGCTGATTGCC; R, TCAGGGTGATCTTGCTGTGAG
m-qTardbp: PrimeTime qPCR Primers, Mm.PT.58.31054883
m-actin_F, GGCTGTATTCCCCTCCATCG; R, CCAGTTGGTAACAATGCCATGT.
Experiments with SH-SY5Y TDP-43 12Q/N
Cloning
The TDP-43 12QN expression plasmid was constructed by replacing the hNIL cassette from the PB-TO-hNIL BSD-mApple piggyBac vector (Addgene, 182313) with human TDP-43 (AA 1-407) fused to 12 repeats for the Q/N domain of TDP-43 (amino acids 339–369) as previously described86. The 12QN repeat was synthesized (IDT) as three DNA fragments of four repeats each and cloned to the carboxy-terminus of TDP-43.
Stable transfections
SH-SY5Y cells were obtained from ATCC and transfected with TDP-43 12QN piggyBac vector and transposase with Lipofectamine 3000 (ThermoFisher Scientific) using the manufacturer’s protocol. Then, 2 days post transfection, cells were selected with 8 μg ml−1 Blasticidin S (Sigma-Aldrich, SBR00022-1ML) for 5 days. Surviving cells were single-cell plated, and individual clones were tested for TDP-43 12QN expression following doxycycline treatment.
Immunofluorescence
SH-SY5Y cells were treated with 2 μg ml−1 doxycycline hyclate (Sigma-Aldrich, D9891) for 4 days, washed once with PBS and fixed with 4% PFA and 4% sucrose at room temperature (20–22 °C) for 15 min. Cells were permeabilized with 0.25% Triton X-100 in PBS for 15 min and then blocked for 1 h with 3% BSA in PBS. Cells were incubated with primary mouse anti-TDP-43 antibody (Abcam, ab104223) in 3% BSA in PBS at 4 °C overnight with gentle rocking. Cells were washed three times with PBS for 5 min each and incubated with secondary anti-mouse Alexa Fluor 647 (ThermoFisher Scientific) for 1 h at room temperature. Cells were washed three times with PBS for 5 min each, and 1:1,000 DAPI was included in the second wash.
Molecular analysis
RNA was extracted from transfected SH-SY5Y cells treated with 2 μg ml−1 doxycycline hyclate (Sigma-Aldrich, D9891) with the RNeasy mini kit (Qiagen) following the manufacturer’s protocol. The RevertAid First Strand cDNA synthesis kit was used for cDNA synthesis with 500 ng of RNA. To assess KCNQ2 splicing, RT–PCR was performed with 2× Q5 Master Mix (NEB) using KCNQ2_E4_F and KCNQ2_E6_R. Amplicons were resolved by capillary electrophoresis on a 4200 TapeStation system (Agilent). qKCNQ2_skip_F and qKCNQ2_skip_R were used to assess KCNQ2 splicing by qPCR with the PowerUp SYBR Green Master Mix (ThermoFisher Scientific) on a QuantStudio 5 Real-Time PCR System (ThermoFisher Scientific) and quantified using the ΔΔCt method87.
RT–qPCR analysis of mis-spliced KCNQ2 in postmortem tissue
Frontal cortex brain tissues from ALS/FTD cases were provided by the Mayo Clinic Florida Brain Bank (Fig. 2). The study cohort is described in Supplementary Table 2. Cases were confirmed to present TDP-43 pathology, and controls were confirmed to lack TDP-43 neuropathological features. Written informed consent was provided by all participants or their family members, and all protocols were approved by the Mayo Clinic Institutional Review Board and Ethics Committee. Sample size was determined based on the availability of tissue in our brain bank. Levels of KCNQ2 splicing transcript variants were determined using cDNA from 500 ng of RNA (RIN ≥ 7.0) that was available from a previous study88. RT–qPCR was conducted using SYBR GreenER qPCR SuperMix (Invitrogen) for all samples in triplicate in a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems). Relative quantification was determined using the ΔΔCt method and normalized to the endogenous controls GAPDH and RPLP0. To compare RNA levels between controls and TDP-43 pathologically confirmed cases, we used single-variable (unadjusted) and multivariable linear regression models (adjusted). Multivariable models were adjusted for age at death, sex and RIN. KCNQ2 RNA levels were analyzed on a base 2 logarithmic scale owing to their skewed distributions. The regression coefficients and 95% confidence intervals were estimated and interpreted as the difference in the means, on a base 2 logarithmic scale, between all TDP-43 pathologically confirmed cases and controls (reference group). P values of <0.05 were considered statistically significant. For associations between KCNQ2 RNA levels and pTDP-43 protein levels, pTDP-43 values were also analyzed on a base 2 logarithmic scale using single-variable and multivariable linear regression models. The multivariable model examining KCNQ2 RNA and pTDP-43 protein levels was adjusted for age, sex and genotype. The model evaluating TDP-43 and age at disease onset was adjusted for sex, RIN and mutation status; the model assessing associations between KCNQ2 RNA and age of onset was adjusted for sex, RIN and mutation status; and the model assessing associations between KCNQ2 RNA and disease duration was adjusted for sex, age at onset, RIN and mutation status. P values of <0.0167 were considered significant after adjusting for multiple comparisons.
Generation of KCNQ2 expression plasmids
For the SyncroPatch experiments in Fig. 3 and Extended Data Fig. 4, full-length human KCNQ2 splice isoform 3 (NM_004518.5) was engineered in the mammalian expression vector pIRES2-EGFP or a modified vector in which EGFP was substituted for CyOFP1. Site-directed mutagenesis of KCNQ2 was performed as previously described54 using Q5 Hot Start High Fidelity DNA polymerase (New England Biolabs) to generate KCNQ2ΔE5 by deleting amino acids 231–272; mutagenic primer sequences were F, CCACAGCAAGATCACGCTGACCACCATTG; R, CAGCGTGATCTTGCTGTGGGCATAGACC. Plasmids were transiently transfected into KCNQ3 stably expressing CHO cells by electroporation using the MaxCyte STX system, as reported previously54 for automated SyncroPatch experiments, or using Lipofectamine 2000 for manual patch experiments. A 1 μg aliquot of plasmid DNA was transfected for the homozygous configuration, and 1 μg of mutant-IRES2–GFP or WT-IRES2–GFP was co-transfected with 1 μg of WT-IRES2–OFP plasmid DNA in the heterozygous configuration.
For the manual whole-cell patch-clamp recordings shown in Extended Data Fig. 4, we created a KCNQ2 fusion protein with enhanced GFP or mCherry (mCh) by placing the reporters before the amino terminus of the KCNQ2 subunit (GFP–KCNQ2WT, mCh–KCNQ2WT, GFP–KCNQ2ΔE5). An NheI-EcoRI cassette of KCNQ2 was generated, encoding for the full-length human KCNQ2 splice isoform 3 (NM_004518.5). A flexible linker sequence (SGRTQISSSLAQQQQQQQ) was introduced by PCR immediately before the translation start codon (ATG) to increase protein flexibility89, using the following oligonucleotides: F, 5′-AGCTCGCTAGCCCAGCAGCAGCAGCAGCAGCAGATGGTGCAGAAGTCGCGCAACGGCGGCGTATACC-3′ and R, 5′-GCCGAATTCAGGCACCGTGCTGAGGAGGGCCG-3′.
Electrophysiological recordings in CHO cells
CHO cell voltage-clamp recordings
Whole-cell voltage-clamp recordings were made from visually identified GFP-expressing (either bicistronically or fused to KCNQ2) CHO cells using an inverted Olympus IX-73 microscope equipped with a ×40 objective. Recording pipettes were made of glass capillaries using a horizontal Sutter P-1000 puller, yielding a 2–3.5 MΩ resistance pipette when filled with standard intracellular solution, containing 120 mM KMeSO4, 10 mM KCl, 10 mM HEPES, 10 mM Na2-phosphocreatine, 4 mM Mg-ATP and 0.4 mM Na3-GTP; the pH was adjusted with KOH to 7.3, and osmolality was 285−290 mOsm kg−1. The bath solution contained 135 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.2 mM MgSO4, 10 mM glucose, 2.4 mM CaCl2 and 10 mM HEPES, pH 7.3, and was heated to 35 °C using a stage-mounted heating device (ALA Scientific Instruments). Whole-cell voltage-clamp recordings were acquired using an Axopatch 200B amplifier (Molecular Devices) and filtered at 5 kHz, with a gain of 0.5 mV pA−1 after a >1 GΩ seal was made on a cell. Whole-cell currents were elicited from a holding potential of −80 mV using 1,000 ms depolarizing pulses (from −80 mV to +50 mV in +10 mV steps). Currents were measured in the final 100 ms of the voltage steps.
SyncroPatch recordings
Automated voltage-clamp recordings of KCNQ3 + KCNQ2 currents were performed at room temperature using a SyncroPatch 384PE (Nanion Technologies) as described previously54. The contribution of background currents was determined by recording before and after the addition of XE991 (25 mM, TOCRIS). Only XE991-sensitive currents were used for analysis. Whole-cell currents were elicited from a holding potential of 80 mV using 1,000 ms depolarizing pulses (from 80 mV to +40 mV in +10 mV steps every 20 s) followed by a 300 ms step to 0 mV to analyze tail currents. Cells with seal resistance of ≥0.5 GΩ and series resistance of <20 MΩ (access resistance compensation set to 80%) were used for analysis. Peak currents were measured 999 ms after the start of the depolarizing voltage pulse, and tail currents were measured 5 ms after changing the membrane potential to 0 mV. The time constant of activation was determined by fitting currents elicited by voltage steps between 30 mV and +40 mV (50–1,000 ms after the start of the voltage step) to a single exponential.
Surface biotinylation assays in CHO cells
For surface biotinylation experiments in Fig. 3 and Extended Data Fig. 4 characterizing subcellular localization of KCNQ2ΔE5, we constructed plasmids to express KCNQ2 under the control of the human PGK (hPGK) promoter by inserting N-terminal EGFP-tagged or mCherry-tagged KCNQ2WT or KCNQ2ΔE5 coding sequences (generated by PCR from the KCNQ2 plasmids described above) into a hPGK-CFP vector (Addgene, 200429) following linearization using BamHI/BsrGI restriction enzymes and Gibson Assembly (NEBuilder HiFi DNA Assembly Master Mix; New England Biolabs, E2621S). KCNQ3 stably expressing CHO cells were plated onto 10 cm plates (two million cells per plate), and these plasmids (mCherry-KCNQ2WT and EGFP-KCNQ2WT or EGFP-KCNQ2ΔE5) were transiently transfected using Lipofectamine 3000 (7.5 μg DNA per plasmid). Cell surface biotinylation was performed 48 h after transfection.
The Pierce Cell Surface Protein Isolation Kit (Thermo Fisher, 89881) was used to isolate cell surface proteins according to the manufacturer’s instructions with slight modifications. In brief, cells were incubated with biotin solution for 30 min at 4 °C with gentle agitation following two quick washes with ice-cold PBS. Following biotinylation, cells were washed twice with ice-cold Tris-buffered saline (TBS) and incubated with quenching solution for 15 min at 4 °C with gentle agitation. Cells were then gently scraped and collected into 50 ml conical tubes, and plates were washed with 10 ml of ice-cold TBS before centrifugation for 3 min at 500g. Pellets were quickly washed with 5 ml ice-cold TBS and centrifuged again for 3 min at 500g. The resulting pellets were then lysed using 500 ml of lysis buffer (containing protease/phosphatase inhibitors) and incubated on ice for 30 min. Samples were sonicated every 15 min (10% power, 5 × 1 s sonication) and vortexed every 5 min of the 30 min incubation to enhance membrane protein solubilization. Samples were then centrifuged for 2 min at 10,000g and 4 °C, and clarified lysate was transferred to a fresh 1.5 ml tube. Following washing of NeutrAvidin columns, 400 μl of clarified lysate was incubated in the columns for 60 min at room temperature with end-over-end mixing (100 μl of lysate set aside for total/input protein analysis). Flow-through lysate was then collected, and columns were washed according to the manufacturer’s instructions. Next, 1× SDS sample buffer (80 mM Tris, 2% SDS, 10% glycerol, 50 mM DTT) was used for elution of cell surface fractions for 5 min at 95 °C before analysis by SDS–PAGE and western blot. Loading of cell surface fractions was normalized to the concentration of input fractions (determined by Pierce BCA Protein Assay).
For analysis of cell surface fractions, 20 μg of input fraction was loaded (along with twice the corresponding volume of cell surface fraction) into 4–20% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad) and separated by SDS–PAGE before wet transfer to nitrocellulose membranes. Membranes were washed with water and TBS before 1 h blocking in 5% nonfat dry milk (LabScientific) in TBS with 0.1% Tween (TBS-T). Membranes were then incubated overnight at 4 °C with gentle agitation with primary antibodies prepared in 5% BSA in TBS-T. The following day, membranes were washed three times in TBS-T before a 1–2 h incubation at room temperature with horseradish peroxidase-conjugated secondary antibodies prepared in 5% nonfat dry milk in TBS-T. After three additional washes with TBS-T, membranes were washed twice with TBS, and signal was detected using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher) and ChemiDoc XRS+ imaging system (Bio-Rad). Densitometry analysis was performed in Image Lab software (Bio-Rad). For cell surface fractions, the relative intensity of both the upper and lower bands was measured together. Primary antibodies and dilutions used were as follows: rabbit anti-GFP (Thermo Fisher, A-11122; 1:1,000), rabbit anti-mCherry (Invitrogen, PA5-34974; 1:1000), mouse anti-Transferrin receptor (Thermo Fisher, 13-6800; 1:500), rabbit anti-Histone H3 (Proteintech, 17168-1-AP; 1:1,000). Secondary antibodies and dilutions used were as follows: goat anti-mouse HRP (Li-Cor, 926-80010; 1:5,000) and goat anti-rabbit HRP (Li- Cor, 926-80011; 1:5,000).
Generation of a homozygous KCNQ2 ∆E5 cell line
Four guide RNAs (gRNAs) targeting introns 4 and 5 of KCNQ2 (Extended Data Fig. 5) were designed using the CRISPR wizard with default settings on Benchling (2021, https://benchling.com). gRNA oligonucleotides were purchased from IDT with BbsI sticky ends to clone into an expression vector driven by the human U6 promoter (custom synthesis, Broad Institute). Oligonucleotides were phosphorylated using T4 PNK (NEB, M0201S) and ligated into the BbsI-linearized pUC6-gRNA vector using T7 Ligase (NEB, M0318S). Clones containing gRNAs were verified by Sanger sequencing. The KCNQ2∆E5 engineered cell line is a derivative of WA01 human embryonic stem cells (Alias: H1, WiCell). H1 cells were transfected using the 100 μl Neon Transfection System (ThermoFisher Scientific, MPK10025). H1 cells growing in mTeSR Plus media (StemCell Technologies, 100-0276) with 10 μM ROCK inhibitor Y-27632 were dissociated using Accutase (Gibco, A11105) treatment for 5 min at 37 °C. Cells were resuspended in buffer R at a concentration of 2.5 × 106 cells per ml. Cells were added to a vial containing 1.4 μg of each of four gRNA vectors and 7 μg of the pSpCas9n(BB)-2A-Puro (PX462) V2.0, a gift from F. Zhang (Addgene). The electroporated cells were immediately fed mTeSR Plus media containing ROCK inhibitor in a 10 cm dish. Then, 24 h after transfection, puromycin selection was started. Media was replaced with mTeSR containing 2 μg ml−1 of puromycin. After 24 h of selection, the medium was aspirated and replaced. Cells were cultured for 10 days before colony picking into a 24-well plate for expansion.
Genomic DNA was extracted from puromycin-selected colonies using the DirectPCR Lysis Reagent (Viagen Biotech, 102-T) and PCR-screened to confirm the presence of the intended deletion in the KCNQ2 gene. PCR products were separated and analyzed after electrophoresis on a 1% agarose gel. Clonal cells with KCNQ2 exon 5 deletion and control cells with intact exon 5 were verified using allele-specific qPCR assays and characterized by karyotyping at Cell Line Genetics. Oligonucleotides used for gRNA cloning included KCNQ2 gRNA1, CTGTGCCCGGAGGCTCAGGG; KCNQ2 gRNA2, GCCCAGGCCAGGAGGCTCAG; KCNQ2 gRNA3, TGGTCCCAGCACAGGGACAG; and KCNQ2 gRNA4, CCCGTGGTCATGATGGCTTG. Genotyping primers for confirmation of exon 5 deletions were KCNQ2_genot_F1: GTGAACGAGCCTCCCTCCCCTT seq_KCNQ2_rvs: GGCCAGATCTGTTCAGACCTG
Immunocytochemistry in iPS cells and embryonic stem cell-derived neurons
Stem cell-derived neurons were co-cultured with primary mouse glia on PDL and laminin-coated glass coverslips placed into the wells of a 24-well plate. Glial cells (around 80,000 glial cells per well) were plated at least 3 days before thawing of neurons (around 45,000 neurons per well). Coverslips were fixed with 2–3.7% formaldehyde in 4% sucrose and PBS for 15 min at room temperature and then washed three times with cold PBS before simultaneous permeabilization and blocking in 0.1% PBST with 5% normal goat serum for 1 h at room temperature. Coverslips were incubated overnight at 4 °C with combinations of the following primary antibodies: anti-GFP (Abcam, ab6673; 1:10,000), anti-MAP2 (Millipore, MAB3418, AB_94856; 1:1,000), anti-KCNQ2 (Thermo, PA1-929; 1:200), anti-Calnexin (SantaCruz, 46669), anti-ANK-G (Neuromab, 73-146; 1:200), anti-TDP-43 (R&D Systems, MAB7778; 1:200) and DAPI (Invitrogen, 33342; 1:1,000). The following day, coverslips were washed three times with cold PBS and then incubated with secondary antibodies for 1 h at room temperature. Secondary antibodies were typically used at 1:10,000 (Thermo Fisher Scientific). Primary and secondary antibodies were diluted in PBS containing 5% normal goat serum. Cells were washed three times in PBS and then briefly in distilled water and mounted onto microscope slides with Fluoromount-G mounting media (Southern Biotech, 0100-01). Analysis of KCNQ2 signal in neurons was done in Nikon Elements (Nikon NIS Elements Software, v.5.41.00) and ImageJ software (v.1.53t) using maximum intensity z-stack projections. Regions of interest were manually defined for each cell and used to compute KCNQ2 signal metrics. For each neuron, KCNQ2 signal in the soma was assigned to one of two categories: diffuse KCNQ2 signal or accumulated KCNQ2 signal. Subsequently, the KCNQ2 signal intensity parameters were measured in Nikon Elements (NIS Elements Software, v.5.41.00).
MEA recordings
MEA recordings of NGN2 neurons were obtained with the Axion Biosystems MEA system (Axion Biosystems, 48-well format with 16 electrodes per well). Plates were coated with polyethylenimine and laminin according to the manufacturer’s protocols. Mouse glial cells and neurons were plated into the well as a droplet, ensuring coverage of the electrode grid. Each well received around 85,000 neurons and 35,000 glia. Spontaneous activity was recorded using Axion Biosystems Maestro 768-channel amplifier and Axion Integrated Studios (AxIS) software. The amplifier recorded from all channels simultaneously using a gain of 1,200× and a sampling rate of 12.5 kHz per channel. After passing the signal through a Butterworth band-pass filter (300–5,000 Hz), online spike detection (threshold = 6 × the root-mean-square of noise on each channel) was performed with the AxIS spike adaptive spike detector. All recordings were conducted at 37 °C in 5% CO2 and 95% O2. All recordings were performed for 5–10 min, starting on day 8 of culture. Starting on day 9, neurons were electrically stimulated with 20 pulses at 0.5 and 0.25 Hz after spontaneous recordings were made.
The MEA results reflect well-wide averages, with the number of wells per condition represented by n values. Total wells were combined from three replicate MEA plates. Active electrodes were defined as those having at least one spike per minute. The weighted mean firing frequency (Hz) was calculated as the total number of spikes divided by the number of active electrodes over the recording duration (300 s). The inter-spike interval coefficient of variation was calculated as the standard deviation divided by the mean inter-spike interval on active electrodes. Bursts were detected according to a Poisson distribution. The algorithm is adaptive to the mean firing rate on each electrode according to a ‘surprise’ threshold, adapted from a previous publication90. The burst % was calculated as the percentage of all spikes that occurred in bursts. Acute treatment with the Kv7 agonist ICA-069673 (1 μM; Tocris Bioscience) was conducted on day 43 in culture. After 5 min of baseline activity (pre), ICA-069673 (diluted in a small volume of NBM media) was added directly to the media in each well. Following a brief adjustment period (approximately 5 min), spontaneous activity was analyzed in 5 min intervals, for up to 50 min ‘post ICA’. Spontaneous activity recorded from each well (pre and post-ICA) was normalized to its respective baseline (pre ICA).
Identification of KCNQ2 smASOs
smASOs were manually designed using sequence-based predictions of TDP-43 binding upstream of KCNQ2 exon 5 (ASOs 1–8) and regions of predicted high RNA structural accessibility downstream of KCNQ2 exon 5 (ASOs 9–17). ASOs were synthesized on a Dr. Oligo 48 Synthesizer (Biolytic) using 2′-O-methoxyethyl RNA phosphoramidites coupled for 8 min in the presence of ethylthiotetrazole activator. ((Dimethylamino-methylidene)amino)-3H-1,2,4-dithiazoline-3-thione was used to generate inter-nucleotide phosphorothioate linkages. Phosphoramidites and reagents were purchased from ChemGenes. Oligonucleotides were cleaved and deprotected in concentrated aqueous ammonia (30% in water) at 55 °C for 16 h and characterized by liquid chromatography–mass spectrometry. Final desalting was affected by diafiltration (3× water wash) in a 3 kDa cutoff Amicon centrifugal filter (Millipore). All ASO sequences, including chemical modifications, are provided in Supplementary Table 4.
MNs differentiated from iPS cell lines 18a and KOLF2.1J were cultured to day 24 with half-media changes made three times each week. On days 25 and 28, they were treated with scrambled or TDP-43 siRNAs (final concentration, 50 nM) using lipofectamine RNAiMAX (ThermoFisher, 13778075). ASOs were added to the media 24 h after siRNA treatment. Cells were collected on day 32 for total RNA collection (Macherey-Nagel, 740955.50). A total of 1 μg of total RNA was used to generate cDNAs (Bio-Rad, 170-8814). RT–qPCR was performed using SYBR green (ThermoFisher, A25777) with conditions of 95 °C for 2 min, 40 cycles of 95 °C for 10 s and 60 °C for 35 s, and a final melt curve step from 65 °C to 95 °C in increments of 0.5 °C per 5 s. Two technical replicates were included for each sample, and samples with a standard deviation of >0.5 for technical replicates were excluded from the final data analysis. Expression was normalized to RPLP0 or GPI/GAPDH.
Primer sequences used for Fig. 5a and Extended Data Fig. 6b,c were as follows: GPI_F:GTGTACCTTCTAGTCCCGCC, GPI_R:GGTCAAGCTGAAGTGGTTGAAGC, GAPDH_F:TCAAGGCTGAGAACGGGAAG, GAPDH_R:CGCCCCACTTGATTTTGGAG, RPLP0_F:TCTACAACCCTGAAGTGCTTGT, RPLP0_R:CAATCTGCAGACAGACACTGG, KCNQ2_skip_F:TATGCCCACAGCAAGATC, KCNQ2_skip_R:AGACACCGATGAGGGTG, TDP-43_F:GTGGCTCTAATTCTGGTGCAT and TDP-43_R:CAC AACCCCACTGTCTACATT.
Electrophysiological patch-clamp recordings in iPS cell-derived neurons
Northwestern University
Whole-cell current-clamp recordings were made from visually identified neurons using an inverted Olympus IX51 microscope equipped with a ×40 objective. Recording pipettes were made of glass capillaries using a horizontal Sutter P-1000 puller, yielding a 2–4 MΩ resistance pipette when filled with standard K-methyl sulfate intracellular solution, containing 120 mM KMeSO4, 10 mM KCl, 10 mM HEPES, 10 mM Na2-phosphocreatine, 4 mM Mg-ATP and 0.4 mM Na3-GTP, with pH adjusted to 7.35 with KOH and osmolality of 285–290 mOsm kg−1. Neurons were continuously perfused with oxygenated artificial cerebrospinal fluid bath solution containing 125 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 1 mM MgSO4, 22 mM glucose, 2 mM CaCl2, pH 7.35 at 32–35 °C and osmolality 310–315 mOsm kg−1. Current-clamp recordings were acquired using a Multiclamp 700B Amplifier (Molecular Devices) and digitized at 10 kHz (filtered at 3 kHz). The capacitance (pF) of each cell was reported from the value automatically calculated in the Multiclamp software experiment log. RMP and spontaneous APs were recorded immediately after establishing the whole-cell patch-clamp configuration in a 15 s gap-free protocol. RMP values were corrected for the liquid junction potential, calculated to be −8.0 mV. Spontaneous AP properties were measured for all spontaneous spikes occurring within the first 15 s after establishing whole-cell configuration and then averaged for each cell. AP properties were computed using an algorithm that measures properties by first identifying the peak (mV) of each spike. The threshold (mV) was identified using the first derivative and is defined as the voltage at which the first sample of the first derivative meets or exceeds 3.00 mV ms−1, within a search region of 20 ms before the AP peak (mV). AP amplitude (mV) was calculated by subtracting the threshold value (mV) from the peak (mV). AP kinetics were calculated with 200 kHz interpolation of the data to improve accuracy. The maximal slope for the rising and decaying phase of each AP was calculated as a regression over two sample points. AHP values were defined as the baseline (mV) minus the minimum value (mV) detected within a search region following the AP peak. The AHP search window was between 0 and 20 ms after AP peak (mV) for the fast AHP and up to 40 ms for the mAHP. Input resistance was determined from the change in voltage (ι1Vm) and current (ι1Im) in a protocol using 800 ms current steps from −80 pA to −10 pA (10 pA steps). The input resistance was calculated as the slope of a linear ordinary least squares fit, as x = ι1Im and y = ι1Vm. For post-burst AHP measurement, all neurons were held at −65 mV. Post-burst mAHP and sAHP were induced by a 50 Hz train of 25 APs evoked by 2 ms per 1.2 nA current injection pulses. The mAHP was calculated as the difference between baseline and the most negative peak following the offset of the last current pulse. The post-burst sAHP was the voltage measured 1 s after the offset of the last current pulse. Neurons were excluded from analysis if they failed to meet the following criteria: (1) formation of a GΩ seal (or greater) before going whole-cell; (2) a series resistance that remained <15 MΩ throughout the duration of recording; (3) a RMP that was more negative than −35mV (after liquid junction potential correction, measured immediately after going whole-cell); and (4) the neuron was capable of firing an AP (determined by the occurrence of spontaneous APs or from APs elicited using 500 ms depolarizing current injection ramps, ranging from 10 pA or greater). Data were analyzed using Pclamp/Clampfit (Molecular Devices, RRID: SCR_011322) and Easy Electrophysiology software (Easy Electrophysiology, RRID:SCR_021190). Statistical significance for all electrophysiological parameters was determined using ordinary one-way ANOVAs.
University College London
Whole-cell patch-clamp recordings were carried out in extracellular solution containing 136 mM NaCl, 2.5 mM KCl, 10 mM HEPES, 1.3 mM MgCl2, 2 mM CaCl2, 10 mM glucose, pH 7.3 and 300 mOsm, and intracellular solution containing 125 mM KMeSO4, 5 mM MgCl2, 10 mM EGTA, 10 mM HEPES, 0.5 mM NaGTP, 5 mM Na2ATP, pH 7.4 and 290 mOsm. Cells were held at −70 mV using a Multiclamp 700B Amplifier (Molecular Devices), and the data were acquired using a Digidata 1440A Digitizer (Molecular Devices). All recordings were carried out on a heated stage set to 37 °C. Data were acquired with Clampex software (Molecular Devices) and Axon Multiclamp Commander software (Molecular Devices), sampled at a rate of 20 kHz and filtered at 10 kHz. To measure M-current deactivation kinetics, cells were held in voltage-clamp at −70mV then depolarized to +20 mV for 1 s, then repolarized in −20 mV increments for 500 ms to record tail currents. Tail currents at the −50 mV increment were used to measure deactivation kinetics. To measure RMP and spontaneous AP properties, cells were held in passive current-clamp mode without current injection for 1 min within the first 2 min after break-in. Intrinsic excitability measurements and AP properties were subsequently recorded in current clamp while using a steady-state current injection to maintain cells at −60 mV. The mAHPs were determined from single AP rheobase recordings at 500 ms current injections at 5 pA increments, 50 ms following AP peak amplitude relative to 50 ms before AP peaks. Passive membrane properties of input resistance, membrane capacitance and series resistance were measured by applying sub-threshold voltage steps. Series resistance was calculated as proportional to the current response amplitude; membrane capacitance was calculated from the transient time constant by fitting current transients to an exponential function; and input resistance was calculated from the steady-state response following the current transients. Passive membrane properties were measured at the start and end of all recordings to ensure that proper access (series resistance of >35 mΩ) was maintained. XE991 was added to the bath at 10 μM for 5 min to inhibit the M-current. Electrophysiological measurements were analyzed using Clampfit 10.6 software and custom MATLAB scripts. Cells with a series resistance greater than 30 MΩ were rejected.
Immunohistochemistry and image analysis in postmortem patient tissue
Northwestern University
Paraffin-embedded postmortem tissue sections, collected from the lumbar spinal cord of three control individuals and three patients with sporadic ALS, were obtained from the Department of Veterans Affairs Biorepository Brain Bank and analyzed by immunohistochemistry (Supplementary Table 5). Immunohistochemistry was performed as previously described91. Sections were first deparaffinized and rehydrated through sequential incubation at room temperature in the following solutions: three times for 10 min in xylene, three times for 5 min in 100% ethanol, three times for 3 min in 95% ethanol, once for 2 min in 75% ethanol, once for 2 min in 50% ethanol and five times for 1 min in deionized water. Sections were then moved into 1× Antigen Decloaker (Biocare Medical), and antigen retrieval was performed within an electronic pressure cooker (decloaking chamber) for 10 min at high pressure (10.2–11.6 psi) and 125 °C. Samples were then cooled to room temperature for 30 min before five washes with deionized water. Blocking of sections was performed using 1% BSA in PBS containing 0.3% Triton X-100 for 1 h at room temperature before five washes with deionized water and overnight incubation with primary antibodies diluted in PBS containing 1% BSA at 4 °C. Primary antibodies and dilutions used were as follows: rabbit polyclonal anti-KCNQ2 (Thermo Fisher, PA1-929; 1:100), mouse monoclonal anti-TDP-43 (Proteintech, 60019-2-Ig; 1:500), mouse monoclonal anti-Ubiquitin (Santa Cruz, sc-8017; 1:100) and chicken polyclonal anti-MAP2 (Abcam, ab5392; 1:5,000). Following five additional washes with deionized water, sections were incubated with secondary antibodies diluted in PBS containing 1% BSA for 1 h at room temperature. Secondary antibodies and dilutions used were as follows: donkey polyclonal anti-rabbit, Alexa Fluor 488 (Invitrogen, A21206; 1:250), donkey polyclonal anti-mouse, Alexa Fluor 568 (Invitrogen, A10037; 1:250), donkey polyclonal anti-chicken, Alexa Fluor 647 (Jackson Immuno, 703-606-155; 1:250). Samples were then washed five times with deionized water before a 45 s incubation with 0.3% Sudan Black in 70% ethanol and 15 additional rinses with deionized water. Sections were then briefly air-dried and mounted using ProLong Diamond Antifade Mountant with DAPI (Invitrogen) and allowed to cure overnight at room temperature before imaging by confocal microscopy (see ‘Image acquisition and analysis’ for details). Mounted slides with postmortem tissue were imaged using a Nikon Ti2 inverted microscope system equipped with a Yokogawa CSU-W1 spinning disk for confocal imaging and Hamamatsu Flash 4 cameras. All images were captured using a 60x oil-immersion objective (Plan Apo VC, Nikon). Neurons in postmortem tissue were selected within the ventral horn of the spinal cord. Z-stacks were acquired using 0.2–0.4 μm intervals. Images were blinded before analysis, and regions of interest were drawn to encompass the entire neuronal soma using MAP2 as a marker. KCNQ2 intensity variation within the neuronal soma was calculated using Nikon Elements (Nikon NIS Elements Software, v.5.41.00) and plotted as fold change from the mean intensity variation of all three control patients. The two-tailed nested t-test (Graphpad Prism) was used to compare mean KCNQ2 signal variation between control and ALS patient groups.
Columbia University
Postmortem tissue was obtained within 6–30 h after death (Supplementary Table 5). Brain and spinal cord samples fixed in buffered formalin (n = 4 familial ALS-C9ORF72; n = 11 sporadic ALS; n = 3 age-matched non-neurological controls) were obtained from the archives of the Department of Neuropathology, Amsterdam University Medical Center, University of Amsterdam. All patients fulfilled the diagnostic criteria for ALS (El Escorial criteria)92 as reviewed independently by several neuropathologists. The controls included in the present study were adult individuals without any history of neurological disease, based on their last clinical evaluation. Relevant demographic details are shown in Supplementary Table 5. Double immunofluorescence staining was performed as described previously93,94. In brief, deparaffinized tissue sections were heated in citrate buffer (pH 6, Dako) for 20 min in a pressure cooker for antigen retrieval. Sections were then blocked with ready-to-use 10% normal goat serum (Life Technologies) for 1 h at room temperature to avoid non-specific binding and then incubated with the primary antibody at 4 °C overnight. The primaries used were mouse anti-Ubiquitin (Millipore, MAB1510-I-100UG; 1:150), mouse anti-pTDP-43 (Cosmo Bio, TIPPTD-M01; 1:1,000), mouse anti-GRP78 (BD Biosciences, 610978; 1:150) and rabbit anti- KCNQ2 (Thermo, PA1-929; 1:150). After a 10 min wash in TBS-T, the sections were incubated with an Alexa-conjugated secondary antibody (1:500 in TBS-T) at room temperature for 2 h. Sections were washed again in TBS-T (twice for 10 mi) and stained for 10 min with 0.1% Sudan Black in 80% ethanol to suppress endogenous lipofuscin autofluorescence. Finally, the sections were washed for 5 min in TBS-T and mounted with Vectashield Mounting Medium (Vector Laboratories) containing DAPI. The ventral horns of spinal cord sections were imaged either with a 40x oil or 63x oil objective using a confocal laser scanning microscope (LSM Zeiss 780). Images were processed using the Zen Microscopy software: we first collapsed stacks using the orthogonal projection tool. Projections were exported as high-quality, single-channel TIFFs. The KCNQ2 channel was converted to greyscale, and large spinal MNs were identified and labeled in Adobe Photoshop. KCNQ2 staining was analyzed in different locations of each labeled spinal MN, excluding the nucleus and regions quenched by Sudan Black. The average intensity and standard deviation was recorded in at least four distinct regions of each cell using a 35 × 35 pixel box. The average KCNQ2 intensity and standard deviation for each neuron was calculated as the mean of these distinct recordings. Each spinal MN was also investigated for TDP-43 pathology by examining the pTDP-43 channel for established forms of pathological manifestations (granules, aggregates and skein-like patterns). Altogether, for each cell, we derived the following metadata: sample ID, cell ID, average KCNQ2 intensity, KCNQ2 intensity standard deviation and TDP-43 pathology, as well as other clinical information regarding these samples, including ALS class (non-neurological control, sporadic or familial) and genetics (C9orf72 repeat expansion). Normalized KCNQ2 intensity was derived as follows: the average KCNQ2 intensity from all control neurons was calculated and set as the normalization factor. Each neuron from each case was divided by this normalization factor to yield normalized KCNQ2 intensity. Cell-level KCNQ2 intensity standard deviation was divided by average KCNQ2 intensity to obtain the coefficient of variation. Neurons were grouped and plotted as indicated in the figure panels using Graphpad Prism. Statistical tests and multiple comparisons (where relevant) are described in figure panels. All procedures involving human participants, including the use of postmortem tissue samples, were performed by the ethical standards of the institutional and national research committees and the 1964 Helsinki Declaration and its later amendments.
Statistics and reproducibility
All the relevant information on study design, statistics and experimental sample sizes is reported within each figure legend, relevant methods section and reporting summary. No statistical methods were used to pre-determine sample sizes. Our sample sizes were determined based on sample availability and prior experimental experience and are similar to those reported in previous publications. Data distribution was assessed by Prism in most cases. For some datasets, distribution was assumed to be normal, but this was not formally tested in all cases unless otherwise stated. No data were excluded from the analyses except for Fig. 2j,k, where one out of the seven RNA-seq datasets was excluded because it lacked sufficient read coverage for KCNQ2 and some neurons that did not meet recording criteria for manual patch-clamp recordings (see Methods). The experiments were not randomized. The Investigators were blinded to allocation during experiments and outcome assessment for most experiments, including the immunohistochemical analysis presented in Fig. 7 and Extended Data Fig. 8.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All RNA sequencing datasets used in this study were previously published and described in the Methods. FACS-purified MNs depleted of TDP-43 studies were reported by the Eggan lab (GSE121569); purified TDP-43-high and TDP-43-low neuronal nuclei datasets were reported by the Lee lab (GSE126543); i3Neurons, SH-SY5Y and SK-N-DZa TDP-43 depletion studies were reported by the Fratta and Ward labs (PRJEB42763); mouse TDP-43 depletion studies were reported by the Cleveland lab (GSE27394); and TDP-43 iCLIP from SH-SY5Y cells were reported by the Fratta and Ward labs and downloaded from E-MTAB-10297. We obtained access to 1,124 ALS and control RNA-seq datasets from the Target ALS/New York Genome Center ALS Consortium. Although these are not publicly available, access to genomics datasets may be sought at https://www.targetals.org/resource/genomic-datasets. GRCh38.p13 human genome and gene annotation files are available at https://www.gencodegenes.org/human. Raw experimental data generated here are provided in supplementary tables and source files. All other data are available upon request. Source data are provided with this paper.
Code availability
Custom code for RNA-seq and manual patch-clamp analysis has been deposited at GitHub (https://github.com/recursivesplicing-beep/KCNQ2_2025).
References
Taylor, J. P., Brown, R. H. Jr. & Cleveland, D. W. Decoding ALS: from genes to mechanism. Nature 539, 197–206 (2016).
Ghasemi, M. & Brown, R. H. Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 8, a024125 (2018).
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R. & Gitler, A. D. ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842 (2020).
Estades Ayuso, V. et al. TDP-43-regulated cryptic RNAs accumulate in Alzheimer’s disease brains. Mol. Neurodegener. 18, 57 (2023).
Meneses, A. et al. TDP-43 pathology in Alzheimer’s disease. Mol. Neurodegener. 16, 84 (2021).
Tziortzouda, P., Van Den Bosch, L. & Hirth, F. Triad of TDP43 control in neurodegeneration: autoregulation, localization and aggregation. Nat. Rev. Neurosci. 22, 197–208 (2021).
Kabashi, E. et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 (2008).
Sreedharan, J. et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 (2008).
Brown, A. L. et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022).
Ma, X. R. et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 603, 124–130 (2022).
Klim, J. R. et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179 (2019).
Melamed, Z. et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190 (2019).
Prudencio, M. et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 130, 6080–6092 (2020).
Dittman, J. S. Unc13: a multifunctional synaptic marvel. Curr. Opin. Neurobiol. 57, 17–25 (2019).
van Es, M. A. et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat. Genet. 41, 1083–1087 (2009).
Menon, P. et al. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. 14, 478–484 (2015).
Vucic, S., Nicholson, G. A. & Kiernan, M. C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131, 1540–1550 (2008).
Geevasinga, N. et al. Axonal ion channel dysfunction in C9orf72 familial amyotrophic lateral sclerosis. JAMA Neurol. 72, 49–57 (2015).
Mogyoros, I., Kiernan, M. C., Burke, D. & Bostock, H. Strength-duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain 121, 851–859 (1998).
Benussi, A. et al. TMS for staging and predicting functional decline in frontotemporal dementia. Brain Stimul. 13, 386–392 (2020).
Benussi, A. et al. Transcranial magnetic stimulation distinguishes Alzheimer disease from frontotemporal dementia. Neurology 89, 665–672 (2017).
García-Cabrero, A. M. et al. Hyperexcitability and epileptic seizures in a model of frontotemporal dementia. Neurobiol. Dis. 58, 200–208 (2013).
Kanai, K. et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 83, 734–738 (2012).
Shibuya, K. et al. Motor cortical function determines prognosis in sporadic ALS. Neurology 87, 513–520 (2016).
Enterzari-Taher, M., Eisen, A., Stewart, H. & Nakajima, M. Abnormalities of cortical inhibitory neurons in amyotrophic lateral sclerosis. Muscle Nerve 20, 65–71 (1997).
Foerster, B. R. et al. Decreased motor cortex γ-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 78, 1596–1600 (2012).
Scekic-Zahirovic, J. et al. Cortical hyperexcitability in mouse models and patients with amyotrophic lateral sclerosis is linked to noradrenaline deficiency. Sci. Transl. Med. 16, eadg3665 (2024).
Bae, J. S., Simon, N. G., Menon, P., Vucic, S. & Kiernan, M. C. The puzzling case of hyperexcitability in amyotrophic lateral sclerosis. J. Clin. Neurol. 9, 65–74 (2013).
Harley, P. et al. Aberrant axon initial segment plasticity and intrinsic excitability of ALS hiPSC motor neurons. Cell Rep. 42, 113509 (2023).
Huang, X. et al. Downregulation of the silent potassium channel Kv8.1 increases motor neuron vulnerability in amyotrophic lateral sclerosis. Brain Commun. 6, fcae202 (2024).
Dyer, M. S. et al. Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 157, 1300–1315 (2021).
Reale, L. A. et al. Pathologically mislocalised TDP-43 in upper motor neurons causes a die-forward spread of ALS-like pathogenic changes throughout the mouse corticomotor system. Prog. Neurobiol. 226, 102449 (2023).
Kiskinis, E. et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 14, 781–795 (2014).
Wainger, B. J. et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7, 1–11 (2014).
Bean, B. P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465 (2007).
Moakley, D. et al. Pharmacological profiling of purified human stem cell-derived and primary mouse motor neurons. Sci. Rep. 9, 10835 (2019).
Masegosa, V. M., Navarro, X. & Herrando-Grabulosa, M. ICA-27243 improves neuromuscular function and preserves motoneurons in the transgenic SOD1G93A mice. NeuroTherapeutics 21, e00319 (2024).
Wainger, B. J. et al. Effect of ezogabine on cortical and spinal motor neuron excitability in amyotrophic lateral sclerosis: a randomized clinical trial. JAMA Neurol. 78, 186–196 (2021).
Simkin, D. et al. Dyshomeostatic modulation of Ca2+-activated K+ channels in a human neuronal model of KCNQ2 encephalopathy. Elife 10, e64434 (2021).
Vaquero-Garcia, J. et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5, e11752 (2016).
Liu, E. Y. et al. Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 27, 1409–1421.e6 (2019).
Nehme, R. et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 23, 2509–2523 (2018).
Limone, F. et al. Efficient generation of lower induced motor neurons by coupling Ngn2 expression with developmental cues. Cell Rep. 42, 111896 (2022).
Tollervey, J. R. et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 (2011).
Polymenidou, M. et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 (2011).
Brown, D. A. & Passmore, G. M. Neural KCNQ (Kv7) channels. Br. J. Pharmacol. 156, 1185–1195 (2009).
Cooper, E. C. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192 (2011).
Devaux, J. J., Kleopa, K. A., Cooper, E. C. & Scherer, S. S. KCNQ2 is a nodal K+ channel. J. Neurosci. 24, 1236–1244 (2004).
Tzingounis, A. V. & Nicoll, R. A. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl Acad. Sci. USA 105, 19974–19979 (2008).
Nappi, P. et al. Epileptic channelopathies caused by neuronal Kv7 (KCNQ) channel dysfunction. Pflug. Arch. 472, 881–898 (2020).
Steinlein, O. K. & Jentsch, T. J. KCNQ2, the first gene found to be mutated in human generalized idiopathic epilepsy. Pathol. Biol. (Paris) 46, 683–684 (1998).
Biervert, C. et al. A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406 (1998).
Goto, A. et al. Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia 60, 1870–1880 (2019).
Vanoye, C. G., et al. High-throughput evaluation of epilepsy-associated KCNQ2 variants reveals functional and pharmacological heterogeneity. JCI Insight 7, e156314 (2022).
Rasmussen, H. B. et al. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J. Cell Sci. 120, 953–963 (2007).
Ziller, M. J. et al. Dissecting the functional consequences of de novo DNA methylation dynamics in human motor neuron differentiation and physiology. Cell Stem Cell 22, 559–574.e9 (2018).
Stein, C. A. et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 38, e3 (2010).
Cooper, E. C. & Jan, L. Y. M-channels: neurological diseases, neuromodulation, and drug development. Arch. Neurol. 60, 496–500 (2003).
Fernandopulle, M. S. et al. Transcription factor-mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol. 79, e51 (2018).
Keuss, M. J. et al. Loss of TDP-43 induces synaptic dysfunction that is rescued by UNC13A splice-switching ASOs. Preprint at bioRxiv https://doi.org/10.1101/2024.06.20.599684 (2024).
Neumann, M. et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
Seddighi, S. et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Sci. Transl. Med. 16, eadg7162 (2024).
Polymenidou, M. et al. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 1462, 3–15 (2012).
Baughn, M. W. et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science 379, 1140–1149 (2023).
Guerra San Juan, I. et al. Loss of mouse Stmn2 function causes motor neuropathy. Neuron 110, 1671–1688.e6 (2022).
Simkin, D. et al. ‘Channeling’ therapeutic discovery for epileptic encephalopathy through iPSC technologies. Trends Pharmacol. Sci. 43, 392–405 (2022).
Gunthorpe, M. J., Large, C. H. & Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 53, 412–424 (2012).
Ihara, Y. et al. Retigabine, a Kv7.2/Kv7.3-channel opener, attenuates drug-induced seizures in knock-in mice harboring Kcnq2 mutations. PLoS ONE 11, e0150095 (2016).
Workman, M. J. et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 111, 1191–1204.e5 (2023).
Ho, R. et al. ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks. Nat. Neurosci. 19, 1256–1267 (2016).
Agra Almeida Quadros, A. R. et al. Cryptic splicing of stathmin-2 and UNC13A mRNAs is a pathological hallmark of TDP-43-associated Alzheimer’s disease. Acta Neuropathol. 147, 9 (2024).
Mehta, P. R., Brown, A.-L., Ward, M. W. & Fratta, P. The era of cryptic exons: implications for ALS–FTD. Mol. Neurodegener. 18, 16 (2023).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience https://doi.org/10.1093/gigascience/giab008 (2012).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Boulting, G. L. et al. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 29, 279–286 (2011).
Pantazis, C. B. et al. A reference human induced pluripotent stem cell line for large-scale collaborative studies. Cell Stem Cell 29, 1685–1702.e22 (2022).
Ortega, J. A. et al. Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron 106, 90–107.e13 (2020).
Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
Schildge, S., Bohrer, C., Beck, K. & Schachtrup, C. Isolation and culture of mouse cortical astrocytes. J. Vis. Exp. https://doi.org/10.3791/50079 (2013).
Vierbuchen, T. et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010).
Hockemeyer, D. et al. A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell 3, 346–353 (2008).
Zufferey, R. et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 72, 9873–9880 (1998).
Wichterle, H. & Peljto, M. Differentiation of mouse embryonic stem cells to spinal motor neurons. Curr. Protoc. Stem Cell Biol. https://doi.org/10.1002/9780470151808.sc01h01s5 (2008).
Budini, M., Romano, V., Quadri, Z., Buratti, E. & Baralle, F. E. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 24, 9–20 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Prudencio, M. et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 26, 3421–3431 (2017).
Soldovieri, M. V. et al. Decreased subunit stability as a novel mechanism for potassium current impairment by a KCNQ2 C terminus mutation causing benign familial neonatal convulsions. J. Biol. Chem. 281, 418–428 (2006).
Legendy, C. R. & Salcman, M. Bursts and recurrences of bursts in the spike trains of spontaneously active striate cortex neurons. J. Neurophysiol. 53, 926–939 (1985).
Deng, H. X. et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215 (2011).
Ludolph, A. et al. A revision of the El Escorial criteria—2015. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 291–292 (2015).
Jesse, C. M. et al. ALS-associated endoplasmic reticulum proteins in denervated skeletal muscle: implications for motor neuron disease pathology. Brain Pathol. 27, 781–794 (2017).
Dreser, A. et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 24, 1655–1671 (2017).
Schirmer, L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Acknowledgements
We thank G. Ntermentzaki for thoughtful suggestions related to the TDP-43 CLIP experiments, T. Tzingounis for thoughtful suggestions related to recording experiments and O. Al Dalahmah for the use of lab equipment related to human pathology. We are grateful to the following funding sources: US National Institutes of Health (NIH) National Institute on Neurological Disorders and Stroke (NINDS) and NIH National Institute on Aging R01NS104219 (E.K.), NINDS NS108874 (A.L.G., E.K.), the Les Turner ALS Foundation (E.K.), the New York Stem Cell Foundation (E.K. and B.J.), ALS Scholars in Therapeutics Fellowship through the Healey Center at MGH (B.J. and J.R.M.), UK MRC SCF and MNDA LEWF (MR/S006508/1 to P.F.), ALS Stitching (ALS Tissue Bank–NL AV20220011; E.A.), NIH R01NS125018, R35GM145279 (C.Z.), Project ALS (H.W.), NIH RF1NS120992 and the Brightfocus foundation (M.P.), NIH U54NS123743 (P.F. and L.P.), Wellcome Trust (215508/Z/19/Z; J.B.), Target ALS (M.P. and L.P.), NIH R35NS097273 (L.P.), NIH T32 AG020506 (W.C.) and NIH S10OD034362 (A.L.G.). Postmortem patient specimens were provided by the Department of Veterans Affairs Biorepository (VA Merit review BX002466), the Mayo Clinic Brain Bank and Amsterdam University Medical Center (approved by the local Ethical Committee: W11073). B.J. is a New York Stem Cell Foundation–Druckenmiller fellow. E.K. is a New York Stem Cell Foundation–Robertson Investigator. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
B.J.J., K.A.M., P.H., P.F., K.E. and E.K. conceptualized the study. B.J.J., K.A.M., P.H., J.R.M., F.A., C.G.V., W.C., M.P., D.S., T.K., R.R.D., M.J.K., S.B., M.Z., P.R.M., J.-M.D., F.L., J.L., A.-L.B., M.F.L.-J., L.A.N., I.G.S.J., E.A. and A.G. conducted the formal analyses and investigations. M.P., J.B., C.Z., H.W., L.P., J.K.W., A.L.G., P.F., K.E. and E.K. acquired funding. B.J.J., K.A.M., P.H., P.F. and E.K. constructed the methodology. B.J.W., M.S., J.B., C.Z., H.W., L.P., J.K.W., A.L.G., P.F., K.E. and E.K. were responsible for project administration and supervision. E.A., N.A.S., D.W.D., P.F. and E.K. acquired resources. P.H., P.F., T.-T.K., A.G., E.A. and N.A.S. validated the work. B.J.J., K.A.M. and P.H. constructed visualizations. B.J.J., K.A.M. and E.K. wrote the original draft; all authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
P.F. is an academic founder of Trace Neuroscience, consults and is a Scientific Advisory Board member for VectorY. B.J.W. is a Scientific Advisory Board member of Quralis. K.E. is a cofounder of Q-State Biosciences, Quralis and Enclear Therapies and is currently employed at BioMarin Pharmaceuticals. J.K.W. and E.K. are academic cofounders of NuCyRNA Therapeutics; E.K. is also an academic cofounder of NeuronGrow and a Scientific Advisory Board member of Axion Biosystems, ResQ Biotech and Synapticure. Named companies were not involved in this project. The other authors declare no competing interests.
Peer review
Peer review information
Nature Neuroscience thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Identification and prioritization of TDP-43-dependent KCNQ2 mis-splicing.
(a) Schematics of distinct classes of alternative splicing events. (b) Heatmap and classification of splicing events that are altered with TDP-43 depletion. Percent Spliced Index (PSI) quantifies proportion of reads mapping to one splice variant relative to all splice variants from the same locus. de novo junctions are splice variants that are not normally expressed at detectable levels. (c) Overlap of 125 differential local splice variants (LSVs) detected in purified MNs after TDP-43 knockdown11, with other datasets of TDP-43 depletion reported in Liu et al.41 and Brown et al.9. (d) Related to Fig. 1b. Left: of the 522 genes with altered splicing in neuronal datasets, 47 are involved in the “regulation of membrane potential” and “synaptic signaling”. Right: list of 47 genes. Those with de novo events, such as cryptic exon de-repression are shown in red. (e-f) Genes involved in the regulation of membrane potential and synaptic signaling are not commonly differentially expressed (DE) in neurons depleted of TDP-43. (e) Total number of DE genes in each set of neuronal datasets. (f) Left: number of DE genes in each set, involved in the “regulation of membrane potential” and “synaptic signaling” and Right: Overlap of DE genes. (g) Sashimi plot depicting HISAT2-mapped sequencing coverage and junction spanning reads of alternatively spliced locus in CACNA1E. Inclusion of annotated cassette exon increases following TDP-43 loss. (h) Top: KCNQ2 gene model, including location of skipped exon 5. Bottom: exon 5 is deeply conserved. Graphic is generated using the UCSC genome browser, where each dot indicates conservation of the related amino acid. (i) Amino acid sequence alignment for WT and KCNQ2∆E5. A fragment of the peptide is shown, and individual domains are boxed, shaded, and annotated. Exon 5 codes for transmembrane domain 5, extracellular components and a portion of the intramembrane pore-forming domain. (j) Confirmation of mis-spliced KCNQ2 by Sanger sequencing.
Extended Data Fig. 2 Identification and prioritization of TDP-43-dependent KCNQ2 mis-splicing.
(a) Verification of mis-spliced KCNQ2 in day 40 human iPSC-MNs. Representative image of n > 3 biological replicates; marker in base pairs. (b) KCNQ2∆E5 abundance increases with TDP-43 knockdown level. Comparison of KCNQ2∆E5 and TDP-43 level in reported datasets9,11. (c-e) Upregulation of KCNQ2∆E5 and decrease of KCNQ2WT in Dox-inducible TDP-43 aggregation model in SH-SY5Y cells. (c) Detection of KCNQ2∆E5 upon Dox induction of TDP-43 containing 12 Q/N domain repeats; marker in base pairs. (d) Percent Spliced Index (PSI) of KCNQ2 (exon 5 inclusion or skipping) in control versus aggregation-prone TDP-43 cells. Points represent n = 3 independent biological replicates; values are mean +/- SEM; p-values are the result of unpaired two-tailed t-test. (e) Immunofluorescence of TDP-43 aggregates in SH-SY5Y 12Q/N cells; NT=not treated control. (f) Conservation of genomic locus of KCNQ2 exons 4-6 between human and mouse. Mouse conservation is reflected on a black to white color scale where black is highly conserved and white is not conserved. Human KCNQ2 exonic sequences are conserved, while intronic sequences are poorly conserved in mice. The 1-kb long UG repeat found upstream of human exon 5 is not present in the mouse genome. (g-h) Splicing of murine Kcnq2 is not regulated by TDP-43; data from Polymenidou et al.45. (g) TDP-43 depletion in mouse brains treated with Tardbp targeting ASOs. (h) Sashimi plot depicting HISAT2-mapped sequencing coverage and junction spanning reads of syntenic mouse Kcnq2 locus. There is no exon skipping following TDP-43 reduction (TDP-43 antisense oligonucleotide; ASO). (i) Comparison of KCNQ2∆E5 abundance in human and mouse stem-cell derived neurons with TARDBP knockdown. qRT-PCR-based relative expression of KCNQ2∆E5 and TARDBP. Circles represent relative expression of KCNQ2∆E5 or TARDBP from n = 3 independent biological replicates of TARDBP knockdowns in MNs. Data are presented as mean +/- SEM; p-values are the result of unpaired t-test. (j) Top: Sashimi plot depicting HISAT2-mapped sequencing coverage and junction spanning reads of the entire mouse Kcnq2 gene. Bottom: gene models. TDP-43 depletion did not induce alternative splicing in this gene; Data from Polymenidou et al.45.
Extended Data Fig. 3 Expression of KCNQ2∆E5 is highly specific to ALS/FTD and TDP-43 pathology.
(a) Reads mapping to KCNQ2∆E5 in RNA-seq datasets stratified by disease status and CNS tissue source. Violin plots shown, each point represents a unique sample, and the score indicates number of unique KCNQ2∆E5 reads. P-values are the result of unpaired one-sided Wilcoxon Rank Sum tests. (b) Boxplots displaying abundance of KCNQ2, STMN2 and UNC13A in CNS regions. Points reflect gene expression from individual patient CNS samples from Target ALS/NYGC. Boxes show the interquartile range of gene expression in TPMs, whiskers reflect those beyond the interquartile range. Number of unique samples indicated within the panel. (c) Relative abundance of TDP-43 targets KCNQ2, STMN2 and UNC13A in human cortex and spinal cord using the same datasets employed for splicing analyses in Fig. 2a-e. Data shown as violin plots with underlying gene expression values from postmortem samples (datapoints). P-values are the result of unpaired, two-sided t-tests. (d) Proportion of regional CNS tissue samples with aberrant splicing events. Left: KCNQ2∆E5; Middle: cryptic truncation of STMN2; Right: cryptic exon inclusion in UNC13A. Numbers of samples with and without detection of events are included (detected: not detected). (e) Expression of KCNQ2, STMN2 and UNC13A in different cell types in the human cortex. Data from Schirmer et al.95 and Velmeshev et al.96. (f) Related to Fig. 2f-i. Correlation between KCNQ2∆E5 abundance by qRT-PCR and disease duration. P-values are the result of linear regression models adjusted for age at death, sex, and RIN. For additional statistics see Supplementary Table 2.
Extended Data Fig. 4 KCNQ2∆E5 is a loss-of-function allele exerting dominant negative activity.
(a) Structural predictions generated by Alpha Fold for single KCNQ2 subunits, WT (top left) and KCNQ2∆E5 (bottom left); and heterotetrametric channels consisting of WT (x2) + KCNQ3 (x2) (top right); or WT + KCNQ2∆E5 + KCNQ3 (x2) (bottom right). (b) Peak current density recorded at -30mV to +40 mV steps (same cells and n as in Fig. 3f plotted as a percentage of WT (x2) current (mean ± SEM); p-values: one-way ANOVA, pairwise-multiple comparisons by Holm-Sidak method. (c) Voltage-dependence of activation measured in heterozygous experiments for WT (x2) (V½= -27.4 ± 0.8, k = 11.8 ± 0.3, n = 98), WT + KCNQ2∆E5 (V½= -27.3 ± 0.8, k = 11.9 ± 0.3, n = 107), WT + KCNQ2T274M (V½= -25.7 ± 0.7, k = 10.9 ± 0.3, n = 129), WT + KCNQ2G279S (V½= -27.8 ± 0.9, k = 9.6 ± 0.2, n = 98). (d-g) Peak current density values plotted as a percentage of WT (x1) for (d-e) or WT (x2) for (f-g). Data separated into replicate experiments. Each circle is one cell, bars are mean ± SEM. P-value between WT and KCNQ2 variants determined by one-way ANOVA, multiple comparisons with Tukey’s test. (d-e) Data for homozygous KCNQ2-expressing cells was collected from one replicate experiment. Cells were held at voltage steps ranging from -30mV to +40 mV (same cells and n as shown in Fig. 3e). Data for (d) - 20 mV and (e) + 40 m voltage steps shown (1 value out of visual range for replicate experiment #1 at -20mV, KCNQ2WT: 324.8%). (f-g) Data for heterozygous KCNQ2-expressing cells was collected from n = 3 replicate experiments. Cells were held at voltage steps ranging from -30mV to +40 mV (same cells and n as in Fig. 3h and Ext. Figure 4b-c). Data for (e) -20mV and (f) + 40 mV voltage steps shown (2 values out of visual range for replicate experiment #2 at -20mV, WT: 335.5% and at +40 mV, KCNQ2∆E5: -51.6%). (h) Manual whole-cell voltage clamp recordings of fused (GFP or mCherry) hKCNQ2 transgenes used in cell surface biotinylation experiments. Current recorded from CHO-Kv7.3 cells expressing: GFP- KCNQ2WT (n = 6), GFP-KCNQ2∆E5 (n = 5), mCh-KCNQ2WT (n = 5), KCNQ2WT-IRES-GFP (n = 6). Summary data (mean ± SEM) for outward current density as a function of command voltage. Fusion does not cause significant alterations in current (KCNQ2WT-IRES-GFP vs. GFP- KCNQ2WT: p = 0.4695 and KCNQ2WT-IRES-GFP vs. mCh-KCNQ2WT: p = 0.0815; both determined by repeated measures ANOVA. (i-j) WB replicates #2 and #3 of Fig. 3j-l.
Extended Data Fig. 5 KCNQ2∆E5 accumulates in the ER and causes hyperexcitability in edited iPSC- derived neurons.
(a) Details of CRISPR mutagenesis of KCNQ2. The four gRNAs targeting KCNQ2 are presented along with details of deletions induced in and KCNQ2∆E5/∆E5 cells. (b) KCNQ2 allele copy number assay for unrelated iPSC cell line, isogenic control and KCNQ2∆E5/∆E5 ESCs. (c) Karyotype results for isogenic control and KCNQ2∆E5/∆E5 ESCs. (d) Representative images of NGN2 cortical neurons stained with DAPI, GFP, and MAP2. (e) Representative images of NGN2 cortical neurons stained with DAPI, MAP2 and KCNQ2. Scale bar: 25 μm. Yellow dashed line outlines neuronal cell body. Letters signify individual neurons for which greyscale images of MAP2 and KCNQ2 signal are magnified. Scale bar: 10 μm. (f) Quantification of the mean KCNQ2 signal was significantly higher in KCNQ2∆E5/∆E5 neurons (p < 0.0001). (g) Quantification of the max KCNQ2 signal was also significantly higher in KCNQ2∆E5/∆E5 neurons (p = 0.0035). Statistical significance for (f-g) was determined by unpaired, two-tailed student’s t-test. Data are shown as mean ± SEM, each circle corresponds to one neuron (control: n = 51, KCNQ2∆E5/∆E5: n = 79).
Extended Data Fig. 6 TDP-43 depleted iPSC-derived spinal motor neurons exhibit hyperexcitability that can be rescued by splice-modulating KCNQ2 ASOs.
(a) Schematic illustrating splice modulating ASO (smASO) screen strategy in iPSC-MNs. (b) qRT-PCR of TARDBP expression in iPSC-MNs at day 32 of the screen; KD: knockdown. Data are presented as mean ± SEM; Statistical significance was determined by 2-tailed, unpaired t-test. Circles represent a biological replicate. (c) qRT-PCR of mis-spliced KCNQ2∆E5 after treatment with smASO-KCNQ2 candidate #2 at three doses (1μM, 3μM, 10μM). Data were presented as mean ± SEM; p value is the result of an unpaired, 2-tailed t-test. Circles represent a biological replicate. (d) Representative ICC images of iPSC-derived MNs stained for DAPI, TDP-43 and MAP2. Scale bar: 10 μm. (e) Cell capacitance recorded from all 3 groups; number of neurons: TDP-KD+smCTRL n = 55, TDP-KD+smCTRL n = 60, TDP-KD+smKCNQ2 n = 39 and (f) Input resistance; number of neurons: TDP-KD+smCTRL n = 49, TDP-KD+smCTRL n = 53, TDP- KD+smKCNQ2 n = 27. Statistical significance determined by a one-way ANOVA; no significant differences between groups. Data represented as mean ± SEM. (g) Representative traces for spontaneous APs recorded from neurons for all 3 groups. Scale bar: 10 mV/5 ms. (h-n) Spontaneous AP properties from neurons for all 3 groups; number of neurons: CTRL+smCTRL n = 51, TDP-KD+smCTRL n = 57, TDP-KD+smKCNQ2 n = 34. Each circle represents the average of all spontaneous APs recorded from one cell over the first 15 seconds of whole cell configuration. Cells recorded from n = 4 independent biological replicates. Statistical significance determined by a one-way ANOVA followed by a Tukey’s multiple comparisons test when applicable. Data presented as the mean ± SEM.
Extended Data Fig. 7 TDP-43 depleted iPSC-derived cortical Halo-iNeurons exhibit hyperexcitability that can be rescued by splice-modulating KCNQ2 ASOs.
(a) Experimental schematic of HaloTag TDP-43 iPSC line and degradation via ubiquitin-proteasomal pathway. (b) WB analysis of TDP-43 in KD experiments. (c) Top: schematic of treatment groups. Bottom: RT-PCR assay reveals some mis-splicing of KCNQ2 because of tagging TDP-43 and significant enhancement upon TDP-43 KD. Right: qRT-PCR analysis of mis-spliced KCNQ2∆E5. Data are mean ± SEM; p-value: one way ANOVA (F(2,9) = 77.97), 13 control vs. Halo+DMSO, I3 vs. Halo+PROTAC, Halo+DMSO vs. Halo+PROTAC. Circles represent 1 of n = 4 biological replicates. (d) Cell capacitance and (e) Input resistance was not significantly different between groups, CTRL (n = 29, n = 28), TDP-43 KD (n = 37, n = 34), TDP-43 KD+smCTRL (n = 21, n = 19) and TDP43 KD+smKCNQ2 (n = 23, n = 22). Each circle represents one cell. Data are mean ± SEM; p-value: unpaired, two-tailed t-test (ns: not significant, p > 0.05 in all cases). (f) Left: mAHP measured from single APs recorded from CTRL (n = 34) and TDP-43 KD neurons (n = 27). Each circle represents one cell. Data are mean ± SEM; p-value: unpaired, two-tailed t-test. Right: Representative traces and quantification of mAHPs of XE991 treatment. Data presented as pre-and post- XE991 for individual cells from CTRL (n = 7) and TDP-43 KD neurons (n = 7); p-value: paired two-tailed t-test. (g) Left: mAHP measured from single APs recorded from TDP-43 KD+smCTRL (n = 25) and TDP-43 KD+smKCNQ2 (n = 27). Each circle represents one cell. Data are mean ± SEM; p-value: unpaired, two-tailed t-test. Right: Representative traces and quantification of mAHPs of XE991 treatment for TDP-43 KD+smCTRL experiments. Data presented as pre-and post-XE991 for individual cells from TDP-43 KD+smCTRL (n = 8) and TDP-43 KD+smKCNQ2 (n = 8); p-value: paired, two-tailed t-test.
Extended Data Fig. 8 KCNQ2 protein forms focal accumulations in postmortem ALS patient spinal MNs.
(a-d) Samples analyzed at Northwestern University (NU cohort). (a-b) Related to Fig. 7a; representative images from control and sALS spinal cord sections stained for DAPI (blue), MAP2 (white), KCNQ2 (green), TDP-43 (red). (c-d) Related to Fig. 7c; representative images from control and sALS spinal cord sections stained for DAPI (blue), MAP2 (white), KCNQ2 (green), Ubiquitin (red). (e-h) Samples analyzed at Columbia University (CU cohort). (e-f) Related to Fig. 7d; representative images from healthy control and ALS spinal cord sections stained for DAPI (blue), KCNQ2 (green) and phospho-TDP-43 (red). (f) Note selective signal of phospho-TDP-43 in ALS samples. (g) Higher normalized KCNQ2 intensity in ALS samples relative to control samples. Neuron counts; control n = 29 from 3 individuals, ALS n = 142 from 15 patients; p-value determined by unpaired, two-tailed t-test with Welch’s correction. (h) Representative image from ALS spinal cord section stained for DAPI (blue), KCNQ2 (green) and Ubiquitin (orange). Note cytoplasmic overlap between KCNQ2 accumulations and Ubiquitin; n = 3 sALS and control cases were assessed. Scale bars for NU: 20 μm and CU: 15 μm.
Supplementary information
Supplementary Tables 1–5
Supplementary Tables 1–5.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 1
Uncropped blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Uncropped blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Uncropped blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Uncropped blots.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 6
Uncropped blots.
Source Data Fig. 7
Statistical source data.
Source Data Extended Fig. 1
Statistical source data.
Source Data Extended Fig. 2
Statistical source data.
Source Data Extended Fig. 2
Uncropped blots.
Source Data Extended Fig. 3
Statistical source data.
Source Data Extended Fig. 4
Statistical source data.
Source Data Extended Fig. 4
Uncropped blots.
Source Data Extended Fig. 5
Statistical source data.
Source Data Extended Fig. 6
Statistical source data.
Source Data Extended Fig. 7
Statistical source data.
Source Data Extended Fig. 7
Uncropped blots.
Source Data Extended Fig. 8
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Joseph, B.J., Marshall, K.A., Harley, P. et al. TDP-43-dependent mis-splicing of KCNQ2 triggers intrinsic neuronal hyperexcitability in ALS/FTD. Nat Neurosci 28, 2476–2492 (2025). https://doi.org/10.1038/s41593-025-02096-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41593-025-02096-w
