
Abstract
Post-translational modification of proteins by SUMO (small ubiquitin-like modifier) regulates fundamental cellular processes and occurs through the sequential interactions and activities of three enzymes: E1, E2 and E3. SUMO E1 activates SUMO in a two-step process involving adenylation and thioester bond formation, followed by transfer of SUMO to its dedicated E2 enzyme, UBC9. This process is termed E1–E2 thioester transfer (or transthioesterification). Despite its fundamental importance, the molecular basis for SUMO E1–UBC9 thioester transfer and the molecular rules governing SUMO E1–UBC9 specificity are poorly understood. Here we present cryo-EM reconstructions of human SUMO E1 in complex with UBC9, SUMO1 adenylate and SUMO1 thioester intermediate. Our structures reveal drastic conformational changes that accompany thioester transfer, providing insights into the molecular recognition of UBC9 by SUMO E1 and delineating the rules that govern SUMO E1–UBC9 specificity. Collectively, our structural, biochemical and cell-based studies elucidate the molecular mechanisms by which SUMOylation exerts its essential biological functions.
Similar content being viewed by others


Main
Reversible post-translational modification by small ubiquitin-like modifier (SUMO) regulates essential cellular processes, including DNA repair, mitosis, transcription and chromatin organization1. SUMOylation alters protein conformation, localization or modification state, thereby affecting function and stability2. Dysregulation of SUMO enzymes is linked to oncogenesis through effects on cell cycle, DNA repair and apoptosis3,4,5. Consequently, the SUMO pathway is an emerging target for cancer therapeutics, with several inhibitors showing promise6,7,8,9.
SUMOylation proceeds through the sequential action of E1, E2, and E3 enzymes10. SUMO E1 is a heterodimer of SAE1 and UBA2 and harbors multiple domains uniquely suited to their specific mechanistic roles11,12,13,14. Active and inactive adenylation domains (AAD and IAD, respectively) are responsible for initial SUMO recognition and subsequent adenylation of the SUMO C-terminal glycine15,16. Following adenylation, a catalytic cysteine in the E1 cysteine domain forms a thioester bond with the SUMO C terminus in the second activation step15,16. The cysteine domain of SUMO E1 is split into first and second catalytic cysteine half-domains (FCCH and SCCH, respectively). The SCCH domain is structured and harbors the catalytic cysteine of SUMO E1 and loop elements such as the crossover, reentry and flexible ‘cys cap’, which coordinate domain positioning during catalysis and protect the active-site cysteine7,16. In contrast, the FCCH domain is intrinsically disordered with no known function15,16. For clarity, we refer to the adenylated form of SUMO as SUMO(a) and the thioester-linked form as SUMO(t) throughout the manuscript. Lastly, the ubiquitin-fold domain (UFD), flexibly tethered to the AAD, recruits the SUMO E1 cognate E2 (UBC9) for thioester transfer15,17. Despite structural similarity with other ubiquitin-like (Ubl) systems16,18,19,20,21,22,23,24,25, each Ubl pathway is functionally distinct, requiring strict E1–E2 specificity26,27.
Although SUMO activation by E1 is well understood16, structural insight into how E1 transfers SUMO to UBC9 is lacking as no structures of human SUMO E1–UBC9 complexes have been reported. Modeling UBC9 onto E2-free SUMO E1 based on Ub, NEDD8 and ISG15 systems23,28,29,30,31,32,33 places the catalytic cysteines more than 60 Å apart, indicating that large conformational changes must occur during thioester transfer. However, the nature of these rearrangements and the interaction networks involved remain unknown. Lastly, although maintaining fidelity within the SUMOylation pathway is essential, the mechanism by which SUMO E1 selectively discriminates against E2 enzymes from other ubiquitin and Ubl systems, ensuring the specific transfer of activated SUMO to its cognate E2, remains unknown.
Here, we present cryo-electron microscopy (cryo-EM) structures of human SUMO E1 in complex with UBC9, SUMO1 adenylate and a SUMO1 thioester mimetic, capturing the complex in a state primed for thioester transfer. The structures reveal a dramatic 175° rotation of the UFD, which aligns the active sites of UBA2 and UBC9. This rearrangement is accompanied by a switch in interaction networks; contacts stabilizing the inactive UFD conformation are broken and replaced by new ones in the active state. Additional conformational changes within the SCCH domain expose the catalytic cysteine and reconfigure the cys cap to accommodate UBC9. The structure also defines the molecular basis of UBC9 recognition and reveals key determinants of E1–E2 specificity. Supported by biochemical and cell-based assays, our findings illuminate the mechanism of SUMO E1–E2 thioester transfer, highlight conserved features across Ubl pathways and uncover SUMO-specific adaptations that ensure pathway fidelity.
Results
Overall structure of a SUMO E1–UBC9/SUMO1(a) complex
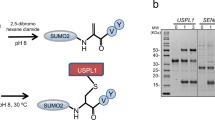
To investigate how human SUMO E1 recruits UBC9 and catalyzes thioester transfer, we used a previously described crosslinking strategy to stabilize the complex through a disulfide bond between the catalytic cysteines of SUMO E1 (UBA2 C173) and UBC9 (C93) (Fig. 1a,b)28,29,30,31,32,34. This approach enabled us to obtain a cryo-EM reconstruction of SUMO E1 in complex with UBC9 and SUMO1 adenylate (SUMO E1–UBC9/SUMO1(a)). Gold-standard Fourier shell correlation (FSC) curves calculated from two independently refined half-maps indicated an overall resolution of 2.7 Å at FSC = 0.143 (Fig. 1c,d, Extended Data Figs. 1 and 2 and Table 1). Overall, the complex exhibits a conserved architecture, with the IAD and AAD engaging SUMO1(a) and UBC9 positioned between the UFD and SCCH domains of SUMO E1 (Fig. 1c,e). UBC9 is recognized through combined contacts from the UFD, SCCH domain and the crossover loop linking the AAD and SCCH domains, burying ~2,600 Å2 of UBC9 surface area at the interface (Fig. 1c,e). The C-terminal G97 of SUMO1 is located in the active site, with continuous density consistent with an acyl adenylate intermediate. Density for the pyrophosphate leaving group is absent, indicating that the structure captures the post-adenylation state of SUMO1 (Extended Data Fig. 1d). As expected, clear density for the disulfide bond between UBA2 C173 and UBC9 C93 is visible (Extended Data Fig. 1d). Lastly, as seen in prior crystal structures7,15,16, most of the FCCH domain remains unstructured in our reconstruction, likely because of conformational flexibility (Fig. 1c,d).

a, Schematic of the SUMO E1–E2 thioester transfer reaction. The ‘inactive’ state is shown, where the catalytic cysteines of SUMO E1 and UBC9 are distal. Upon conformational change, both the catalytic cysteines come in proximity and form a tetrahedral intermediate with SUMO1(a). SUMO E1 in complex with adenylated SUMO1(a) is termed as ‘singly loaded’. b, Schematic (left) and purification (right) of a trapped SUMO E1–UBC9/SUMO1(a) complex. c, Left, cryo-EM map of SUMO E1–UBC9/SUMO1(a) complex colored and labeled as indicated. Right, cartoon representation of the model. The catalytic cysteines of SUMO E1 and UBC9 are shown as yellow spheres. Adenosine monophosphate (AMP) and zinc are shown as spheres and colored in lime and gray, respectively. d, Domain organization of heterodimeric SUMO E1 (UBA2 and SAE1) with domains colored as in c. e, SUMO E1 atoms contacting UBC9 colored by domain, as in c. Aden., adenylation; Conf., conformational; PURE, purified; DTT, dithiothreitol.
Conformational changes accompanying SUMO E1–E2 thioester transfer
In all previous structures of SUMO E1 determined in the absence of UBC9, the UFD adopts a thioester transfer-inactive conformation, where the E2 binding surface points away from the SUMO E1 active-site cysteine7,15,16 (Extended Data Fig. 3a). Docking UBC9 onto a representative E2-free structure shows that the catalytic cysteines of E1 and E2 are separated by ~67 Å, too far for thioester transfer to occur (Fig. 2a). In our SUMO E1–UBC9/SUMO1(a) structure, the UFD undergoes a ~175° rotation and 17-Å center-of-mass translation, aligning the active sites and enabling thioester transfer (Fig. 2a–c).

a, Left, UBC9 docked onto E2-free SUMO E1 (PDB 1Y8R). E1 and E2 catalytic cysteines are shown as yellow spheres. Right, network of intramolecular contacts that stabilize the thioester transfer-inactive conformation of the UFD. Interacting residues are shown as sticks and hydrogen bonds are indicated by dashed lines. UFD residues stabilizing inactive and active UFD conformations are shown as red and green spheres, respectively. b, The SUMO E1–UBC9/SUMO1(a) structure presented as in a. c, Left, conformational changes of the SUMO E1 UFD accompany E1–E2 thioester transfer. The adenylation domains of apo SUMO E1 (PDB 1Y8R) and the SUMO E1–UBC9/SUMO1(a) structures were superimposed and the UFDs are shown as cartoon representations. Middle, magnified view of the UFD linker region highlighting the bending region that accompanies the UFD conformational change. Right, differences in phi and psi angles of residues in the UFD linker region of inactive versus active UFD. d, SUMO E1–E2 thioester transfer assay of the indicated mutants of SUMO E1. e, Comparison of the cys cap of SUMO E1–UBC9/SUMO1(a) (this study; left) and E2-free SUMO E1 (PDB 1Y8R; middle). Right, superposition of the two structures with the E2-free structure colored gray. f, Left, SUMO E1–E2 thioester transfer assays with WT and Δcys cap variants of SUMO E1. Right, representative SYPRO-stained gel for the E1–E2 thioester transfer assay. Data are presented as bar graphs with the mean ± s.d. of three technical repeats displayed as a percentage of the WT value and individual replicates shown as black dots (d,f, left).
Active and inactive UFD conformations are stabilized by distinct intramolecular contact networks involving the UFD, AAD–UFD linker and AAD (Fig. 2a,b). Interestingly, UFD residues that mediate these contacts are on opposite surfaces, which are repositioned by the UFD’s ~175° rotation. In the inactive state, Y442 in the linker engages a hydrophobic pocket formed by V450, F473 and V546 and forms a backbone hydrogen bond with Y526 (Fig. 2a). R127 of the AAD forms a hydrogen bond with the backbone carbonyl of A474, while H124 interacts with Q524 (Fig. 2a). During thioester transfer, all of these UFD residues are displaced from the AAD–UFD linker and AAD because of the ~175° rotation of the UFD and are replaced with UFD residues that stabilize the active UFD conformation. R127 now interacts with E483 and D484 of the UFD and P99 and D100 (AAD) form hydrogen bonds with R512 (UFD) (Fig. 2b). The importance of contacts stabilizing the active UFD conformation is demonstrated by a ~10-fold reduction in thioester transfer activity for an E483R;R512A mutant with no observed defect in SUMO E1 activation of SUMO1 (Fig. 2d and Extended Data Fig. 4a). Notably, R127 was the only residue that made contacts with the AAD domain in both states and its substitution compromised SUMO activation (Extended Data Fig. 4a).
The zinc-binding motif15, comprising C158 and C161 in the crossover loop and C441 and C444 in the AAD–UFD linker, remains intact in our UBC9-bound structure (Fig. 2a,b). A structural comparison indicates that the UFD rotation pivots around a hinge centered on S446 and K447, located in the AAD–UFD linker just after the zinc-binding motif (Fig. 2c). The importance of this hinge in thioester transfer is underscored by the A445G;S446G;K447G triple mutant, which reduces thioester transfer ~4-fold without affecting SUMO1 activation (Fig. 2d and Extended Data Fig. 4a). Rigidifying the hinge with A445P or S446P;K447P substitutions similarly reduces activity by ~50% (Extended Data Fig. 4e,i), highlighting its essential role in enabling UFD mobility. These findings underscore the critical role of the flexible hinge region in enabling the conformational dynamics required for efficient thioester transfer.
Although the SCCH domain remains in an open conformation in both E2-free and UBC9-bound structures (Fig. 2a,b and Extended Data Fig. 3b), the cysteine cap within the SCCH undergoes substantial remodeling during thioester transfer (Fig. 2e). In E2-free structures, residues 205–218 of the cysteine cap are ordered and occlude the E1 active site, while residues 219–239 are disordered7,15,16. In our active-state structure, residues 205–218 shift, unmasking the catalytic cysteine, and residues 219–239 become ordered and engage UBC9 (Fig. 2e and Extended Data Fig. 6a,b). Together, these conformational changes unmask the SUMO E1 catalytic cysteine, prevent steric clashes between the cysteine cap and UBC9 and position cysteine cap residues for productive interactions with UBC9 that likely account for its ordering. The importance of the cysteine cap is demonstrated by a substantial reduction in thioester transfer activity when residues 219–239 are deleted from SUMO E1 (Fig. 2f and Extended Data Fig. 4f).
Molecular recognition of UBC9 by SUMO E1
Our structure reveals that SUMO E1 engages UBC9 through three distinct interfaces: the UFD (site I), the crossover loop (site II) and the SCCH domain (site III) (Fig. 3a,c). The UBC9–UFD interface buries ~900 Å2 and features extensive electrostatic interactions between a conserved acidic patch on the UFD centered on D479, E494, E497 and D520 and basic residues on UBC9 helix A (Fig. 3a and Extended Data Fig. 5a). R13 forms hydrogen bonds with S492 and E497, R17 forms hydrogen bonds with S493 and K14 forms a hydrogen bond with the carbonyl oxygen of D521 (Fig. 3a). F522 inserts into a pocket formed by R13, K14, R17 and K18 of UBC9.

a, Left, network of contacts between SUMO E1 UFD and UBC9 with involved residues shown as sticks with red oxygen atoms, blue nitrogen atoms and yellow sulfur atoms. Hydrogen bonds are indicated by dashed lines. Right, electrostatic surface representation of SUMO E1 UFD with UBC9 overlaid as on the left. b, E1–E2 thioester transfer assays of the indicated mutants for UBC9 and SUMO E1 UFD, respectively. c, Left, interaction network between the SUMO E1 crossover loop and UBC9. Right, network of contacts between the SCCH domain and cysteine cap of SUMO E1 with UBC9. d, E1–E2 thioester transfer assays of the indicated mutants for UBC9 and SUMO E1 crossover loop interface, respectively. e, E1–E2 thioester transfer assays of the indicated mutants for SUMO E1 SCCH domain and cys cap and UBC9, respectively. f, E1–E2 thioester transfer assays of the indicated charge swapped mutants for SUMO E1 and UBC9, respectively. Data are presented as bar graphs with the mean ± s.d. of three technical repeats, displayed as a percentage of the WT value. Individual replicates shown as black dots.
Additional contacts involve the UBC9 β1–β2 loop. K30 forms a hydrogen bond with the carbonyl oxygen of E497, while M36 engages in van der Waals contacts with I489, E497 and T498 (Fig. 3a). Backbone-mediated hydrogen bonds between M36 and L38 (UBC9) and G487 and I489 (UFD) further stabilize the interface (Fig. 3a). The crucial importance of these networks of interactions is evidenced by drastically reduced thioester transfer activities of UBC9 mutants R13E, K14E, R17E, K30E and M36D and in reciprocal UFD mutants (Fig. 3b and Extended Data Fig. 4b–d). To exclude loss of activity because of mutation-induced instability, we performed thermal shift assays for several SUMO E1 and UBC9 mutants. Some mutants diverged in terms of stability when compared to the wild-type (WT) SUMO E1 and UBC9; hence, although we cannot with absolute certainty exclude potential mitigating effects because of reduced stability, our combined biochemical and structural data indicate that the observed effects arise because of compromised interactions (Extended Data Fig. 7a,b).
A yeast UFD–UBC9 crystal structure previously revealed a similar topology17 but structural alignment shows a ~23° rotation of UBC9 between yeast and human complexes (Extended Data Fig. 6c), shifting helix A and altering its contacts with the acidic UFD surface (Extended Data Fig. 6d). This may reflect distinct reaction stages, with the yeast structure capturing early recognition and the human structure poised for catalysis. The acidic patch in yeast differs in both sequence and spatial arrangement, leading to more clustered interactions, including a species-specific contact from Q10 (A10 in human UBC9) (Extended Data Fig. 6d,e and Extended Data Fig. 7d). These differences support the idea of evolutionary tuning of E2 recruitment mechanisms.
At site II, the SUMO E1 crossover loop engages UBC9 helix A and two loops (β3–β4 and β7–hB) near the UBC9 active site (Fig. 3c, left). S2 and I4 of the N terminus of UBC9 contact a hydrophobic patch formed by E160 and P163 at the base of the crossover loop. Substitutions I4A in UBC9 or E160A;P163A in SUMO E1 reduce thioester transfer by over 50% (Fig. 3d and Extended Data Fig. 4e). Additional contacts include K65, D66 and D67 (β3–β4 loop) and E98 and E99 (β7–hB loop), which interact with T166, Q167, T169 and P171 of the SUMO E1 crossover loop (Fig. 3c, left). The T166A;Q167A mutant further impairs activity (Fig. 3d and Extended Data Fig. 4e).
At site III (Fig. 3c, right), the catalytic cysteines of SUMO E1 (UBA2 C173) and UBC9 (C93) are within disulfide bonding range. R176 and N177 of SUMO E1 interact with UBC9 S89, T91 and P128. M269 (SCCH domain) fits into a hydrophobic patch with UBC9 P128 and A129, while Y134 and N139 (UBC9 helix C) form hydrogen bonds with D270 and T220 of the SUMO E1 SCCH domain (Fig. 3c, right). Proximal to this, a surface of the SUMO E1 cysteine cap comprising a stretch of residues from E205 to D209 forms a pocket into which Y87 and P88 of UBC9 insert. The importance of site III interactions is demonstrated by substitutions in UBC9 (S89F and Y134A) and SCCH (N177E, R237E and M269D), which substantially reduce thioester transfer activity (Fig. 3e and Extended Data Fig. 4f,g).
Lastly, several cys cap residues in SUMO E1 become ordered upon UBC9 binding and contribute to site III. Although the P219–D234 region of the cysteine cap is poorly resolved, an α-helix comprising E221–S229 packs into a complementary surface of UBC9 (Fig. 3c, right, and Extended Data Fig. 5b, middle). D232 and R237 of the cysteine cap are near UBC9 K154 and E132 of UBC9, respectively, allowing electrostatic interactions (Extended Data Fig. 5b, right). D232K and E132F substitutions reduce thioester transfer activity, whereas R237E results in complete loss of activity (Fig. 3e and Extended Data Fig. 4f,g). Notably, a double charge swap (R237E SUMO E1 + E132K UBC9) partially rescues activity, highlighting the role of electrostatic complementarity (Fig. 3f and Extended Data Fig. 4h). Together with the drastic reduction in activity observed for a Δcys cap SUMO E1 mutant (Fig. 2f), our data suggest that the cys cap contributes to the SUMO E1–UBC9 interface through shape and charge complementarity and possibly by properly orienting the incoming UBC9 for productive thioester transfer. Interestingly, deletion of the analogous region in UBA1 impairs thioester transfer activity24, although this region has not been visualized in UBA1–E2 structures to date28,29,30,34. Consistent with their functional importance, many interface residues mediating SUMO E1–UBC9 recognition are evolutionarily conserved (Extended Data Fig. 7c,d).
Determinants of SUMO E1 specificity for UBC9
Of the eight human E1 enzymes for ubiquitin and Ubl systems, five are considered canonical, sharing a multidomain architecture that includes active and inactive adenylation domains, FCCH and SCCH domains and a UFD35. These include SUMO E1, NEDD8 E1, UBA1 (Ub), UBA6 (Ub/FAT10) and UBA7 (ISG15). Each enzyme selectively transfers its cognate Ubl to specific E2s. Because these cascades regulate distinct biological processes, fidelity at the E1–E2 step is a key determinant of specificity. We used our structure to investigate how SUMO E1 achieves selectivity for UBC9.
A key feature of SUMO E1–UBC9 recognition is an electrostatic interface between basic residues in UBC9 helix A and a bipartite acidic patch on the SUMO E1 UFD. This patch spans both sides of the UFD and engages basic residues on opposite faces of UBC9 helix A, including R13 and R17 on one side and K14 and K18 on the other (Fig. 4a,e,f). R13 and R17 are unique to UBC9 and contact acidic surfaces that are specific to SUMO E1. In contrast, the acidic patch of UBA1 lies on one face of the UFD and interacts with R14 and R17 of CDC34 (Fig. 4b,e,f). A similar arrangement is seen in the UBA7–UBE2L6 complex, where a smaller acidic patch contacts R6 and K9 of UBE2L6 (Fig. 4c,e,f). NEDD8 E1 lacks a comparable acidic patch, precluding analogous electrostatic interactions (Fig. 4c,e,f).

a, Left, electrostatic surface representation of the SUMO E1 UFD with UBC9-interacting residues overlaid. Right, network of contacts between the SUMO E1 UFD and UBC9 with involved residues shown as sticks with red oxygen atoms, blue nitrogen atoms and yellow sulfur atoms. b, The UBA1–CDC34/Ub(a) (PDB 6NYA) structure presented as in a. c, The UBA7/UBE2L6 (PDB 8SV8; left) and NEDD8 E1/UBC12 (PDB 2NVU; right) interfaces presented as in a. d, A UBA1/UBC9 model was generated by docking UBC9 onto CDC34 from the UBA1–CDC34/Ub(a) structure (PDB 6NYA; left). A UBA7/UBC9 model was generated by docking UBC9 onto UBE2L6 from the UBA7–UBE2L6–ISG15(t)/ISG15(a) structure (PDB 8SV8; right). The models are presented as in a. e, Structure-based sequence alignment of the UFD-interacting regions of Ubl E2s with secondary-structure assignments mapped above the sequence and basic residues within helix A (hA) indicated by blue spheres. Periods indicate gaps in the alignment. Shaded boxes indicate E2 residues that interact with E1 UFDs in the corresponding E1–E2 complexes. Structures used to create the alignment were as follows: SUMO E1/UBC9 (this study), NEDD8 E1/UBC12 (PDB 2NVU), UBA7/UBE2L6 (PDB 8SV8), UBA1/CDC34 (PDB 6NYA), UBA1/UBC15 (PDB 5KNL) and UBA1/UBC4 (PDB 4II2). f, Structure-based sequence alignment of the E2-interacting region of Ubl E1 UFDs with secondary-structure assignments mapped above the sequence and acidic residues within the acidic patch indicated by red spheres. Shaded boxes indicate E1 residues that interact with E2 in the corresponding E1–E2 complexes. Acidic residues comprising the acidic patch of Ubl E1 UFDs are indicated by red boxes around their sequences. g, The UFD–E2 interfaces of the SUMO E1/UBC9, UBA1/CDC34 and UBA7/UBE2L6 complexes are shown as cartoons, with key structural elements underlying E1–E2 specificity across different Ubl systems highlighted and labeled. NT, N-terminal.
Secondary-structure features also contribute to specificity. UBC9 contains a distinctive insertion in its β1–β2 loop that engages SUMO E1 (Figs. 3a and 4e and Extended Data Fig. 5c,d). This loop is one to four residues longer than that in E2s for UBA1 and UBA6 and four residues longer than that in UBE2L6 (Fig. 4e and Extended Data Fig. 5c). When UBC9 is docked onto UBA1 or UBA7 E1 structures, the extended β1–β2 loop clashes with a long α-helix (H33) that is conserved in both UBA1 and UBA7 (Fig. 4d,f,g and Extended Data Fig. 5e). The corresponding helix in SUMO E1 (H18) is five residues shorter, providing space for UBC9’s loop and enabling the productive interface observed in our structure (Figs. 3a and 4a,f,g). In UBA1 and UBA7 complexes, the longer H33 interacts with shorter β1–β2 loops of their respective E2s (Fig. 4e–g). These elements form a structurally complementary pair that contributes to E1–E2 selectivity.
Another structural feature influencing Ubl E1–E2 specificity is a loop near the bottom of the UFD: β12–β13 in SUMO E1, β30–β31 in UBA1 and UBA6 and β31–β32 in UBA7. This loop lies close to E2 in known E1–E2 complexes (Fig. 4g). As noted above, UFD charge patterns vary widely among E1s (Fig. 4a–d) and most acidic residues forming the UBA1, UBA6 and UBA7 acidic patches are located within this loop (Fig. 4f). The number and arrangement of acidic residues within this loop differ across E1s and correspond to distinct conformations in E2-bound structures (Fig. 4f,g). NEDD8 E1 contains only one acidic residue in this region, consistent with its largely uncharged UFD surface (Fig. 4c,f). SUMO E1 also has a single acidic residue in its loop but includes additional acidic residues on β10 (D479) and the β11–H18 loop (E494 and E497), creating a distinct charge distribution (Fig. 4a–c,f). By contrast, UBA1, UBA6 and UBA7 each contain five partially overlapping but nonidentical acidic residues within their loops (Fig. 4f). These variations in charge and structural features support selectivity by promoting favorable electrostatic and steric interactions between E1 UFDs and E2 helix A regions while disfavoring noncognate pairs.
Altogether, our analysis reveals three primary determinants of SUMO E1–UBC9 specificity: (1) the pattern of acidic residues in the UFD; (2) the arrangement of basic residues on E2 helix A; and (3) distinct structural elements within the UFD and E2 that form the basis for structural complementarity. These features shape the geometry of helix A docking and prevent unfavorable contacts, reinforcing fidelity in Ubl E1–E2 recognition.
Distinct UFD transitions accompany thioester transfer across Ubl systems
We examined how conformational changes during SUMO E1–UBC9 thioester transfer compare to those in other canonical Ubl E1–E2 systems. UBA1 is the only other canonical E1 with structures in both E2-free and E2-bound thioester transfer states20,24,28,29,30,34. Modeling E2s onto E2-free UBA1 and SUMO E1 structures reveals markedly different UFD movements during thioester transfer (Fig. 5a–d). In UBA1, the active-site cysteines of E1 and E2 are separated by ~17 Å and are brought into proximity by a ~20° rotation and 6-Å shift of the UFD (Fig. 5c,d). In contrast, the SUMO E1 UFD rotates ~175° and translates 17 Å to close a ~67-Å gap between UBC9 and SUMO E1 in the transition to the active state (Fig. 5a,b). In the Ub system, the UFD rotates toward the E1 active site, projecting the E2 downward to bring catalytic cysteines into proximity (Fig. 5c,d). SUMO E1 instead rotates the UFD away from the active site, sweeping UBC9’s catalytic cysteine laterally by over 65 Å to align it with the E1 active site (Fig. 5a,b). A similar sweeping UFD movement occurs in the NEDD8 E1–NEDD8(t)/UBC12/NEDD8(a) structure, where the UFD rotates ~110° and shifts 18 Å to reduce the distance between active sites from ~55 Å to ~23 Å (ref. 33) (Extended Data Fig. 8a–c). As seen in E2-free structures, the cys cap of UBA1 covers the active-site cysteine20,24,28 and remains ordered in the double-loaded UBA1–Ub structure36. In UBA1–CDC34, UBA1–UBC4 and UBA1–UBC15 thioester-active complexes, the cap becomes disordered to accommodate the E2 (refs. 28,29,30,34). In contrast, the SUMO E1 cap is remodeled; a previously disordered segment becomes ordered and contacts UBC9 during transfer, while steric clashes are avoided (Extended Data Fig. 6a,b).

a, Left, UBC9 docked onto E2-free SUMO E1 (PDB 1Y8R). The distance between E1 and E2 catalytic cysteines, shown as yellow spheres, is indicated. The β12–β13 loop of the UFD is colored red and boxed. The disordered region of the cysteine cap is indicated by semitransparent hot-pink spheres. Right, the thioester transfer-active SUMO E1–UBC9/SUMO1(a) complex (this study) presented as on the left. b, Conformational changes of the SUMO E1 UFD accompanying thioester transfer. The adenylation domains of E2-free SUMO E1 (PDB 1Y8R) and the SUMO E1–UBC9/SUMO1(a) structures were superimposed and the UFDs are shown as cartoon representations. Right, axis of rotation and direction of translation. c, Left, CDC34 docked onto E2-free UBA1 (PDB 3CMM) with E1 and E2 catalytic cysteines shown as yellow spheres. The distance between catalytic cysteines is indicated. The β30–β31 loop of the UFD is colored red. Disordered region of the cysteine cap is indicated by semitransparent hot-pink spheres. Right, the thioester transfer-active UBA1–CDC34/Ub(a) complex (PDB 6NYA) presented as on the left. d, Conformational changes of the UBA1 UFD accompanying thioester transfer. The adenylation domains of E2-free UBA1 (PDB 3CMM) and the thioester transfer-active UBA1–CDC34/Ub(a) complex (PDB 6NYA) were superimposed and the UFDs are shown as cartoon representations. Right, axis of rotation and direction of translation. e, Network of interactions stabilizing the inactive UFD conformation of apo UBA1 (PDB 3CMM). f, Network of interactions stabilizing the active UFD conformation of UBA1 in the UBA1–CDC34/Ub(a) structure (PDB 6NYA). The structure is shown in the same view as E2-free UBA1 in e to highlight similarities in the network of intramolecular interactions stabilizing inactive and active UFD conformations.
The networks stabilizing UFD conformations also differ. In SUMO E1, inactive and active UFD states are supported by totally different surfaces of the UFD engaging a common AAD or AAD–UFD linker region, rearranged because of the ~175° rotation (Fig. 2a,b). In UBA1, the ~20° UFD rotation preserves most linker contacts (Fig. 5c–f). Here, a β-hairpin (β25–β26) before β27 forms a rigid unit with the UFD, rotating by a hinge at R930–K934. A conserved tryptophan (Trp anchor) just after the hairpin inserts into a hydrophobic pocket formed by the AAD and crossover loop (Fig. 5e,f and Extended Data Fig. 6f). This trp anchor, along with contacts between the hairpin, UFD and AAD, reduces UFD flexibility. In SUMO E1, six flexible residues after the zinc-binding motif within the AAD–UFD linker allow for the broader conformational range of its UFD.
Architecture of double-loaded SUMO E1–E2 mimetic and comparison to other Ubls
Double-loaded E1–E2 complexes, with one Ubl noncovalently bound at the E1 adenylation site and a second covalently linked through a thioester bond to the E1 catalytic cysteine, are most efficient at E2 recruitment and catalysis37,38. Structural studies of such double-loaded complexes have been limited by the instability of the thioester bond, low E1–E2 affinity and the transient nature of the transfer intermediate. While our high-resolution SUMO E1–UBC9/SUMO1(a) structure provided insights into UBC9 recognition and thioester transfer, the transferred SUMO1(t) molecule was absent because of the stabilization strategy used for cryo-EM analysis.
We previously developed a two-step approach that enabled visualization of Ub(t) and ISG15(t) in complex with UBA1 and UBA7, respectively31,34. Building on that success and related work in SUMO E2–E3 systems39,40,41, we applied this strategy to SUMO to visualize SUMO1(t) during transfer. Briefly, we substituted A129 near the UBC9 catalytic cysteine to lysine (A129K) to promote SUMO1 conjugation and create a stable UBC9–SUMO1(t) mimetic. To stabilize the complex further, we crosslinked UBC9 C93 to the E1 catalytic cysteine (UBA2 C173) (Extended Data Fig. 9a,b). This allowed us to trap the SUMO E1–UBC9–SUMO1(t)/SUMO1(a) double-loaded mimetic and obtain a cryo-EM reconstruction at 3.6-Å resolution (Fig. 6a,b, Extended Data Figs. 9c–g and 10 and Table 1). Comparison to the SUMO E1–UBC9/SUMO1(a) structure showed no major architectural changes upon the addition of SUMO1(t) (Extended Data Fig. 9h). SUMO1(t) is clearly resolved and placed into cryo-EM density using secondary-structure features (Extended Data Fig. 9g), although the local resolution is moderate (Extended Data Fig. 9e). We focus here on how SUMO1(t) integrates into the overall complex and describe its stabilizing interactions.

a, Left, schematic of a trapped SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex. Middle, cryo-EM map of double-loaded SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex. Right, cartoon representation of the double-loaded SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex model. Inset, relative distances among UBA2 C173, UBC9 C93 and SUMO1(t) G97 within the active sites. b, Double-loaded SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex shown as a surface representation. E1 and E2 are colored gray and black, respectively. SUMO1(a) and SUMO1(t) are colored shades of yellow. c, E2-free double-loaded UBA1 (PDB 4NNJ; left), double-loaded UBA1–CDC34 model with Ub(t) in the open conformation (PDB 7K5J; middle) and double-loaded UBA1–CDC34 model with Ub(t) in the closed conformation (PDB 7K5J; right) are shown as surface representations. E1 and E2 are colored gray and black, respectively. Ub(a) is colored cyan, Ub(t)OPEN is colored aqua blue and Ub(t)CLOSED is colored dark blue. The double-loaded models were created by docking Ub(a) onto the Ub(a) binding site of UBA1 in the states preceding thioester transfer (UBA1–CDC34–Ub(t)OPEN) and following thioester transfer (UBA1–CDC34–Ub(t)CLOSED). d, Double-loaded NEDD8 E1–NEDD8(t)/UBC12/NEDD8(a) is shown as a surface representation in two views rotated 90° about the x axis. E1 and E2 are colored gray and black, respectively. NEDD8(a) and NEDD8(t) are colored shades of green. Right, catalytic cysteines of NEDD8 E1 and UBC12 are colored hot pink and the ~23-Å distance between them is highlighted. e, Double-loaded UBA7–UBE2L6–ISG15(t)/ISG15(a) complex shown as a surface representation in two views rotated 90° about the x axis. E1 and E2 are colored gray and black, respectively. ISG15(a) and ISG15(t) are colored shades of pink.
SUMO1(t) is positioned on the front face of the complex, the same side as SUMO1(a), where UBC9 engages the helical surface of SUMO1(t) (Fig. 6a,b). The interface involves the α1–β3 and β4–α2 loops of SUMO1(t)’s β-grasp fold and the β7–hB and hB–hC loops of UBC9 (Fig. 6a and Supplementary Fig. 1a). Density for the flexible SUMO1 C terminus extends into the active sites of E1 and UBC9, placing G97 near UBA2 C173 and UBC9 C93 (Extended Data Fig. 9f). Although the interface between SUMO1(t) and E1–UBC9 buries only ~750 Å2, the contacts are sufficient to orient SUMO1(t) at the front of the complex.
Previous UBA1–E2 structures captured two distinct Ub(t) conformations corresponding to pre- and post-transfer states (Fig. 6c)34. In the pre-transfer open state, Ub(t) sits on the UBA1 FCCH domain with minimal E2 contact (Fig. 6c, middle), similar to a UBA1–Ub(t) structure lacking E2 (ref. 36) (Fig. 6c, left). During transfer, Ub(t) adopts a closed conformation that loses E1 contacts and forms extensive interactions with E2, driving the reaction forward34,42 (Fig. 6c, right). In contrast, SUMO1(t) adopts a unique conformation in the SUMO E1–UBC9–SUMO1(t)/SUMO1(a) structure, distinct from the open or closed Ub(t) states in structures such as UBA1–CDC34–Ub(t), UBA1–UBC4–Ub(t)/Ub(a) or E2-free UBA1–Ub(t)/Ub(a) (Fig. 6b,c and Supplementary Fig. 1a,b)34,36,42. SUMO1(t) and SUMO1(a) do not contact each other in this structure. Notably, no cryo-EM classes exhibited open or closed SUMO1(t) conformations resembling those in Ub systems (Extended Data Fig. 10)34,36,42.
We propose that the differing conformations of SUMO1(t) and Ub(t) in their respective E1 complexes arise in part from differences in the FCCH domains. UBA1 contains a well-structured ~100-residue FCCH domain that is clearly resolved in structural studies. In contrast, the SUMO E1 FCCH domain is only ~40 residues and has not been visualized, likely because of intrinsic disorder (Extended Data Fig. 3b). In the double-loaded UBA1–Ub(t)/Ub(a) structure, both Ub(a) and Ub(t)—the latter linked to the UBA1 catalytic cysteine—interact with the FCCH domain, which may help stabilize the open Ub(t) conformation36. The double-loaded NEDD8 E1–NEDD8(t)/UBC12/NEDD8(a) structure, captured in an inactive state with E1 and E2 catalytic sites more than 20 Å apart33, shows NEDD8(t) positioned at the front of the complex. Its conformation is distinct from that of SUMO1(t) and Ub(t) and involves direct contacts with UBC12 (Fig. 6d and Extended Data Fig. 8a). Lastly, the architectures of SUMO1 and Ub double-loaded complexes differ markedly from the UBA7–UBE2L6–ISG15(t)/ISG15(a) structure31, where ISG15(t) is located at the back of UBA7 and wedged between the UFD and SCCH domains (Fig. 6e). The reason for this architectural divergence is not yet understood.
Molecular determinants of SUMOylation in cells
We next tested the functional relevance of key contacts observed in our SUMO E1–UBC9 structures using human cells. Guided by biochemical assay results (Fig. 3b,e,f), we generated mutants targeting the UFD–UBC9 (site I) and SCCH–UBC9 (site III) interfaces and assessed their effects on cell proliferation, UBC9–SUMO1 thioester formation and global SUMOylation in HCT-116 cells (Fig. 7). Using a previously described approach43,44, we established inducible short hairpin RNA (shRNA) and small interfering RNA (siRNA) systems to deplete endogenous SUMO E1 and UBC9, respectively (Fig. 7a,c). Colony formation assays revealed that mutations impacting residues at the UFD–UBC9 interface impaired proliferation and underscored the role of UBC9 R17, which contacts SUMO E1 S493 (Fig. 7b,d and Supplementary Fig. 2a–c). Likewise, mutations disrupting the SCCH–UBC9 interface also reduced proliferation, highlighting the contribution of UBC9 E132, which interacts with SUMO E1 R237 and Y265 (Fig. 7b,d). Consistent with these observations, UBC9 mutants R17E, E132A and Y134A showed markedly reduced SUMO1 charging in thioester formation assays compared to WT (Fig. 7e). These mutants also caused a substantial drop in global SUMOylation levels in cells (Fig. 7f). Lastly, we examined SUMOylation of c-Myc, a known SUMO target45,46. c-Myc modification was abrogated in cells expressing the UBC9 mutants but not in those expressing WT UBC9 (Supplementary Fig. 2d,e). Altogether, these results define critical molecular interactions required for SUMOylation in human cells.

a, HCT-116 cells expressing IPTG-inducible UBA2 shRNA (shRNA1 clones 1 and 3, shRNA2 or both) showed reduced endogenous UBA2 levels after 72 h of IPTG induction (1 mM), confirmed by western blot. Data shown are from one independent experiment. b, Colony formation assays assessed the impact of UBA2 mutations on proliferation. KD, knockdown. Top, representative images of colonies. Bottom, data are presented as bar graphs with the mean ± s.e.m. of three technical repeats, displayed as a percentage of colony area relative to parental cells. Individual replicates are shown as dots. c, Effectiveness of UBC9-specific siRNAs in downregulating endogenous UBC9, tested by western blot in HCT-116 cells. Data shown are from one independent experiment. d, Impact of UBC9 mutations on HCT-116 cell proliferation, assessed by colony formation assays (quantification as in b). Statistical analysis was performed using a one-way analysis of variance and Sidak’s multiple-comparisons test with a single pooled variance. Data are presented as the mean ± s.e.m. of three technical repeats, displayed as a percentage of colony area relative to parental cells. Individual replicates are shown as dots. e, Western blot analysis of WT and mutant UBC9 ability to form UBC9–SUMO thioester intermediate in UBC9-knockdown cells. Experiments were independently repeated twice with similar results. f, Global SUMOylation in HCT-116 cells using UBC9 mutants, siRNA1 and siRNA3, tested by immunoblotting with SUMO1 antibody after 72 h. Experiments were independently repeated twice with similar results. IPTG, isopropyl β-D-1-thiogalactopyranoside.
Discussion
In this study, we report cryo-EM structures of the full-length human SUMO E1 enzyme in complex with its cognate E2, UBC9, in both single-loaded and double-loaded states. Together, these structures define the conformational and molecular basis by which SUMO E1 coordinates activation and transfer of SUMO1 and achieves specificity for UBC9. The structural analysis reveals three key features that distinguish SUMOylation from other Ubl modification systems: a sweeping ~175° UFD rotation that repositions UBC9 for catalysis, an unexpected conformational reorganization and ordering of the SCCH cysteine cap to engage UBC9 and a distinct, noncanonical architecture of SUMO1(t) at the E2 interface.
Although the general organization of the SUMO E1–UBC9 complex resembles that of canonical Ubl E1–E2 pairs, the path to catalysis is marked by unique domain dynamics. Whereas UBA1 and UBA7 undergo relatively modest conformational changes (~20°–30° UFD rotation) to align the E1 and E2 catalytic centers, SUMO E1 achieves this through a dramatic ~175° UFD rotation coupled with hinge-driven repositioning to close a ~67-Å gap between active-site cysteines. This sweeping motion is enabled by a highly flexible AAD–UFD linker that lacks the conserved trp anchor found in UBA1, UBA6 and UBA7, which otherwise stabilize the UFD through contacts with the AAD and crossover loop. In contrast, SUMO and NEDD8 E1s share a zinc-binding motif near the hinge and lack the trp anchor, features that may facilitate greater UFD mobility. Our structural data indicate that UBC9 binding correlates with UFD repositioning, although it remains unclear whether E2 engagement alone drives this transition or whether additional local rearrangements, such as those within the crossover loop or Zn2+-binding subdomain, also contribute. These findings support a model in which canonical Ubl E1s fall into mechanistic subclasses defined by hinge architecture, with SUMO and NEDD8 E1s relying on flexible linkers for large-scale repositioning, in contrast to the more restrained movements seen in UBA1, UBA6 and UBA7.
The UFD–E2 interface (site I) is conserved in overall topology but reveals several mechanistically important differences when comparing our full-length human SUMO E1–UBC9 structure to the previously reported yeast UFD–UBC9 fragment. Structural alignment shows a ~23° rotation in UBC9 orientation, which repositions helix A and alters its engagement with the acidic surface of the UFD. This shift results in a broader and more distributed electrostatic interface in the human complex, involving multiple basic residues on UBC9 that contact distinct acidic surfaces across the UFD. While some of these differences likely reflect primary sequence divergence, they may also correspond to distinct stages of the E1–E2 reaction cycle, consistent with the idea that the yeast fragment captures an early recognition state, whereas the conformation observed here is poised for thioester transfer. Biochemical and thermal stability assays confirm the functional relevance of this interface, with charge-reversal mutants at key contact points disrupting SUMO transfer without compromising protein stability. Notably, our full-length structure also reveals additional contacts absent from prior fragment models, including interactions with the crossover loop (site II) and ordered cysteine cap residues (site III), which likely contribute to productive E2 engagement and catalytic alignment.
Our structure-guided mutational analysis demonstrates that site II, formed by the SUMO E1 crossover loop and UBC9 helix A and β-loop elements, has an essential role in catalysis. Mutations of either partner disrupt thioester transfer, confirming site II as an active and previously underappreciated element of E1–E2 recognition. Similarly, site III, located at the SCCH–E2 junction, includes specific cysteine cap–E2 interactions that orient UBC9 for catalysis. While the cys cap is disordered in prior SUMO E1 and other Ubl E1 structures, our model reveals ordering of previously unresolved elements that contact UBC9 directly. Deletion of this region impairs SUMO transfer, underscoring its architectural and catalytic importance.
To place our findings in the broader context of Ubl pathways, we compared the conformational transitions of SUMO E1 to those of other E1–E2 systems, with a focus on doubly loaded complexes that capture key thioester transfer intermediates. In our SUMO E1–UBC9–SUMO1(t)/SUMO1(a) structure, SUMO1(t) adopts a single, well-defined conformation on the front of the complex, engaging the helical face of UBC9 and projecting its C terminus toward both catalytic centers. This orientation contrasts with the open and closed Ub(t) states observed in UBA1 structures, which are stabilized by a structured FCCH domain. SUMO E1 lacks an ordered FCCH domain and the modest buried surface area of SUMO1(t) may further contribute to differences in Ubl(t) stabilization. Comparisons to other systems reveal additional diversity; NEDD8(t) also localizes to the front but adopts a distinct conformation from SUMO1(t) and Ub(t), while ISG15(t) occupies a position on the back of UBA7, wedged between the UFD and SCCH domains. These observations suggest that Ubl identity, E1 architecture and catalytic stage together dictate the spatial and mechanistic diversity of thioester transfer intermediates across Ubl pathways.
Altogether, this work defines the structural and mechanistic basis of specificity and thioester transfer in the SUMOylation pathway while also highlighting key distinctions from other Ubl systems. The principles emerging from this study, including electrostatic tuning, conformational control and modular flexibility, underscore how E1 architecture is adapted to the unique requirements of its cognate E2. These findings provide a framework for understanding fidelity across E1–E2 systems and set the stage for future investigations into regulation, inhibition and engineering of Ubl pathways.
Methods
Cloning
All the constructs used in this study were previously described. Briefly, the DNA fragment encoding human SAE1 (residues 1–349) was cloned in pET11c vector and the DNA fragment encoding human UBA2 (residues 1–550) was cloned into pET28b to introduce a thrombin-cleavable 6×His tag at the N terminus of UBA2. Full-length human UBC9 and SUMO1 were cloned in pET28b vector with N-terminal 6×His tags. All the mutations reported here were either introduced using PCR-based site-directed mutagenesis or gBlocks from Integrated DNA Technologies and ligated into the respective plasmids used for WT constructs. All the primer pairs used are described in Supplementary Table 1.
Protein expression and purification
All proteins were recombinantly expressed in Escherichia coli BL21 (DE3) Codon Plus strain (Agilent, 230280). For SUMO E1, the cells were grown at 37 °C to an optical density at 600 nm (OD600) of 0.8 and induced by the addition of IPTG to 1 mM for 4 h at 30 °C. For UBC9 and SUMO1 expression, cells were grown at 37 °C to an OD600 of 1.0 and induced by adding 0.3 mM IPTG for overnight at 18 °C. The cell pellet was resuspended in lysis buffer (50 mM Tris-Cl pH 8.0, 300 mM NaCl, 5% glycerol and 5 mM β-mercaptoethanol) with 10 mM imidazole and lysed using sonication. The lysate was cleared by centrifugation at 35,000g for 30 min at 4 °C and applied to Ni-NTA agarose (Qiagen) beads and the protein was eluted in elution buffer (50 mM Tris-Cl pH 8.0, 200 mM NaCl and 5 mM β-mercaptoethanol) and 300 mM imidazole. For SUMO E1, the eluted protein was directly injected on the gel-filtration chromatography with Superdex 200 26/600pg columns (GE Healthcare) followed by anion-exchange chromatography (MonoQ 10/100; GE Healthcare). For UBC9 and SUMO1, the eluted protein was subjected to gel-filtration chromatography with Superdex 75 26/600pg columns (GE Healthcare). All the proteins were concentrated to 5–10 mg ml−1, aliquoted and snap-frozen in liquid nitrogen in a final buffer composition of 20 mM Tris-Cl pH 8.0, 200 mM NaCl and 3 mM DTT. A similar purification protocol was used for all the mutants used in the study.
E1–E2 thioester transfer assays
All E1–E2 thioester transfer assays were performed at 25 °C. Briefly, 0.1 μM SUMO E1, 1 μM UBC9, 5 μM SUMO1, 5 mM MgCl2, 2 mM adenosine triphosphate (ATP), 20 mM HEPES (pH 7.5) and 50 mM NaCl were mixed together. Reactions were initiated by the addition of ATP and incubated for 1 min, before terminating it with the addition of nonreducing urea SDS–PAGE buffer. The reactions were subjected to SDS–PAGE and run for 45 min at a constant voltage of 180 V on Any kD Mini-PROTEAN TGX precast gels (Bio-Rad). Gels were stained with Sypro ruby (Bio-Rad) and imaged using a Bio-Rad GelDoc Go gel imaging system (12009077EDU). The intensity of the UBC9–SUMO1 band was quantified by densitometry in ImageJ (version 1.53t) and further processed in GraphPad Prism 7a. Densitometry values were normalized to the percentage of the WT control from the same gel. All SUMO E1 and UBC9 mutants were analyzed under identical experimental conditions. Results are shown as the mean of three technical replicates, with error bars representing the s.d. Uncropped images of representative gels used in biochemical assays are included in the Source data.
Generation of UBC9–SUMO1 thioester mimetic and purification
The UBC9–SUMO1(t) thioester mimetic was generated as described previously39,40 with minor modifications. Briefly, 200 μM UBC9-F22A;G23Q;V25S;A129K;C138S;K153R and 400 μM SUMO1-C52A were added in a reaction mixture containing 20 mM Bis–Tris propane pH 10.0, 50 mM NaCl, 10 mM MgCl2, 0.1% Tween-20, 3 mM ATP and 1 μM SUMO E1 and incubated for 30 min at 30 °C. The UBC9–SUMO1(t) mimetic was purified by anion-exchange chromatography (MonoQ 10/100; GE Healthcare). The purity of the peak fractions (containing UBC9–SUMO1(t)) was assessed by SDS–PAGE, concentrated to 5 mg ml−1 and snap-frozen in liquid nitrogen in a final buffer composition of 20 mM HEPES pH 7.5, 150 mM NaCl and 2 mM DTT.
Thermal shift assay
The thermal shift assay was performed as previously described47. Briefly, SUMO E1, UBC9 and their mutants were analyzed using the GloMelt thermal shift protein stabilization kit (Biotium) as per the manufacturer’s protocol. Each protein sample (10 µl at 2.0 mg ml−1) was combined with 89.5 μl of assay buffer (10 mM HEPES and 150 mM NaCl, pH 7.4) and 0.5 μl of GloMelt thermoshift dye. The mixtures were distributed into a 96-well qPCR plate (three wells with 20 μl each). Protein denaturation was monitored using a Quantstudio 3 thermal cycler (Thermo Fisher) with a melt curve from 25 to 95 °C at a rate of 0.02 °C s−1. Fluorescence derivatives were plotted against temperature and the temperature corresponding to the minimum derivative was taken as the melting temperature.
Trapping of the single-loaded and double-loaded SUMO E1
To generate the single-loaded complex, SUMO E1–UBC9/SUMO1(a), we needed to crosslink SUMO E1 active C173 and UBC9 active C93 using 2,2′-dipyridyldisulfide, as previously described28,29,30. Briefly, purified UBC9-C138S;K153R (150–200 µM) was desalted into crosslinking buffer (20 mM HEPES pH 7.5 and 75 mM NaCl) using a HiPrep 26/10 desalting column. Purified SUMO E1 (50–100 µM) was also desalted into the crosslinking buffer. To the desalted UBC9-C138S;K153R protein, an equal volume of ‘activating buffer’ (20 mM HEPES pH 7.5, 75 mM NaCl, 2.5 mM 2,2′-dipyridyldisulfide and 2.5% DMSO) was added and incubated at 25 °C for 10–15 min. This ‘activated’ UBC9-C138S;K153R sample was subjected to a desalting column equilibrated with crosslinking buffer to remove excess 2,2′-dipyridyldisulfide and DMSO. The activated UBC9-C138S;K153R protein was mixed with the desalted SUMO E1 and incubated at 25 °C for 30 min in a molar ratio of 2:1. The crosslinking mixture was subjected to a MonoQ anion-exchange column (GE healthcare) to isolate purified crosslinked SUMO E1–UBC9-C138S;K153R complex. To generate the single-loaded complex, the SUMO E1–UBC9-C138S;K153R complex was incubated with a 1.2 molar excess of free SUMO1, ATP and Mg2+ for 10 min at 4 °C. This mixture was further concentrated to 3 mg ml−1 for EM grid preparation. For the double-loaded complex, SUMO E1–UBC9–SUMO1(t)/SUMO1(a), a similar strategy was used except that UBC9–SUMO1(t) mimetic was used instead of UBC9-C138S;K153R generated earlier.
Cryo-EM sample preparation and data collection
First, 3 μl of freshly prepared SUMO E1–UBC9/SUMO1(a) complex was applied onto cryo-EM Au grids (UltrAuFoil 1.2/1.3 300-mesh, EM Sciences) that were glow-discharged at 20 mA for 30 s in a Quorum EMS glow discharger. Grids were vitrified with a Vitrobot Mark IV (Thermo Fisher Scientific) maintained at 8 °C and 100% humidity. A blot force of 0 and blot time of 3 s was applied before plunge-freezing into liquid ethane. The data were collected using a Titan Krios G3 microscope (Thermo Fisher Scientific) operating at 300 kV at the University of Texas (UT) Austin with a Gatan biocontinuum imaging filter and a K3 direct electron detector Gif+DED. We collected 15,560 videos with a 0.8332-Å pixel size at a nominal ×105,000 transmission EM (TEM) magnification. A total dose of 52 e− per Å2 with 101 frames and a defocus range of −2.5 to −1.0 nm were used during data collection by SerialEM.
For the SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex, a similar strategy was used but the data collection was carried out at 200 kV on a Glacios cryo-TEM instrument equipped with a Falcon IV camera and Selectris energy filter (slit width: 10 eV) at the UT Health San Antonio cryo-EM facility. Imaging was acquired using Thermo Fisher Scientific’s EPU software with an AFIS model. We collected 26,154 videos with a 0.87-Å pixel size at a nominal ×130,000 TEM magnification. The total dose of 53 e− per Å2 with 1,809 EER frames and defocus range of −2.0 to −0.8 nm was used during data collection.
Cryo-EM data processing
Data processing was performed in cryoSPARC (version 3.3.2)48. For the single-loaded structure, 15,560 videos were motion-corrected using patch motion correction followed by contrast transfer function (CTF) estimation using patch CTF estimation. Blob picker was used to pick an initial 19,319,480 particles with a minimum particle diameter of 70 Å and maximum particle diameter of 120 Å. Particles were extracted with a box size of 256 pixels. Three rounds of two-dimensional (2D) classification were performed to remove junk particles and noise. A five-class ab initio classification was performed to generate initial volumes. Several rounds of heterogenous refinement were conducted to get rid of junk particles. The best class showing clear features of all components of single-loaded SUMO E1 complex was processed with homogeneous refinement followed by local refinement and then local refinement with CTF refinement, yielding a map of 2.74 Å. A full processing tree is shown in Extended Data Fig. 2.
Processing for the double-loaded structure followed an essentially similar path. A total of 26,154 videos were motion-corrected using patch motion correction followed by CTF estimation using patch CTF estimation. The same blob picking and extraction parameters were used. After multiple rounds of 2D refinement, 3,792,237 particles were kept, of which 411,645 particles were used to create six initial volumes using ab initio reconstruction. This was followed by three sequential rounds of heterogeneous refinement, where the particles from the best class and volumes of all classes were used in subsequent rounds. A final round of 3D classification was performed with ten volumes of the best class from the previous round of heterogeneous refinement and particles from the best class. Particles from classes 9 and 10, which showed density for both SUMO molecules, were used for homogeneous refinement followed by nonuniform refinement and local refinement, yielding a 3.59-Å map. A full processing tree is shown in Extended Data Fig. 10.
Model building and refinements
For SUMO E1–UBC9/SUMO1(a) complex, PDB 3KYC (SUMO E1), PDB 2GRN (UBC9) and PDB 4WJQ (SUMO1) models were used to perform rigid-body docking into the cryo-EM map using UCSF ChimeraX (version 1.5)49. Further model building and manual inspection were carried out using Coot (version 0.9.8.7)50 and refinements of stereochemistry and secondary structure were carried out in PHENIX (version 1.20.1-4487)51 using phenix.real_space_refine. Iterative rounds of real space refinement and model building were conducted to generate optimized coordinates for the complex. Figures for the cryo-EM map and models were prepared with ChimeraX version 1.5 and PyMOL version 2.0 (Schrödinger). For the SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complex, the above-generated model and PDB 4WJQ (SUMO1) were used to dock into the cryo-EM map using UCSF ChimeraX version 1.5. A similar strategy to that described above was used to perform refinement and model building for this complex.
Generation of UBA2-knockdown and UBC9-knockdown cells and mutant-expressing cells
Two unique shRNA plasmids that uniquely target UBA2 were created as IPTG-inducible lentiviral vectors (pLV(shRNA)-LacI:T2A:Puro-U6/2×LacO-hUBS2) were custom-synthesized using Vector Builder. The shRNA sequences were as follows: UBA2 shRNA1, CTGATTGATCTGGATACTATT; UBA2 shRNA2, GCTGCCCGAAACCATGTTAAT. Stable cell lines were established by introducing lentiviral transfection to express UBA2 shRNA1 or shRNA2 or both shRNA1 and shRNA2. Pooled clones were generated using puromycin selection. Each individual UBA2 shRNA was capable of decreasing UBA2 expression by approximately 30–40%. Hence, we used a simultaneous transfection of shRNA1 and shRNA2, resulting in a notable 70–80% decrease in UBA2 expression. The UBA2 shRNA expression was stimulated by the addition of IPTG (1 mM) and its efficacy in decreasing UBA2 was confirmed using western blotting after 72 h. UBA2 lentiviral expression (pLV[Exp]-EGFP:T2A:Bsd-EF1A>3×Flag–UBA2) plasmids (encoding 3×Flag–UBA2 WT and S492A;S493A, R237E, Y265D;M269D, 445GGG447 and R127D mutants) were custom-synthesized using Vector Builder and sequence-verified. Stable expression of the specified 3×Flag–UBA2 mutants was achieved in HCT-116 cells (CCL-247EMT, American Type Culture Collection) expressing IPTG-inducible UBA2 shRNA1 and shRNA2 using the transfection of lentiviral plasmids and pooled clones were selected using blasticidin S. The confirmation of UBA2 mutant expression was achieved by western blotting using both anti-UBA2 antibody and anti-Flag antibody. The UBA2 expression plasmids were specifically engineered to contain a silent mutation (wherein the change in the DNA sequence of the gene has no effect on the amino acid sequence) in the shRNA-coding region. Therefore, shRNA1 and shRNA2 do not possess the capability to degrade Flag–UBA2 constructs but they do effectively suppress the expression of endogenous UBA2.
For the generation of UBC9-knockdown cells, we used an siRNA approach. Four distinct siRNAs that target UBC9 were procured from Sigma-Aldrich. The siRNAs were transiently transfected into HCT-116 cells using RNAiMAX agent and their effectiveness in reducing UBC9 was verified using western blotting 72 h later. The siRNA sequences were as follows: UBC9 siRNA1, GGCAAGAACUUGUUUACAA; UBC9 siRNA2, CCUAUUAGGAAUACAGGAA; UBC9 siRNA3, GAAGGGAUUGGUUUGGCAA; UBC9 siRNA4, CAAACAGAUCCUAUUAGGA. UBC9 lentiviral expression (pLV-EGFP:T2A:Bsd-EF1A>1×FLAG/(UBC9) plasmids (encoding 1×Flag WT and E132A, Y134A and R17E mutants) were custom-synthesized using Vector Builder and sequence-verified. HCT-116 cells that were genetically modified to express WT UBC9 or mutant versions were then subjected to transient transfection with specific siRNA molecules targeting UBC9 (siRNA1 and siRNA3). The impact of specific UBC9 mutations on the ability of HCT-116 cells to form colonies was subsequently assessed after a period of 14 days. The efficacy of siRNA in suppressing UBC9 was verified using western blotting analysis. siRNA1 and siRNA3 only target the noncoding domain of UBC9 and they do not affect the Flag-tagged UBC WT or UBC9 mutants. These Flag–UBC9 expression vectors exclusively include the UBC9-coding area. Western blotting confirmed that the levels of endogenous UBC9 were reduced by siRNA knockdown, whereas the Flag-tagged UBC9 mutants remained unaffected. The colony formation assay and western blotting using radioimmunoprecipitation assay buffer were conducted using a published protocol52.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Atomic coordinates for the SUMO E1–UBC9/SUMO1(a) and SUMO E1–UBC9–SUMO1(t)/SUMO1(a) complexes reported in this study were deposited to the PDB under accession codes 9DRJ and 9DQB and the corresponding cryo-EM maps were deposited to the EM Data Bank under accession numbers EMD-47127 and EMD-47110. Source data are provided with this paper.
References
Hay, R. T. Decoding the SUMO signal. Biochem. Soc. Trans. 41, 463–473 (2013).
Vertegaal, A. C. O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 23, 715–731 (2022).
Flotho, A. & Melchior, F. SUMOylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 82, 357–385 (2013).
Seeler, J. S. & Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 17, 184–197 (2017).
Han, Z. J., Feng, Y. H., Gu, B. H., Li, Y. M. & Chen, H. The post-translational modification, SUMOylation, and cancer (review). Int. J. Oncol. 52, 1081–1094 (2018).
He, X. et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 13, 1164–1171 (2017).
Lv, Z. et al. Molecular mechanism of a covalent allosteric inhibitor of SUMO E1 activating enzyme. Nat. Commun. 9, 5145 (2018).
Berger, A. J. et al. Pharmacodynamic evaluation of the novel SUMOylation inhibitor TAK-981 in a mouse tumor model. Cancer Res. 79, 3079 (2019).
Biederstädt, A. et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 69, 1472–1482 (2020).
Streich, F. C. Jr & Lima, C. D. Structural and functional insights to ubiquitin-like protein conjugation. Annu. Rev. Biophys. 43, 357–379 (2014).
Desterro, J. M., Rodriguez, M. S., Kemp, G. D. & Hay, R. T. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 274, 10618–10624 (1999).
Johnson, E. S., Schwienhorst, I., Dohmen, R. J. & Blobel, G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 16, 5509–5519 (1997).
Okuma, T., Honda, R., Ichikawa, G., Tsumagari, N. & Yasuda, H. In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem. Biophys. Res. Commun. 254, 693–698 (1999).
Gong, L., Li, B., Millas, S. & Yeh, E. T. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 448, 185–189 (1999).
Lois, L. M. & Lima, C. D. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 24, 439–451 (2005).
Olsen, S. K., Capili, A. D., Lu, X., Tan, D. S. & Lima, C. D. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463, 906–912 (2010).
Wang, J. et al. Crystal structure of UBA2ufd–Ubc9: insights into E1–E2 interactions in SUMO pathways. PLoS ONE 5, e15805 (2010).
Walden, H., Podgorski, M. S. & Schulman, B. A. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature 422, 330–334 (2003).
Walden, H. et al. The structure of the APPBP1–UBA3–NEDD8–ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 12, 1427–1437 (2003).
Lv, Z., Williams, K. M., Yuan, L., Atkison, J. H. & Olsen, S. K. Crystal structure of a human ubiquitin E1–ubiquitin complex reveals conserved functional elements essential for activity. J. Biol. Chem. 293, 18337–18352 (2018).
Hann, Z. S. et al. Structural basis for adenylation and thioester bond formation in the ubiquitin E1. Proc. Natl Acad. Sci. USA 116, 15475–15484 (2019).
Lv, Z. et al. Domain alternation and active site remodeling are conserved structural features of ubiquitin E1. J. Biol. Chem. 292, 12089–12099 (2017).
Yuan, L. et al. Crystal structures reveal catalytic and regulatory mechanisms of the dual-specificity ubiquitin/FAT10 E1 enzyme Uba6. Nat. Commun. 13, 4880 (2022).
Lee, I. & Schindelin, H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 134, 268–278 (2008).
Truongvan, N., Li, S., Misra, M., Kuhn, M. & Schindelin, H. Structures of UBA6 explain its dual specificity for ubiquitin and FAT10. Nat. Commun. 13, 4789 (2022).
Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 458, 422–429 (2009).
Kerscher, O., Felberbaum, R. & Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180 (2006).
Olsen, S. K. & Lima, C. D. Structure of a ubiquitin E1–E2 complex: insights to E1–E2 thioester transfer. Mol. Cell 49, 884–896 (2013).
Lv, Z. et al. S. pombe Uba1–Ubc15 structure reveals a novel regulatory mechanism of ubiquitin E2 activity. Mol. Cell 65, 699–714 (2017).
Williams, K. M. et al. Structural insights into E1 recognition and the ubiquitin-conjugating activity of the E2 enzyme CDC34. Nat. Commun. 10, 3296 (2019).
Afsar, M. et al. Cryo-EM structures of Uba7 reveal the molecular basis for ISG15 activation and E1–E2 thioester transfer. Nat. Commun. 14, 4786 (2023).
Wallace, I. et al. Insights into the ISG15 transfer cascade by the UBE1L activating enzyme. Nat. Commun. 14, 7970 (2023).
Huang, D. T. et al. Basis for a ubiquitin-like protein thioester switch toggling E1–E2 affinity. Nature 445, 394–398 (2007).
Yuan, L., Lv, Z., Adams, M. J. & Olsen, S. K. Crystal structures of an E1–E2-ubiquitin thioester mimetic reveal molecular mechanisms of transthioesterification. Nat. Commun. 12, 2370 (2021).
Schulman, B. A. & Harper, J. W. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (2009).
Schafer, A., Kuhn, M. & Schindelin, H. Structure of the ubiquitin-activating enzyme loaded with two ubiquitin molecules. Acta Crystallogr. D 70, 1311–1320 (2014).
Haas, A. L., Bright, P. M. & Jackson, V. E. Functional diversity among putative E2 isozymes in the mechanism of ubiquitin–histone ligation. J. Biol. Chem. 263, 13268–13275 (1988).
Pickart, C. M., Kasperek, E. M., Beal, R. & Kim, A. Substrate properties of site-specific mutant ubiquitin protein (G76A) reveal unexpected mechanistic features of ubiquitin-activating enzyme (E1). J. Biol. Chem. 269, 7115–7123 (1994).
Goffinont, S. et al. Structural insights into the regulation of the human E2 approximately SUMO conjugate through analysis of its stable mimetic. J. Biol. Chem. 299, 104870 (2023).
Streich, F. C. Jr & Lima, C. D. Capturing a substrate in an activated RING E3/E2-SUMO complex. Nature 536, 304–308 (2016).
Varejao, N. et al. Structural basis for the E3 ligase activity enhancement of yeast Nse2 by SUMO-interacting motifs. Nat. Commun. 12, 7013 (2021).
Kochanczyk, T. et al. Structural basis for transthiolation intermediates in the ubiquitin pathway. Nature 633, 216–223 (2024).
He, X. et al. Characterization of the loss of SUMO pathway function on cancer cells and tumor proliferation. PLoS ONE 10, e0123882 (2015).
Filice, F., Blum, W., Lauber, E. & Schwaller, B. Inducible and reversible silencing of the Pvalb gene in mice: an in vitro and in vivo study. Eur. J. Neurosci. 50, 2694–2706 (2019).
Gonzalez-Prieto, R., Cuijpers, S. A., Kumar, R., Hendriks, I. A. & Vertegaal, A. C. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 14, 1859–1872 (2015).
Kalkat, M. et al. Identification of c-Myc SUMOylation by mass spectrometry. PLoS ONE 9, e115337 (2014).
Nayak, D. et al. Development and crystal structures of a potent second-generation dual degrader of BCL-2 and BCL-xL. Nat. Commun. 15, 2743 (2024).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF ChimeraX: ctructure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Viswanadhapalli, S. et al. EC359: a first-in-class small-molecule inhibitor for targeting oncogenic LIFR signaling in triple-negative breast cancer. Mol. Cancer Ther. 18, 1341–1354 (2019).
Acknowledgements
We thank members of the S.K.O., E.V.W. and R.K.V. laboratories for helpful discussions. Research reported in this publication was supported by the National Institutes of Health (NIH) R01 GM115568, R01 GM128731 and Cancer Prevention and Research Institute of Texas (CPRIT) RR200030 (S.K.O.), NIH R01 CA262757 (R.K.V.), NIH RO1 CA267893-01A1 (S.V.), NIH R35 CA241801 (P.S.), NIH R01AI136697 and The Welch Foundation Research Grant AQ-1996-20190330 (D.N.I.), and NIH R00 GM140264 and CPRIT RR220068 (E.V.W.). E.V.W. is supported by a Voelcker Fund Young Investigator Award. K.M.N. was supported by Epigenetics, DNA Repair and Genomics (EDGe) training program T32CA279363. Cryo-EM screening and initial data collection were conducted at the UT Health San Antonio cryo-EM facility on a Glacios TEM equipped with a Falcon IV camera and Selectris energy filter, which were purchased with the support of UT Science and Technology Acquisition and Retention awards 402-1288 (P.S.) and 402-1317 (S.K.O.). We thank A. Brilot and E. Schwartz for collecting Krios cryo-EM datasets used for final reconstructions of the single-loaded complex reported in this study at the UT Austin Sauer Structural Biology Laboratory (RRID:SCR_022951). This research used resources of the Structural Biology Core Facilities, part of the Institutional Research Cores at the UT Health Science Center at San Antonio supported by the Office of the Vice President for Research and the Mays Cancer Center Drug Discovery and Structural Biology Shared Resource (NIH P30 CA054174). The high-performance computing cluster used in these studies is funded by NIH Office of Research Infrastructure Programs Shared Instrument Grant S10 OD036251 (S.K.O.). The content of this study is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
Protein purification was conducted by A.N., D.N. and P.S.B. Structural experiments and analysis were conducted by L.J., E.A.R., D.N., A.N., A.A.T., E.V.W. and S.K.O. D.N. and A.N. conducted the biochemical assays. S.V. and K.M.N. conducted the cell-based functional studies under the supervision of R.K.V. C.H.Y., D.N.I., C.M.S., P.E. and P.S. assisted with the experimental design and data interpretation. The figures and manuscript were prepared by D.N., A.N., E.A.R. and S.K.O. with input from all authors.
Corresponding author
Ethics declarations
Competing interests
D.N.I. is a cofounder and a shareholder of E3 Bioscience, a commercial entity that manufactures fluorescent reagents for studies of protein ubiquitylation. The other authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Dimitris Typas, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Quality of the SUMO E1-UBC9/SUMO1(a) complex cryo-EM reconstruction.
a, The final Euler angle distribution of particles used for 3D refinement. b, Fourier Shell Correlation (FSC) plot from two half maps, with an overall resolution of 2.7 Å as determined at 0.143 criterion. c, Local resolution map color-coded from lower (red) to higher resolution (blue). d, Overall map (left) and detailed views of local cryo-EM density for IAD, AAD, UBC9, SUMO1 C-terminal AMP adenylate and E1-E2 disulphide regions of the complex. e, Cryo-EM map of the SUMO E1-UBC9/SUMO1(a) complex with two views related by a 180° rotation about the y-axis (left). Maps of the individual subunits are shown in the right with IAD (slate), AAD (lightpink), FCCH (forestgreen), SCCH (purple), UFD (orange), Cys cap (magenta), UBC9 (cyan) and SUMO1(a)(yellow).
Extended Data Fig. 2 Cryo-EM data processing tree of the SUMO E1-UBC9/SUMO1(a) complex.
Cryo-EM data processing flow chart depicting the major steps of 2D and 3D classification leading to the best map.
Extended Data Fig. 3 Structural features of the SUMO E1 UFD and FCCH domain.
a, Shown here is the location of UBC9-interacting residues on UFD in our study as compared to other published structures. The UFD domain is colored orange. Residues colored in cyan and circled black on UFD interact with UBC9. b, Comparing the FCCH and SCCH domain in SUMO E1 complex from this study with other published SUMO E1 and Uba1 structures. The red circle shows the location of FCCH domain (colored forest) in different published E1 structures. The overlay shows the FCCH and SCCH domains of other published structures colored in grey.
Extended Data Fig. 4 Structure-function analysis of SUMO E1-E2 interfaces.
a, Raw data for thioester transfer assay with UFD mutants presented in Fig. 2d. b-d, Raw data for thioester transfer assay for UFD:UBC9 interface with UBC9 and UFD mutants presented in Fig. 3b. e, Raw data for thioester transfer assay for Crossover loop:UBC9 interface with crossover and UBC9 mutants presented in Fig. 3d. f, g, Raw data for thioester transfer assay with SCCH and UBC9 interface mutants presented in Figs. 3e and 2f. h, Raw data for thioester transfer assay for charge swapped mutants of SUMO E1 and UBC9 presented in Fig. 3f. i, SUMO E1-E2 thioester transfer assay of the indicated mutants of SUMO E1. Data represented as bar graph with mean ± SD of 3 technical repeats displayed as percentage of the WT value and individual replicates shown as black dots. All the gels presented are representative results from n = 3 technical replicates independently repeated with similar results. Source data are provided as source data file.
Extended Data Fig. 5 Determinants of SUMO E1-E2 specificity.
a, Sequence conservation for acidic path residues on SUMO E1 UFD domain shown as red box. b, Shown here is the surface and electrostatic representation of interface shared by UBC9 and cyscap of SUMO E1 complex. HelixD of UBC9, helix(residue 221-229) of cyscap and residues of cyscap interacting with UBC9 are labeled. c, Superimposition of different Ubl E2s onto UBC9. Show here the β1-β2 insertion loop present only in UBC9 colored as teal. The corresponding loops are colored green for UBE2L6, violet for UBCH5b, red for UBC12 and yellow for UBE2Z. d, Comparing the b1-b2 loop present in our structure to other PDBs corresponding to UBC9. The other PDBs are colored coded as shown. e, Docking UBC9 onto UBA7, UBA6 and UBA1 to show clash due to β1-β2 loop of UBC9 with UBA7 and UBA1. β30-β31 of UBA6 clashes with UBC9.
Extended Data Fig. 6 Conformational changes of the SUMO E1 Cys cap and differences between how UFD engages UBC9 for human and yeast.
a, Showing D209-E218 of cys cap, from this study which would otherwise clash when UBC9 is modeled on E2-free structures of SUMO E1. b, SCCH domains from auto-inhibitory SUMO E1 superimposed onto SCCH domain from this study showing major conformational changes corresponding to cys cap including the residues shown in a. c, UFD’s from this study and PDB: 3ONG were aligned to show the differences in arrangement of UBC9 between human (this study) and yeast (PDB:3ONG). d, Electrostatic surface representation of the SUMO E1 UFD with UBC9 interacting residues overlaid in this study (Left) and yeast (PDB:3ONG) (right). e, Structure-based sequence alignemnt to show the UFD-UBC9 interacting residues in this study (green sphere, top) and in yeast (red sphere, bottom). f, Structure-based sequence alignment of the AAD-UFD linker region of Ubl E1s. The green star shows the conserved tryptophan.
Extended Data Fig. 7 Thermal Shift Assays and Sequence Alignments of SUMO E1 and UBC9.
a, Data showing the melting temperature of WT and mutant SUMO E1 proteins. A graph is plotted for derivative vs temperature for each SUMO E1 mutant separately along with the WT for each plot to compare the change in melting temperatures. b, Data is presented as in a for WT and mutant UBC9 proteins. The data presented are representative results from n=2 technical replicates performed independently with similar results c, Sequence conservation for residues on SUMO E1 (UBA2) that interacts with UBC9. The interacting residues are marked as stars and colored salmon, red and cyan for UFD:UBC9, crossover loop:UBC9 and SCCH:UBC9 interfaces, respectively. d, Sequence conservation for residues on UBC9 that interacts with SUMO E1 (UBA2). The interacting residues are marked as stars and colored salmon, red and cyan for UFD:UBC9, crossover loop:UBC9, and SCCH:UBC9 interfaces, respectively.
Extended Data Fig. 8 Conformational changes in SUMO E1 and NEDD8 E1.
a, Left, UBC12 was docked onto apo NEDD8 E1 (PDB:1R4M); distance between catalytic cysteines indicated as spheres and colored in yellow is 55 Å: thioester transfer inactive state. Center, NEDD8 E1/UBC12/NEDD8(a)-NEDD8(t) (PDB: 2NVU); distance between catalytic cysteines is ~ 23 Å: thioester transfer active. Right, Superimposition to show the conformational changes between active and inactive states of UFDs in NEDD8 E1. b, Comparison of SUMO E1-UBC9/SUMO1(a) (this study) and NEDD8 E1/UBC12/NEDD8(a)-NEDD8(t) (PDB: 2NVU), which represent thioester transfer active states, showing conformational changes in their UFDs. c, Same as b, but showing the thioester inactive states and associated conformational changes in their UFDs.
Extended Data Fig. 9 Quality of the SUMO E1-UBC9-SUMO1(t)/SUMO1(a) cryo-EM reconstruction.
a, Schematic representation of SUMO E1-E2 thioester transfer reaction. b, SDS-PAGE of sample preparation steps of the double-loaded SUMO E1-E2 complex used for cryo-EM analysis (right). c, The final Euler angle distribution of particles used for 3D refinement. d, Fourier Shell Correlation (FSC) plot from two half maps, with an overall resolution of 3.6 Å as determined at 0.143 criterion. e, Local resolution map color-coded from lower (red) to higher resolution (blue). f, Overall map (left) and detailed views of local cryo-EM density for SUMO E1, UBC9, SUMO1(a), SUMO1(t), AMP and SUMO E1-UBC9/SUMO1(t) active site regions of the SUMO E1 complex. g, Cryo-EM map of the SUMO E1-UBC9/SUMO1(a)/SUMO1(t) complex with two views related by a 180° rotation about the y-axis (left). Maps of the individual subunits are shown on the right. h, Superimposition of SUMO E1 single loaded (grey) onto SUMO E1 double loaded.
Extended Data Fig. 10 Cryo-EM data processing tree of the SUMO E1-UBC9-SUMO1(t)/SUMO1(a) complex.
Cryo-EM data processing flow chart depicting the major steps of 2D and 3D classification leading to the best map.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2, Table 1 and uncropped scans of Supplementary Fig. 2.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed gels.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed gels.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 7
Unprocessed western blot with colony formation plates.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed gels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nayak, A., Nayak, D., Jia, L. et al. Cryo-EM structures reveal the molecular mechanism of SUMO E1–E2 thioester transfer. Nat Struct Mol Biol 32, 2441–2453 (2025). https://doi.org/10.1038/s41594-025-01681-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41594-025-01681-8
