
Abstract
The flat-headed loach (Oreonectes platycephalus) is a small fish inhabiting headwaters of hillstreams of southern China. Its local populations are characterized by low genetic diversity and exceptionally high differentiation, making it an ideal model for studying small population isolates’ persistence and adaptive potential. However, the lack of Oreonectes reference genomes limits endeavours toward these ambitions. We assembled the first haplotype-resolved chromosome-level genome of the genus Oreonectes using PacBio HiFi and Hi-C technologies. This genome consists of two haplotypes (24 pseudo-chromosomes in each), with sizes of 565.68 Mb (haplotype A) and 521.13 Mb (haplotype B) and scaffold N50 lengths of 22.80 Mb and 21.91 Mb, respectively. Chr01 was identified as the likely sex chromosome pair. After masking repetitive elements which accounted for 34.43% to 36.44% of the genome, there are 27,127 protein-coding genes in haplotype A and 25,576 in haplotype B. The availability of this haplotype-resolved chromosome-level reference genome will facilitate the study of population and conservation genetics of the flat-headed loach and other Oreonectes species.
Similar content being viewed by others


Background & Summary
Species living in isolated habitats may not be able to migrate to new areas when faced with environmental changes. Whether these species can adapt to changing conditions depends on the extent of genetic variation within populations which influences their fitness. Small isolated populations are predicted to have lowered fitness due to inbreeding and loss of genetic diversity, compromising their ability to adapt to new environmental conditions1. The current challenge is to gain insights into how these small isolated populations will handle various stressors, including rising temperatures caused by climate change. The flat-headed loach (Oreonectes platycephalus: Noemacheilidae; Fig. 1) is a small freshwater fish living in the headwaters of hillstreams of southern China2. Using reduced-representation genome sequencing, flat-headed loaches in Hong Kong were found to display an exceptionally high degree of genetic differentiation among local populations with low levels of genetic diversity and very small effective population sizes3. The wide but naturally highly fragmented distribution of this species makes it a highly replicated model system to study the persistence and adaptive potential of small population isolates. However, the lack of a high-quality reference genome in O. platycephalus presents challenges in conducting in-depth population genomic studies on this species. A high-quality genome assembly of O. platycephalus would be an initial step to enable in-depth studies.

Characteristics of the flat-headed loach genome assembly. From outer to inner circle: (a) 24 pairs of pseudo-chromosomes (haplotype A in blue and haplotype B in red); (b) gene density in 1 Mb nonoverlapping windows; (c) repetitive elements abundance in 1 Mb nonoverlapping windows; (d) GC content in 1 Mb nonoverlapping windows; (e) collinear blocks between chromosomes.
PacBio HiFi sequencing, high-throughput chromosome conformation capture technologies (Hi-C) as well as short-read sequencing were applied to generate the first chromosome-level haplotype-resolved genome of the flat-headed loach (Fig. 2). The final assembly consisted of two haplotypes anchored on 24 chromosomes – the genome size of haplotype A was 565.68 Mb with scaffold N50 length of 22.80 Mb and that of haplotype B was 521.13 Mb with scaffold N50 length of 21.91 Mb. Chr01 was identified as the putative sex chromosome pair with the Y chromosome at haplotype A and the X chromosome at haplotype B. Repetitive elements made up 36.44% and 34.43% of haplotype A and haplotype B, respectively. After masking repetitive elements, a total of 27,127 protein-coding genes in haplotype A and 25,576 in haplotype B were predicted. As the first chromosome-level haplotype-resolved genome for genus Oreonectes, this high-quality reference genome not only greatly facilitates the study of population and conservation genetics of the flat-headed loach, but also provides an important resource to understand the genetics and evolution of other species of Oreonectes.

Schematic view of the flat-headed loach genome assembly and annotation workflow.
Methods
Sample collection and sequencing
The fieldwork was conducted with the approval of the Agriculture, Fisheries and Conservation Department (AFCD) of Hong Kong Permission to Make Field Collections for Research Purpose(168 AF GR CON 11/17 Pt.6). A phenotypically identified male flat-headed loach was collected from the wild (Lung Fu Shan: 22.2785 N, 114.1330E) and euthanized in the laboratory with dry ice. Frozen muscle tissue was cut into several pieces and stored in dry ice during transportation to Haorui Genomics (Xi’an, China) where DNA extraction as well as the HiFi (High Fidelity) sequencing, Hi-C (High-throughput chromosome conformation capture) sequencing and short-read sequencing was performed. HiFi sequencing is a technology producing long reads of DNA, based on the Pacbio (Pacific Biosciences Inc., CA, USA) Sequel II platform4. A total of 15 µg DNA was used for SMRT (Single-Molecule Real-Time) library construction through SMRTbell express template prep kit 2.0 (Pacific Biosciences Inc., CA, USA). DNA molecules in SMRTbell library were sequenced in real-time by the DNA polymerase enzyme and a total of 18.08 Gb HiFi data was generated. Hi-C sequencing, a method to examine a genome’s three-dimensional organization5, was used to phase haplotypes and scaffold the genome at the chromosome level. The chromatin was crosslinked using formaldehyde to maintain the genome’s spatial organization and then fragmented with a restriction enzyme (DpnII). The digested fragments were ligated to create a paired-end (PE) 150 sequencing library on the DNBSEQ-T7 (BGI, China) platform. 51.31 Gb clean reads were filtered out from 51.91 Gb raw Hi-C reads by fastp v 0.23.26 with the default setting. The pair-end sequencing (PE150) library was constructed on the MGISEQ-2000 (BGI, China) platform for genome survey, which generated 74.09 Gb clean reads.
Genome survey and assembly
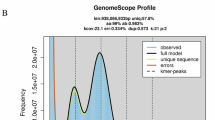
To explore the genomic features of the flat-headed loach, a genome survey based on k-mer analysis was conducted using short-read sequencing data before genome assembly to estimate the genome size and heterozygosity. The 74.09 Gb clean reads underwent 17-mer frequency distribution analysis using KMC v 3.0.07 with the parameters set to “-k17 -ci1 -cs1000000”. Using default parameters GenomeScope8 estimated the genome size of flat-headed loach to be 485.53 Mb with a heterozygosity rate of 0.654%.
HiFi reads were assembled with Hi-C paired-end reads to generate two haplotype contig-level assemblies by Hifiasm v 0.18.99 with parameters of “-D 50 -N 500–hom-cov 32 -s 0.45–n-weight 5–n-perturb 18000–f-perturb 0.3” using the Hi-C integrated assembly mode. The clean Hi-C reads were separately aligned to two sets of contigs by Juicer v 1.610 and utilized to anchor these two contig sets onto pseudo-chromosomes by 3D-DNA11, respectively. Placement and orientation errors exhibiting obvious discrete chromatin interaction patterns were manually adjusted in Juicebox v 2.1512 to generate the final chromosome-level assembly for each haplotype. The statistics of the final assembled genomes were calculated by TBtools-II v 2.09713, which consisted of two haplotypes - haplotype A (565.68 Mb) and haplotype B (521.13 Mb), with scaffold N50 length of 22.80 Mb and 21.91 Mb, respectively (Fig. 1 and Table 1 & S1). About 95.88% and 95.28% of assembled sequences were anchored onto 24 pseudo-chromosomes of haplotype A and B, respectively (Table 1).
Genome annotation
Repetitive elements in the genome assembly were annotated using a combination of de novo and homology-based approaches. In brief, we used high-quality transposable element (TE) sequences from the Actinopteri database (homology-based approach) in RepBase v 23.0814 to identify repeats with RepeatMasker v 4.1.615. The de novo prediction including the LTR discovery pipeline (-LTRStruct) was conducted by RepeatModeler v 2.0.516. According to the annotation, repetitive elements made up 34.43% and 36.44% of haplotype A and haplotype B, respectively (Fig. 2 & Table 2).
Before protein-coding gene annotation, the identified repetitive elements were masked to avoid interference. Similar to TEs annotation, protein-coding gene prediction was also conducted through a combination of de novo and homology-based approaches. Miniprot v 0.12-r23717, in conjunction with CD-HIT v 4.8.118, was used for homology-based predictions, incorporating five non-redundant fish reference genomes from NCBI, including a model species Danio rerio and four related species - Misgurnus anguillicaudatus, Megalobrama amblycephala, Astyanax mexicanus and Triplophysa dalaica. Helixer19 was employed for de novo predictions using a vertebrate deep-learning model. Additionally, we applied a non-overlapping protein method for de novo genes for a gene set. A total of 27,127 and 25,576 genes of haplotype A and haplotype B were annotated, with average gene lengths of 12,150.00 bp and 11,690.75 bp, respectively (Fig. 2 & Table 3).
Functional annotation of the predicted protein-coding genes was performed using public databases including SwissProt20, the NCBI non-redundant protein database21, InterProScan v 5.3622, eggNOG-mapper v 2.1.1223 and TrEMBL v 26.024. Overall, 26,009 and 24,584 functional genes made up 95.88% and 96.74% of the total predicted genes in haplotype A and B respectively, which were successfully annotated by at least one database (Fig. 3 & Table 4).

The unique and shared functional genes in the flat-headed loach genome annotated by different databases.
To obtain the non-coding RNA genes including transfer RNA (tRNA), micro-RNA (miRNA), ribosome RNA (rRNA) and small nuclear RNA (snRNA) genes, two strategies were used: searching against a database and prediction with a model. TRNAs were predicted using tRNAscan-SE v 1.3.125 with default parameters for eukaryotes. The miRNA genes and snRNA genes were detected using Infernal v 1.0.226 searching the Rfam27 database. The rRNA genes and their subunits were predicted using Barrnap v 0.928 with default settings. For haplotype A, the non-coding RNA genes identified in the flat-headed loach genome included 904 miRNAs, 7435 tRNAs, 5070 rRNAs, and 965 snRNAs (Table 5) while there were 629 miRNAs, 6449 tRNAs, 689 rRNAs and 804 snRNAs identified of haplotype B (Table 5).
Mitogenome assembly and annotation
The mitochondrial genome was assembled from HiFi clean reads using MitoHiFi v 3.0.029 with one published flat-headed loach mitochondrial genome as a reference (NCBI accession: NC_031579) and parameters of “-t 32 -o 2”. The assembled mitochondrial genome was annotated by a web software - MitoAnnotator30. The 16,570 bp mitochondrial genome consists of 13 protein-coding genes, two rRNA genes, and 22 tRNA genes (Fig. 4).

Assembled and annotated mitogenome of the flat-headed loach. The inner grey bars depict GC content in 5 bp-window. The location and size of different genes are indicated.
Sex chromosome identification
To identify the sex chromosomes of the flat-headed loach, a population of 15 individuals (seven phenotypically sexed males & eight females) was collected from the wild (Tai Po Kau, TPK, Hong Kong: 22.4159 N, 114.1807E) for sex chromosome validation. The extracted DNA of each sample was sent for DNBseq PE150 short-read sequencing performed by Berry Genomics (Guangzhou, China) with a target coverage of 15X. After quality control by fastp v 0.23.26, a total of 131.74 Gb clean reads were generated (Table S1). They were mapped to haplotype A which had a higher BUSCO completeness using BWA v 0.7.1731 and GATK v 4.5.0.032 was applied to call SNPs. SNPs were hard filtered with parameters “MQRankSum < -12.5 || FS > 60.0 || ReadPosRankSum < -8.0 || MQ < 40.0 || QD < 2.0” via gatk and then custom filtered with parameters “ --maf 0.05 --max-missing 0.5 --minQ 30 --min-alleles 2 --max-alleles 2 --minDP 6 --maxDP 30 --recode --recode-INFO-all” by vcftools v 0.1.1733. Genomic FST between males and females was estimated by vcftools with parameters “ --fst-window-size 500000 --fst-window-step 50000” and the results were visualised through R package ggplot2, indicating that Chr01 contained more divergent variants between males and females than any of the other pseudochromosomes (Fig. 5a). Therefore, Chr01 was indicated to be the sex chromosome in haplotype A. Given the large synteny blocks between the Chr01 pair in two haplotypes (Fig. 1), the Chr01 in haplotype B is likely the other sex chromosome. To identify the sex determination system and which of the haplotypes correspond to X and Y chromosome (or Z and W chromosome), a pair (phenotypically sexed male & female) of flat-headed loaches were collected from Lung Fu Shan and euthanized for HiFi sequencing. Minimap234 was used to map the HiFi reads of the two samples to two haplotypes and bamdst (https://github.com/shiquan/bamdst) was used for mapping depth calculation of the pseudo sex chromosomes which were then visualised with R package ggplot235 in 20 kb windows. The mapping of the female individual showed zero depth in most regions between 0 to 12.5 Mb on Chr01 of the haplotype A whereas the male exhibited high mapping depth across this large region (Fig. 5b). This indicates male heterogametic sex determination system (XX/XY) in the flat-headed loach and that Chr01 of haplotype A is the Y chromosome as the reference individual is a male. By inference, Chr01 of haplotype B is the likely X chromosome which is also supported by mapping results (Fig. 5b). In summary, Chr01 of haplotype A is considered to be the Y chromosome with the size of 32.71 Mb, which is ca. 11 Mb larger than the Chr01 of the haplotype B (21.91 Mb) considered to be the X chromosome (Fig. 5c & Table S2).

The process of sex chromosome identification. (a) genomic FST between males and females in each chromosome as estimated in 500k-windows and 50k-step; (b) Standardized 20 kb-window size mapping depth (standardized depth = window depth/average depth) of female and male HiFi reads mapped on Chr01 of the two haplotypes. (c) The collinearity analysis between Chr X and Chr Y.
Data Records
Sequencing data has been stored in the National Center for Biotechnology Information (NCBI) SRA database36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55 with accession numbers SRR30361771 ∼ SRR30361787 and SRR30361798 ∼ SRR30361800 under the BioProject accession number PRJNA1128407 (Table S3). Genome assembly data on GenBank can be found via accession numbers JBJFMQ00000000056 and JBJFMR00000000057. The genome annotations were openly available from figshare58.
Technical Validation
To evaluate the quality of genome assembly, the consensus quality values (QV) of the assembly were estimated by Merqury v 1.359. The QV reached 59.73 and 58.53 of haplotype A and haplotype B, respectively, demonstrating a high level of assembly accuracy. The Hi-C assemblies were visualized with Juicebox12, which showed strong interactive signals within the diagonal of the pseudo-chromosomes with weak interactive noise outside the diagonal (Fig. 6), indicating the high quality of the two chromosome assemblies.

The Hi-C interactive heatmap of the flat-headed loach.
Benchmarking Universal Single-Copy Orthologues (BUSCO) v 5.4.760 was used to evaluate the completeness of the assembly and annotation with parameters “-l actinopterygii -g genome”. The genome assemblies covered 96.8% and 93.4% complete BUSCOs of haplotype A and haplotype B, respectively (Fig. 7). A total of 97.0% and 94.3% BUSCOs were covered by annotated haplotype A and haplotype B, respectively (Fig. 7). The BUSCO results indicated reliable genome assemblies and annotations.

BUSCO evaluation of genome assemblies and annotations of the two flat-headed loach haplotypes.
Considering that the Chr01 of haplotype B was identified as the likely X chromosome, the obtained clean reads of the 15 TPK individuals were mapped to haplotype B with BWA. SNPs on Chr01 of haplotype B were called by GATK32 and then filtered for principal component analysis (PCA) with the same parameters as applied in FST analysis. Plink v 1.9061 was used for conducting PCA and the results were visualized with R package ggplot235, showing clear separation of males and females (Fig. 8). To sum up, Chr01, as the pseudo sex chromosome pair (Y in haplotype A and X in haplotype B), contains the likely sex-determining region that can separate the sexes of the flat-headed loach.

Principal component analysis (PCA) of SNPs on Chr01 of the haplotype B for male and female flat-headed loaches.
Code availability
All commands and pipelines used were performed according to the manuals or protocols of the tools used in this study. The software and tools used are publicly accessible, with the version and parameters specified in the Methods section. If no detailed parameters were mentioned, default parameters were used. No custom code was used in this study.
References
Lanfear, R., Kokko, H. & Eyre-Walker, A. Population size and the rate of evolution. Trends Ecol Evol 29, 33–41, https://doi.org/10.1016/j.tree.2013.09.009 (2014).
Du, L., Chen, X. & Yang, J. A review of the Nemacheilinae genus Oreonectes Günther with descriptions of two new species (Teleostei: Balitoridae). Zootaxa 1729, 23–36, https://doi.org/10.11646/zootaxa.1729.1.3 (2008).
Wang, X., Reid, K., Chen, Y., Dudgeon, D. & Merilä, J. Ecological genetics of isolated loach populations indicate compromised adaptive potential. Heredity 133, 88–98, https://doi.org/10.1038/s41437-024-00695-0 (2024).
Wenger, A. et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol 37, 1155–1162, https://doi.org/10.1038/s41587-019-0217-9 (2019).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293, https://doi.org/10.1126/science.1181369 (2009).
Chen, S. Ultrafast one‐pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2, e107, https://doi.org/10.1002/imt2.107 (2023).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761, https://doi.org/10.1093/bioinformatics/btx304 (2017).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant 16, 1733–1742, https://doi.org/10.1016/j.molp.2023.09.010 (2023).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 1–6, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152, https://doi.org/10.1093/bioinformatics/bts565 (2012).
Stiehler, F. et al. Helixer: cross-species gene annotation of large eukaryotic genomes using deep learning. Bioinformatics 36, 5291–5298, https://doi.org/10.1093/bioinformatics/btaa1044 (2021).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370, https://doi.org/10.1093/nar/gkg095 (2003).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33, D501–504, https://doi.org/10.1093/nar/gki025 (2005).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829, https://doi.org/10.1093/molbev/msab293 (2021).
Apweiler, R. et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32, D115–119, https://doi.org/10.1093/nar/gkh131 (2004).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33, W686–689, https://doi.org/10.1093/nar/gki366 (2005).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337, https://doi.org/10.1093/bioinformatics/btp157 (2009).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res 31, 439–441 (2003).
Seemann T. Barrnap: BAsic Rapid Ribosomal RNA Predictor https://doi.org/10.1093/nar/gkg006 (2018).
Uliano-Silva, M. et al. MitoHiFi: a python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC bioinformatics 24, 288, https://doi.org/10.1186/s12859-023-05385-y (2023).
Iwasaki, W. et al. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol 30, 2531–2540, https://doi.org/10.1093/molbev/mst141 (2013).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158, https://doi.org/10.1093/bioinformatics/btr330 (2011).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Wickham, H. ggplot2. Wiley interdisciplinary reviews: computational statistics 3, 180–185, https://doi.org/10.1002/wics.147 (2011).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361771 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361772 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361773 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361774 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361775 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361776 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361777 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361778 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361779 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361780 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361781 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361782 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361783 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361784 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361785 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361786 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361787 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361798 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361799 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR30361800 (2024).
NCBI GenBank http://identifiers.org/ncbi/insdc:JBJFMQ000000000 (2024).
NCBI GenBank http://identifiers.org/ncbi/insdc:JBJFMR000000000 (2024).
Wang, X. et al. Chromosome-level haplotype-resolved genome assembly and annotation of the genetically highly structured loach (Oreonectes plathycephalus). figshare https://doi.org/10.6084/m9.figshare.26819455.v2 (2024).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, https://doi.org/10.1086/519795 (2007).
Acknowledgements
We thank Chi Kit Yueng and Sheung Tsz Tam for their help with the fieldwork. The study was supported by Seed Funding from the University Grants Committee awarded to DD, KR and JM (# 202111159018). XW was supported by the Robert Whyte Memorial Postgraduate Fellowship 2024-25.
Author information
Authors and Affiliations
Contributions
Conceived the study: K.R., J.M., D.D.; Data collection: X.W., D.W., H.W.; Analyzed the data: X.W., D.W., H.W.; Led the writing: X.W.; Commented and edited the manuscript: D.W., H.W., D.D., K.R., J.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Wang, D., Wang, H. et al. Chromosome-level haplotype-resolved genome of the tropical loach (Oreonectes platycephalus). Sci Data 12, 29 (2025). https://doi.org/10.1038/s41597-024-04301-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-024-04301-0
