
Abstract
Ischemic stroke constitutes a multifaceted neurological affliction that spans various cellular types. Lack of dynamic chromatin accessibility data after stroke is one of the obstacles to understanding this process. To gain insights into the variations in transcriptional regulation among various cell types subsequent to a stroke, we employed single-nucleus ATAC-seq to curate a chromatin accessibility compendium from the cerebral cortex of mice subjected to middle cerebral artery occlusion/reperfusion (MCAO/R). Tissue samples were collected at various time points including 0, 6, 12, 24 hours, and 7, 14 days post-reperfusion, in addition to Sham control group. We obtained 99,271 high-quality nuclei across nine cell types, thereby establishing the single-nucleus chromatin accessibility atlas. This atlas provides data for interpreting the regulatory mechanisms that pervade the continuum of ischemic stroke. The data presented herein constitutes a valuable resource for the comprehension of regulatory interplays within the pathology-afflicted cerebrum.
Similar content being viewed by others


Background & Summary
Ischemic stroke is the second leading cause of death and disability worldwide, typically resulting in motor and cognitive impairments1,2. Due to the intricate nature of brain structure and the diverse array of cell types present, our understanding of the molecular mechanisms underlying stroke remains incomplete. To decipher the biochemical regulatory alterations and interactions during stroke, researchers have employed a variety of methods including transcriptomic and proteomic sequencing3,4. Single-cell RNA sequencing (scRNA-seq) has emerged as a transformative technology in stroke research, providing valuable insights into the activation of immune response, multicellular interactions, new potential biomarkers, and therapeutic targets5,6. However, these single-cell studies have predominantly focused on transcriptomics, leaving the regulatory mechanisms that control gene expression largely unexplored.
The coordination between chromatin structure and transcription plays a crucial role in gene regulation, influencing processes from embryonic development to the progression of disease7,8. Chromatin accessibility is essential in regulating gene expression and cellular identity, and alterations in accessibility have been implicated in the occurrence and development process of neurological diseases9,10. Single-nucleus epigenomic profiling has been applied across a range of diseases, including coronary artery disease and neurodegenerative diseases. By utilizing single-nucleus assay for transposase-accessible chromatin with high-throughput sequencing (snATAC-seq), researchers can pinpoint cell type-specific, disease-related candidate cis-regulatory elements (CREs) and their candidate target genes, underscoring the significance and potential of this technique in the realm of neurological diseases11. The identification of emerging CREs unveils cell-state-specific modifications, aiding in the discovery of novel putative biomarkers and therapeutic targets12. However, the existing literature lacks a comprehensive view of the dynamic landscape of global chromatin accessibility of stroke.
To investigate the CREs and their dynamic within specific cell types following ischemic stroke, we selected cortical tissues from middle cerebral artery occlusion/reperfusion (MCAO/R) mice at seven different time points after reperfusion: 0 hours (h), 6 h, 12 h, 24 h, 7 days (d), and 14 d, as well as Sham group. We performed snATAC-seq analysis on chromatin accessibility of 99,271 nuclei and constructed a single-nucleus chromatin accessibility atlas, annotating cell types with their respective putative target genes and functions for CREs. The delineation of transcriptional regulatory networks offers a valuable resource for potentially identifying key cell subtypes, transcription factors (TFs) and dynamic alterations pertinent to ischemic stroke, guiding the development of more targeted and efficacious therapeutic interventions for this disease.
Methods
Animal experiments
Male C57BL/6 mice (age: 7-8 weeks, weight: 22–24 g) were employed for this study. All animal procedures were performed in compliance with guidelines for the care and use of animals and were approved by the University Huazhong Agriculture Institutional Animal Care and Use Committee (approval number: HZAUMO-2021-0011). Mice were assigned into groups subjected to MCAO/R with various recovery periods, or into Sham operation.
MCAO/R model establishment
Ischemic stroke was established with MCAO/R surgery on male C57BL/6 mice according to the laboratory’s preliminary experimental methods13. The mice were anesthetized with 2% isoflurane (RWD, R510-22-10), and were placed in the supine position. To induce focal cerebral ischemia, a silicon-coated filament (RWD, MSMC21B120PK50) was inserted into the left middle cerebral artery (MCA) to block blood flow. After 60 minutes later, the filament was extracted for reperfusion. Rectal temperature was maintained at 37 °C during the entire procedure. The cerebral blood flow (rCBF) was detected using a laser doppler flowmetry (Perimed, PERIMED005). A 55% decrease in the rCBF of the ipsilateral hemisphere, as compared to contralateral hemisphere, was considered the threshold for successful establishment of cerebral ischemia. Mice of the sham-operated group were performed the same as the MCAO/R procedure without filament insertion.
Brain tissue dissection
Following the MCAO/R operation, the mice were allowed recovery periods of 0 h, 6 h, 12 h, 24 h, 7 d and 14 d. Both MCAO/R mice and sham-operated mice were euthanized via cervical dislocation. The brains of mice were promptly and meticulously excised. Ophthalmic forceps were used to carefully separate the left cerebral cortex from the rest of the brain without compromising its integrity. All samples were placed on ice and quickly dissected, frozen in liquid nitrogen, and stored at −80 °C until further processing.
Nuclei isolation for snATAC-seq
Nuclei were isolated from cerebral cortex of mice for snATAC-seq according to the mechanical extraction method with slight modification14. In brief, the frozen tissues were thawed from −80 °C storage and 2 mL pre-chilled homogenization buffer (Sigma, CELLYTPN1-1KT) was added to each sample. Each 1 mL mixture was transferred to a 2 mL Dounce homogenizer (Kimble, NO.885300-0002) on ice, and then homogenized with 10–20 strokes of the tight pestle (the B pestle).
Next, the homogenized mixture was filtered into a 1.5 mL tube through 40 µm cell strainers (Falcon, 352340), followed by centrifugation at 1,200 g for 5 minutes at 4 °C to pellet the nuclei, and then suspended in a blocking buffer containing 1% bovine serum albumin (BSA, Sangon Biotech, 68551-06-4) and 0.2 U/μL RNase inhibitor in 1 × PBS. Finally, the nuclei were pelleted for a second time by centrifugation at 500 g for 5 minutes at 4 °C, and resuspended in 1 × PBS containing 1% BSA for following library preparation.
snATAC-Seq library preparation and sequencing
For snATAC-Seq library preparation, DNBelab C Series Single-Cell ATAC Library Prep Set (MGI, 1000021878) was employed. In summary, libraries constructed as the user guide involved in transposition, droplet generation, PCR pre-amplification, emulsion breakage, capture beads collection, DNA amplification, and eventually purification for sequencing on the MGI DNBSEQ-T1 platform, with which we can acquire 109 bp for read 1, 50 bp for read 2, and 10 bp for the sample index. For quality control all of these snATAC-Seq libraries were assayed by Qubit ssDNA Assay Kit (Thermo Fisher Scientific, Q10212) to make sure enough DNA molecule and concentrations for sequencing and assessed the fragment size distribution using Agilent Bioanalyzer 2100 (Agilent Technologies, G2939A).
snATAC-seq raw data processing
First, the raw reads were separated and filtered using PISA (version 1.1)15, then aligned to mouse genome (mm10) with the software BWA (version 0.7.17-r1188)16. The obtained BAM files were further processed by bap2 (version 0.6.2)17 to identify reads with the same cell barcode and create fragment files for downstream bioinformatic analysis.
Quality control
ArchR (version 1.0.2)18 was employed to filter nucleus with low quality based on the following parameters: for each library the value of log10 (unique nuclear fragments) ≥ 3 and transcriptional start site (TSS) enrichment score ≥ 4. Subsequently, the potential doublets were removed from the datasets by applying the ‘addDoubletScores’ function of ArchR with the parameters of filterRatio = 2. The outlier samples are removed based on UMAP integration results. Pearson correlation coefficients between technical or biological replicates across tissues were calculated based on the PeakMatrix. An overview of quality control parameters for the snATAC-seq profiles seen in Table 1.
Latent semantic indexing (LSI) clustering of snATAC-seq data
Considering the sparsity of snATAC-seq data, iterative latent semantic indexing was adopted with ‘addIterativeLSI’ function of ArchR for dimensionality reduction, then library integration was performed by ‘addHarmony’ function and unsupervised clustering was employed using the ‘addClusters’ function on the basis of the Seurat Leiden clustering algorithm19.
The gene activity scores were calculated by ‘addGeneScorelatriy’ function. Marker genes were identified by ‘getMarkerFeatures’ function (parameters: useMatrix = ‘GeneScoreMatix’, groupBy = ‘Clusters’) with metrics of FDR ≤ 0.05 & Log2FC ≥ 0.25, where the statistical test for differential genes was the rank-sum test. Further, cell types was annotated for each cluster in accordance with well-established cell type-specific markers. Additionally, scCODA20 employs a Bayesian model primarily for analyzing differences in cell composition across different sample groups. Consequently, we used the scCODA package to compute the proportions of different cell types at various time points and performed significance testing. In the model construction process (using the function sccoda. utl. cpuana. mod. PositionalAnalysis), the “formula” parameter is set to the mouse injury time “time”. Set the reference_cellotype parameter to “automatic”. Then use the sample_ hmc() function to perform HMC sampling inference and view the results through summay(). The “Final Parameter” displays significance. If the value is 0, the effect is considered statistically unreliable; otherwise, it is considered reliable. In addition, logFC is used to indicate a decrease or increase in abundance (relative to the reference cell type).
Pseudo-bulk: verification of data authenticity
DESeq221,22 is an R package used for differential expression analysis of RNA-seq data. It employs an empirical Bayesian method to estimate the prior values of log2 fold changes and variances, and calculates the posterior values of these statistics.
DEseq2 was used to calculate the differentially expressed genes in microglia (MG) cells at different time points. Specifically, we extracted MG and added peaks with identical biological replicates to form pseudo-bulk data. We then used DESeq2 to calculate the differential pseudo-bulk peaks between M24h and M14d. Subsequently, we conducted functional enrichment analysis of the genes associated with pseudo-bulk peaks in biological processes (BP) pathways. This functional enrichment was used to interpret the functions that MG perform at M24h and M14d, thereby reflecting the authenticity of the data.
Peak calling of snATAC-seq
Given that per-cell snATAC-seq data is essentially binary, we created pseudo-bulk replicates using the ‘addGroupCoverages’ function for each cell type and the ‘addReproduciblePeakSet’ function was executed to call peak with the parameters set as groupBy = ‘annotation’, pathToMACS = pathToMacs2, maxCells = 20,000. Further analysis involved the acquisition of the marker peaks was achieved by the ‘getMarkerFeatures’ function and ‘getMarkers’ functions.
Motif enrichment analysis
With the intention of exploration what motifs enriched in peaks we performed cell type-specific and time-specific motif enrichment analysis through ‘addMotifAnnotations’ function of ArchR to add motif annotations first, specifying the motifSet as ‘cisbp’ and assigning the name ‘Motif’, and then using the ‘peakAnnoEnrichment’ function for enrichment of various motifs, whereby the top 7 cell type-specific and time-specific motifs were shown in the heatmap.
TF footprinting analysis
For the purpose of determining the precise binding location of the TF the ‘getPositions’ function was utilised to obtain the positions of the relevant motifs with default parameters. Afterwards the ‘addGroupCoverages’ function and ‘getFootprints’ function were used to accurately profile TF footprints which were visualized by ‘plotFootprints’ function.
Pathway enrichment analysis
Metascape bioinformatics tool (http://metascape.org)18 was leveraged to perform Gene Ontology (GO) and Kyoto Encylopedia of Genes and Genomes (KEGG) pathway enrichment analyses with the metrics of P value < 0.05, enrichment factor of >1.5 and a minimum count of 3.
We have provided a sheet (Table 2) highlighting the detailed parameters used for each datasets.
Data Records
The data (BAM files and fragment files) generated in this study by snATAC-seq (0 h, 6 h, 12 h, 24 h, 7 d, 14 d and Sham) have been deposited to CNGB Nucleotide Sequence Archive (CNSA)23 of China National GeneBank DataBase (CNGBdb)24 https://db.cngb.org/search/project/CNP0005552/: CNP0005552 (2024)25 and NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP548281 (2024)26. Additionally, the gene score matrix, cell-type peak matrices and metadata had also uploaded to Figshare (https://doi.org/10.6084/m9.figshare.25558785.v4)27.
Technical Validation
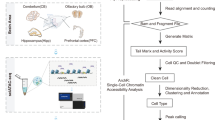
In the present study, we collected the identical left cerebral cortex from mice at seven distinct time points (0 h, 6 h, 12 h, 24 h, 7 d, and 14 d) after MCAO/R, as well as from sham-operated mice (Fig. 1). To ensure data reliability, we generated three biological replicates (comprising 6–12 libraries) for each post-reperfusion time point as well as the Sham group to construct the snATAC-seq library (Methods, Table 1).

An overview of the experimental and data analysis workflow. Schematic shows the collection of samples, sequencing procedures and downstream bioinformatic analyses. Samples from mice at 7 different time points (0 hours [M0h], 6 hours [M6h], 12 hours [M12h], 24 hours [M24h], 7 days [M7d], and 14 days [M14d] post-stroke) and Sham group (Sham) were collected from cerebral cortex for single-nucleus assay for transposase-accessible chromatin with high-throughput sequencing (snATAC-seq).
Initially, we applied Pearson correlation coefficient analysis to the 42 libraries to evaluate the similarity among the libraries at different time points post-stroke. The heatmap revealed that replicates originating from the same time points exhibited the highest correlation, affirming the high reproducibility of both biological and technical replicates (Fig. 2a). For quality control, we excluded cells with a low TSS enrichment score and a small number of unique nuclear fragments (Fig. 2b). Further doublet removal was performed to mitigate their substantial effects on clustering by ArchR addDoubletScores with a filterRatio parameter of 5 (Fig. 2c). After quality control, we obtained 99,271 high-quality cells for across the 7 groups from MCAO/R and sham-operated mice, encompassing 42 snATAC-seq libraries. We calculated the TSS enrichment fraction and the number of unique fragments for each cell nucleus under different time points after injury. The TSS enrichment fraction was mainly within the range of 4–20, while the number of unique fragments was mainly 1,000–32,000 (Fig. 2d and 2e, Table 1). The nuclei numbers of libraries at each time point were counted for further analyses (Fig. 2f). Additionally, the TSS enrichment profiles showed a clear peak around the TSS sites and a smaller shoulder peak in the right of TSS sites, as well as nucleosome periodicity, consistent with previous studies (Fig. 2g)7,18,28.

snATAC-seq data quality control and features. (a) Heatmap showing the similarity among the 42 libraries. The heatmap for the seven time points, from top to bottom, represents Sham group (Sham), 0 hours (M0h), 6 hours (M6h), 12 hours (M12h), 24 hours (M24h), 7 days (M7d), and 14 days (M14d) post-stroke. (b) Scatter plots show bivariate distributions of transcriptional start site (TSS) enrichment vs log10 (unique fragments) of different time points after stroke and Sham group. (c) Uniform manifold approximation and projection (UMAP) of snATAC-seq data shows the doublet enrichment of different time points after stroke. (d) Violin Plots show the unique fragment numbers of 7 different time points and Sham group. (e) Violin Plots show the TSS enrichment scores of 7 different time points and Sham group. (f) Histogram shows the filtered nuclei numbers of 7 different time points and Sham group. (g) Plots show the enrichment of snATAC-seq fragments around TSS.
Upon completion of the quality control process, the nuclei of all groups were integrated and clustered, with gene activity scores utilized to estimate gene expression and annotate cell types. We identified eight different cell types (Fig. 3a and 3b), including Astrocytes (AST, marker gene: Aqp4 and Fgfr3), Endothelial cells (EC, marker gene: Pecam1 and Cldn5)29,30, Fibroblast (FB, marker gene: Col1a2 and Dcn), Microglia (MG, marker gene: P2ry12 and Cx3cr1), Oligodendrocytes (OL, marker gene: Plp1 and Mag), Oligodendrocyte progenitor cells (OPC, marker gene: Pdgfra and Cspg4), Excitatory neurons (Ex_Neuron, marker gene: Rbfox3 and Satb2) and GABAergic neurons (GABA_Neuron1, marker gene: Rbfox3 and Meis2; GABA_Neuron2, marker gene: Rbfox3 and Gad1). Rbfox3 serves as a marker for mature neurons, distinguishing neuronal cells from glial cells31. Subsequently, we identified eight differential accessibility regions (DARs) with cell types specificity (Fig. 3c). Visual inspections on a genome browser of 9 DARs showed that each cell cluster exhibited a unique signature of snATAC-seq peaks. For example, signal peaks detected in the TSS region of marker genes Fgfr3 are specifically enriched in AST, but not observed in other cell types (Fig. 3c). Similarly, the signal peaks detected around Meis2 are significantly enriched in GABA_Neuron1 than in other cell types (Fig. 3c). Furthermore, GO analysis on the differential peaks specific to each cell type revealed a significant correlation between the identified cell types and their corresponding GO pathways (Fig. 3d). Vasculature development related pathways were enriched in EC, while the leukocyte activation and synapse immune response pathways exhibited enrichment in MG. In addition, myelin assembly-related pathways show enrichment in OL (Fig. 3d).

Clustering and annotation of chromatin accessibility in the cerebral cortex of MCAO/R mice. (a) Clustering visualization and annotations of 9 clusters identified by snATAC-seq data. (b) UMAP visualization of cell type–specific gene activity scores. Each dot represents an individual cell colored by cluster assignment. (c) The genome browser view shows aggregated chromatin accessibility profiles of cell type-specific gene loci. (d) The bar plot displays representative enriched pathways specific to the cell types. (e) The stacked chart illustrates the composition of cell proportions in the cerebral cortex for seven different time points of MCAO/R and Sham group, with colours indicate the cell types. (f) GO enrichment pathway analysis of genes highly expressed at M24h in MG. (g) GO enrichment pathway analysis of genes highly expressed at M14d in MG.
By calculating the proportions of each cell population, we observed dynamic changes of nine cell types within the cerebral cortex after MCAO/R. Compared to sham-operated mice, there was an significant increase in the proportion of Ex_Neuron during the acute stage (post-MCAO/R 24 hours, M24h)32, which was accompanied by the significant decrease in the proportion of AST, GABA_Neuron, and OPC (Fig. 3e). As the stroke progresses into the chronic phase (post-MCAO/R 7 days-14days, M7d-M14d)32, the composition of different cell types (Fig. 3e) continues to shift over time. Specifically, there was a significant changes between M7d and M4d in GABA_Neuron1. The proportion of Ex_Neuron exhibits a complex pattern of initial decline, subsequent increase, and eventual decrease. We used scCODA to statistically analyze the proportions of different cell types at various time points. The results indicated significant changes in cell proportions for Ex_Neuron at M24h (logFC = 0.448) and for GABA_Neuron2 at 7d (logFC = −1.187). At M14d, subpopulations such as FB (logFC = 1.742), GABA_Neuron1 (logFC = −4.272), MG (logFC = 2.571), and GABA_Neuron2 (logFC = −1.370) showed significant changes in cell proportions. To verify the authenticity of the data analysis, we used DEseq2 to calculate the differentially expressed genes in MG cells at different time points.
To verify the authenticity of the data, we extracted MG and added peaks with identical biological replicates to form pseudo-bulk data. We then used DESeq2 to calculate the differential pseudo-bulk peaks between M24h and M14d (see Methods section), identifying a total of 503 differential peaks. Subsequently, pathway enrichment analysis was performed on the genes associated with these peaks to validate the accuracy of the results. The results showed that the differentially expressed genes at M24h were enriched in migration and immune pathways (Fig. 3f), which is consistent with the function of microglia in acute injury phase post-stroke33. The differentially upregulated peak-related genes at M14d were enriched in pathways related to adhesion and wound healing (Fig. 3g), which is also consistent with the function of MG in the chronic phase post-stroke34. Additionally, differential peaks were observed in genes such as Serpinf1, Cxcr4, Ctsb and Siglech, all of which have been reported to be involved in the activation of microglia and secretion of inflammatory factors after stroke35,36,37. Therefore, these pieces of evidence demonstrate that our data reflect authentic biological phenomena. These patterns in cell type proportions highlight potential molecular mechanisms that underlie the functionality of different cell types in the context of MCAO/R-induced ischemic stroke.
The TFs binding sites of different cell types (Fig. 4a) and time points (Fig. 4b) are closely related to their corresponding cell functions. TFs related to neurodevelopment and function, such as Neurog1 and Neurod2, are enriched in Ex_Neuron. TFs related to lymphocyte proliferation and differentiation, such as Elf1 and Sfpi1 families, are enriched in MG. OL are specifically enriched with Sox938,39,40. At distinct time points (Fig. 4b), binding motifs associated with Fos, a transcriptional regulator implicated in the modulation of oxidative stress responses and maintenance of mitochondrial homeostasis, exhibited specific enrichment during the acute phase41.

Motif enrichment in each cell type and time point. (a) Heatmaps of motifs enrichment in marker peaks of each cell type. (b) The heatmap illustrates the time point–specific enrichment of motifs. The color represents the normalized motif enrichment score calculated in ArchR using HOMER with the hypergeometric test.
In order to validate the identified motifs, we performed the TF footprinting analysis to confirm the binding of TFs to DNA. The TF footprinting analysis also demonstrated that the identified motifs exhibited active TF binding activity in the corresponding cell types (Fig. 5)42,43,44. In summary, our data reproduce the enrichment patterns of key cell type-specific TFs reported in previous studies, indicating the accuracy of our cell types identification and the high quality of our data.

TF footprinting analysis identifies representative cell type-specific TFs activities in snATAC-seq data. (a) Cell type-specific TFs binding motifs sequence logo of cell types (top), and representative cell type-specific TFs footprint profiles (bottom).
Taken together, our datasets provide a valuable resource for exploration of the epigenetic regulation mechanisms after ischemic stroke in mice.
Usage Notes
The pipeline of the snATAC-seq data processing, including the read mapping, low-quality cells filtering, unsupervised clustering and peak calling were run on the Linux operating system. All R source codes with the optimized parameters used for the downstream data analyses and visualization are provided online (https://doi.org/10.6084/m9.figshare.25558785)27.
Code availability
The codes used to analyze the data in this study were available online (https://doi.org/10.6084/m9.figshare.25558785)27.
References
Collaborators, G. N. S. D. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 23, 344–381 (2024).
Gil-Garcia, C. A. et al. Essential Topics About the Imaging Diagnosis and Treatment of Hemorrhagic Stroke: A Comprehensive Review of the 2022 AHA Guidelines. Curr Probl Cardiol. 47, 101328 (2022).
Euskirchen, P. et al. Cellular heterogeneity contributes to subtype-specific expression of ZEB1 in human glioblastoma. PLoS One. 12, e0185376 (2017).
Zhang, C., Qin, F., Li, X., Du, X. & Li, T. Identification of novel proteins for lacunar stroke by integrating genome-wide association data and human brain proteomes. BMC Med. 20, 211 (2022).
Skinnider, M. A. et al. Cell type prioritization in single-cell data. Nat Biotechnol. 39, 30–34 (2021).
Qiu, M. et al. Cell Heterogeneity Uncovered by Single-Cell RNA Sequencing Offers Potential Therapeutic Targets for Ischemic Stroke. Aging Dis. 13, 1436–1454, (2022).
Lin, X. et al. Single-nucleus chromatin landscapes during zebrafish early embryogenesis. Sci Data. 10, 464 (2023).
Xiong, X. et al. Epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion. Cell. 186, 4422–4437.e4421 (2023).
Turner, A. W. et al. Single-nucleus chromatin accessibility profiling highlights regulatory mechanisms of coronary artery disease risk. Nat Genet. 54, 804–816 (2022).
Brase, L. et al. Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat Commun. 14, 2314 (2023).
Zemke, N. R. et al. Conserved and divergent gene regulatory programs of the mammalian neocortex. Nature. 624, 390–402 (2023).
Hocker, J. D. et al. Cardiac cell type-specific gene regulatory programs and disease risk association. Sci Adv. 7, (2021).
Zhang, R. et al. NSC-derived exosomes enhance therapeutic effects of NSC transplantation on cerebral ischemia in mice. Elife. 12, (2023).
Preissl, S. et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci. 21, 432–439 (2018).
Shi, Q., Liu, S., Kristiansen, K. & Liu, L. The FASTQ+ format and PISA. Bioinformatics. 38, 4639–4642 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25, 1754–1760, (2009).
Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol. 37, 916–924 (2019).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet. 53, 403–411 (2021).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell. 184, 3573–3587.e3529 (2021).
Büttner, M., Ostner, J., Müller, C. L., Theis, F. J. & Schubert, B. scCODA is a Bayesian model for compositional single-cell data analysis. Nat Commun. 12, 6876 (2021).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Gontarz, P. et al. Comparison of differential accessibility analysis strategies for ATAC-seq data. Sci Rep. 10, 10150 (2020).
Guo, X. et al. CNSA: a data repository for archiving omics data. Database (Oxford). 2020, (2020).
Chen, F. Z. et al. CNGBdb: China National GeneBank DataBase. Yi Chuan. 42, 799–809 (2020).
CNGB Nucleotide Sequence Archive https://db.cngb.org/cnsa/project/CNP0005552 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP548281 (2024).
Guo, S. ATAC data and code of mononuclear chromatin structure in mice with ischemic stroke. Figshare https://doi.org/10.6084/m9.figshare.25558785.v4 (2024).
Deng, Y. et al. Spatial profiling of chromatin accessibility in mouse and human tissues. Nature. 609, 375–383 (2022).
Li, H. et al. Acute ischemia induces spatially and transcriptionally distinct microglial subclusters. Genome Med. 15, 109 (2023).
Cao, G. Z. et al. Single-cell RNA sequencing reveals that VIM and IFITM3 are vital targets of Dengzhan Shengmai capsule to protect against cerebral ischemic injury. J Ethnopharmacol. 311, 116439 (2023).
Ma, Y. et al. Single-nucleus chromatin landscape dataset of mouse brain development and aging. Sci Data. 11, 616 (2024).
Qin, C. et al. Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 7, 215 (2022).
Kronenberg, G. et al. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 135, 551–568 (2018).
Jackson, L., Dumanli, S., Johnson, M. H., Fagan, S. C. & Ergul, A. Microglia knockdown reduces inflammation and preserves cognition in diabetic animals after experimental stroke. J Neuroinflammation. 17, 137 (2020).
Butovsky, O. & Weiner, H. L. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 19, 622–635 (2018).
Zhao, S. C. et al. Regulation of microglial activation in stroke. Acta Pharmacol Sin. 38, 445–458 (2017).
Konishi, H. et al. Siglec-H is a microglia-specific marker that discriminates microglia from CNS-associated macrophages and CNS-infiltrating monocytes. Glia. 65, 1927–1943 (2017).
Finzsch, M., Stolt, C. C., Lommes, P. & Wegner, M. Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development. 135, 637–646, (2008).
Pozniak, C. D. et al. Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proc Natl Acad Sci USA. 107, 21795–21800 (2010).
Tran, T. H. et al. Role of canonical Wnt signaling/ß-catenin via Dermo1 in cranial dermal cell development. Development. 137, 3973–3984 (2010).
Mu, Q. et al. Transcriptomic Profiling Reveals the Antiapoptosis and Antioxidant Stress Effects of Fos in Ischemic Stroke. Front Neurol. 12, 728984 (2021).
Cuadros, M. A., Sepulveda, M. R., Martin-Oliva, D., Marín-Teva, J. L. & Neubrand, V. E. Microglia and Microglia-Like Cells: Similar but Different. Front Cell Neurosci. 16, 816439 (2022).
Tchieu, J. et al. NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat Biotechnol. 37, 267–275 (2019).
Motojima, Y., Ueta, Y. & Sakai, A. Analysis of the proportion and neuronal activity of excitatory and inhibitory neurons in the rat dorsal spinal cord after peripheral nerve injury. Neurosci Lett. 749, 135707 (2021).
Acknowledgements
We thank the China National GeneBank for providing sequencing services for this project. Zhiqiang Dong is funded by Key Project of Research and Development of Hubei Province (2022BCE049) and Fundamental Research Funds for the Central Universities (2662022JC002). Ruolin Zhang is funded by the Postdoctoral Fellowship Program of CPSF under Grant Number: GZC20240562.
Author information
Authors and Affiliations
Contributions
Zhiqiang Dong and Chang Liu supervised this work and guided the bioinformatics analysis. Zhiqiang Dong, Chang Liu and Ruolin Zhang designed the project. Ruolin Zhang, Sicheng Guo and Jie Zhou wrote the manuscript. Ruolin Zhang and Ying Wang prepared the samples. Sicheng Guo and Jie Zhou undertaken the main task of data analyses with the assistance of Xiumei Lin and Yiqi Wang. Xiumei Lin and Kaichen Zhao performed the libraries construction of snATAC-seq. Wendai Bao, Ke Shui, Chuanyu Liu and Muyang Li participated in project discussion. Zhiqiang Dong and Chang Liu participated in the manuscript editing and discussion.
Corresponding authors
Ethics declarations
Competing interests
The employees of BGI have stock holdings in BGI.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, R., Guo, S., Zhou, J. et al. Monitoring of single-nucleus chromatin landscape of ischemic stroke in mouse cerebral cortex across time. Sci Data 12, 47 (2025). https://doi.org/10.1038/s41597-025-04367-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04367-4
