
Abstract
Mandarin (Citrus reticulata) is broadly recognized as one of the foremost citrus crops globally. Our study identified the Xingan mandarin (Citrus reticulata ‘Xingan’) as a primitive type found near Maoer Mountain. This report provides a high-resolution, chromosome-scale genome assembly for the Xingan mandarin. The total size of the genome assembly is an impressive 325.12 Mb, including contig N50 and scaffold N50 values of 29.32 Mb and 29.62 Mb, respectively. Notably, we successfully anchored approximately 93.08% of the assembled sequences onto nine pseudochromosomes. Our predictions identified 30,581 protein-coding genes, 166 miRNAs, 415 tRNAs, 728 rRNAs, 325 snRNAs, and 659 snoRNAs. We were able to predict the functions of 27,242 genes, constituting 89.08% of the total protein-coding genes. A notable finding of our study was the high degree of genome synteny between the Xingan mandarin and the Mangshan mandarin (Citrus reticulata ‘Mangshan’), reinforcing their genetic similarity. The acquisition of the chromosome-level genome for the Xingan Mandarin represents a significant milestone, laying an indispensable foundation for rigorous molecular investigations of this species. Moreover, it is poised to invigorate advanced research in comparative genomics within the Citrus genus.
Similar content being viewed by others


Background & Summary
The species Citrus reticulata, categorized within the Rutaceae family, is recognized as one of the three fundamental species of the Citrus genus1. Archaeobotanical evidence and historical records suggest that the cultivation of this species dates back over 4000 years, with early references appearing in ancient texts such as the “Yu Gong” section of the “Xia Shu,” which chronicled the history of the Xia Dynasty2,3. The peelability of mandarin fruits, a trait highly valued by consumers, facilitates easier access to their nutrient-dense flesh. This characteristic, coupled with their high content of vitamin C and dietary fiber, positions mandarins as one of the best-loved fruits4,5. Moreover, studies have demonstrated that certain local or wild mandarin varieties, which are rich in phenolic compounds and antioxidants, exhibit potential for medicinal applications and serve as beneficial food ingredients6,7.
The south-central regions of China are recognized as the primary centers of origin for the genus Citrus8. During the past several decades, numerous wild mandarin indigenous to China have been documented, including Citrus mangshanensis, the Mangshan mandarin, and Citrus daoxianensis8,9,10. Wild Citrus species are known for their high genetic diversity, which constitutes a significant genetic resource for the breeding of Citrus crops. The genetic variability within these species provides a wealth of alleles that can be harnessed to improve the traits of cultivated varieties, such as disease resistance, fruit quality, and adaptability to different environmental conditions11,12. The investigation into wild Citrus species not only sheds light on the evolutionary relationships of the Citrus genus but also furnishes compelling evidence that enhances our understanding of the considerable transformations in key quality traits throughout the domestication of Citrus.
Xingan mandarin is a wild mandarin species discovered on Mao’er Mountain in Xing’an County, located in northern Guilin (Fig. 1a–d). In December 2013, Deng Chongling’s research group from the Guangxi Academy of Specialty Crops first discovered wild Citrus reticulata in Maoer Mountain, the main peak of the Yuechengling Mountains in the Nanling Mountains of Guangxi. This discovery marks the first record of wild mandarin in the Yuechengling Mountains. It is located in Huajiang Yao Township, Xing’an County, Guilin City, Guangxi Zhuang Autonomous Region, with a latitude and longitude of 25° 51′51″N, 110°51′51″E and an altitude of 601 m.

Photo and genomic characteristics of Xingan mandarin. (a) Xingan mandarin tree. (b) Flowers and leaves during the peak flowering period. (c) Anatomical diagram of Xingan mandarin flower. (d) Xingan mandarin mature fruits. (e) Characterization of the Xingan mandarin genome. The circle from inside to outside, respectively, represents Chromosome ideograms (I), TE density (II), SSR density (III), gene density (IV), and GC density (V).
The main traits of Xingan mandarin include: arbor, medium tree vigor, semi-circular crown, upright tree posture, fine branches and spines. The leaves are long oval, the top is acuminate, the base is wedge-shaped, and the leaves of spring shoots are 5.4 cm in length and 2.4 cm in width. The fruit is oblate, with a height of 3.6 cm and a diameter of 4.6 cm, with an average fruit weight of 46.2 g. The peel is yellow and rough, with large, obviously raised oil cells, and the flesh is orange-yellow. The average number of seeds is 9.8, and the seeds are nearly spherical. It exhibits favorable traits such as vigorous growth, strong resistance to pests and diseases, and good fruiting habits. Xingan mandarin shows obvious resistance to infection by Citrus Huanglongbing. After infection with Huanglongbing, the bacterial content in it was significantly lower than that of other materials, and the growth was normal. This species harbors numerous exceptional genes associated with high yield, quality, disease resistance, and abiotic stress tolerance. Thus, it serves as a valuable resource for research on the origins, classification, zoning, genetic breeding, and high-yield cultivation techniques of citrus, representing an important genetic resource for agricultural citrus breeding.
In the scope of the present study, we utilized a comprehensive sequencing strategy, ingeniously combining the capabilities of Illumina short-read sequencing, PacBio long-read sequencing, and Hi-C sequencing technologies. This integrated approach facilitated the assembly, annotation, and anchoring of the Xingan mandarin genome at the chromosome level, as illustrated in Fig. 1e. This genome assembly is poised to enhance the discovery of pivotal genes associated with agronomic traits, positioning it as an indispensable resource with potential medicinal applications.
Methods
Sample collection
All samples designated for sequencing were procured from the Citrus germplasm resource garden at the Guangxi Academy of Specialty Crops (Guilin). We utilized tender leaves from Xingan mandarin trees for Illumina, HiFi, and Hi-C sequencing. To ensure exhaustive capture of transcriptomic data, we collected a variety of tissues from the Xingan mandarin, including roots, stems, leaves, flowers, seeds, as well as fruits at both immature and mature stages. These samples were rapidly frozen in liquid nitrogen and subsequently stored at −80 °C, awaiting the extraction of DNA and RNA.
Library construction and sequencing
Genomic DNA was carefully extracted from the leaf tissue of the Xingan mandarin, employing the established CTAB method. Post extraction, short-read libraries, each with a read length of 350 base pairs, were meticulously constructed using a dedicated library construction kit. Following this, the libraries were sequenced on the HiSeq. 2500 platform (Illumina, CA, USA). The process resulted in an impressive total of 18.89 Gb of data, corresponding to an overall sequencing depth of approximately 64 × of the genome. The GC content was approximately 36.19%, and the Q20 and Q30 ratios surpassed 97.27% and 94.91%, respectively. The cleaned reads obtained were used for genomic surveys, encompassing assessments of genome size, GC content, and heterozygosity.
To obtain HiFi sequencing data, after the samples passed the quality control test, the genomic DNA fragments were selected using BluePippin, then subjected to end repair and A-tailing. Subsequently, adapters were ligated to both ends of the fragments to prepare a DNA library. After the library passed qualification, the sequencing operation was conducted using the PacBio Revio platform (Pacific Biosciences, Menlo Park, CA, USA), guided by the library’s effective concentration and the specifications for data output. PacBio HiFi sequencing generated approximately 12.71 Gb of clean data, with an overall sequencing depth of approximately 46 × of the genome. The reads exhibited an N50 of 16.26 kb and an average read length of 15.99 kb.
For Hi-C sequencing, the library underwent rigorous quality assessment to ensure quality, including library concentration determination, insert size evaluation, and precise determination of library molar concentration. The main methods for Hi-C sequencing include: 1) the initial evaluation of library concentration using Qubit 2.0 (Invitrogen, CA, USA); 2) the assessment of library DNA fragment integrity and insert size facilitated by Agilent 2100 (Agilent Technologies, CA, USA); 3) the exact quantification of the effective library concentration using the qPCR approach. After library qualification, high-throughput sequencing was performed on the Illumina platform, with a sequencing read length of PE150. The project ultimately generated a total of 35.25 Gb of clean data, with an overall sequencing depth of approximately 108 × of the genome, and the Q20 and Q30 ratios surpassed 95.55% and 92.34%, respectively.
Genome survey and assembly
The HIFI long reads, generated from the Pacbio platform, were subjected to a quality filtration process performed using fastp (v0.23.4)13, operating under default parameters. The quality-filtered reads were subsequently used in the critical process of genome size estimation. We used Jellyfish (v2.2.10)14 software to count the 17-mers and assessed the genome characteristics using GenomeScope (v2.0)15 software (Fig. 2). The genome size of Xingan mandarin was estimated to be 328.20 Mb, with approximately 55.62% repeat sequences, a heterozygosity rate of ~1.32%. Thus, the genome of this species is classified as a highly heterozygous and complex genome.

K-mer distribution plot of Xingan mandarin. The presented overview comprehensively displays the frequency distribution of the 17-mer in the Xingan mandarin genome, where the x-axis signifies the k-mer depth, and the y-axis stands for the k-mer frequency that aligns with the aforementioned depth.
The HiFi long reads were assembled using Hifiasm (v0.19.8-r603)16, yielding a total contig length of 340.74 Mb and a contig N50 value of 29.32 Mb. Comparisons were conducted against the NCBI nucleotide sequence database, along with the mitochondrial and plastid databases (https://www.ncbi.nlm.nih.gov/refseq/), to filter out mitochondrial and plastid sequences from the assembled genome. As a result of this procedure, the contig length was recalibrated to 332.40 Mb, while concurrently sustaining a contig N50 value of 29.32 Mb (Table 1).
To anchor contigs into chromosomal scaffolds, we first generated clean read pairs from the Hi-C library and aligned them to the polished Xingan mandarin genome using BWA (v0.7.17)17 with default parameters. Paired-end reads mapping to different contigs were then used for Hi-C-based scaffolding. A stringent filtering step was applied to remove invalid reads, including those derived from random breaks, self-ligation, non-ligation events, and fragments with abnormally large or small sizes. Subsequent ordering and orientation of the filtered contigs were performed using Lachesis (https://github.com/shendurelab/LACHESIS)18. This workflow yielded the first high-quality chromosomal-level assembly of Xingan mandarin, with individual chromosome lengths ranging from 25.43 Mb to 49.65 Mb and collectively accounting for 93.08% of the total assembly length (Fig. 3; Table 1). The final chromosome-scale genome assembly spanned 325.12 Mb, with contig N50 and scaffold N50 values reaching 29.32 Mb and 29.62 Mb, respectively. In this study, the final assembled genome size is found to align closely with both the previously reported citrus genomes and the k-mer-based estimates.

The Hi-C interaction heatmap illustrates the chromosomal interactions in Xingan mandarin.
Repeat element identification
The task of annotating Transposable Elements (TEs) and tandem repeats was performed via a series of well-defined workflows. The identification of TEs was achieved using an integrated approach combining homology-based and de novo strategies. Our initial step involved the construction of a de novo repeat library of the genome, leveraging the capabilities of RepeatModeler (v1.0.5)19, an automated software that efficiently runs two de novo repeat discovery tools, namely RECON (v1.0.8)20 and RepeatScout (v1.0.6)21. The subsequent stages of our methodology focused on identifying and characterizing full-length long terminal repeat retrotransposons (fl-LTR-RTs), a process enabled by both LTRharvest (v1.5.9)22 and LTR_finder (v2.8)23. We then generated high-quality, intact fl-LTR-RTs and a non-redundant LTR library using LTR_retriever (v2.9.0)24.
In an effort to create a species-specific, non-redundant TE library, we combined de novo TE sequences with the well-regarded Dfam database (v3.5). The definitive TE sequences in the Xingan mandarin genome were subsequently identified and classified via a homology search against this library using RepeatMasker (v4.12)25. For the annotation of tandem repeats, we used Tandem Repeats Finder (v4.09)26 and the Microsatellite Identification Tool (MISA, v2.1)27. In the Xingan mandarin genome, we identified 112.96 Mb (34.75% of the genome) as TEs and 39.61 Mb (12.18% of the genome) as tandem repeats. The majority of these repeats (26.27% of the genome) were Class I retrotransposons, with Gypsy elements being the most prevalent (comprising 11.47% of the genome). Class II DNA transposons were also identified, making up 8.47% of the Xingan mandarin genome (Table 2).
Protein-coding genes prediction
The annotation of protein-coding genes within the genome was achieved through the combination of three uniquely effective approaches: de novo prediction, homology search, and transcript-based assembly. The generation of de novo gene models was performed using two leading ab initio gene prediction tools, namely Augustus (v3.1.0)28 and SNAP (v2006-07-28)29. For the homology-based strategy, we used the GeMoMa (v1.7)30 software, leveraging reference gene models derived from other Citrus species.
The transcript-based prediction process involved the alignment of RNA-sequencing data to the reference genome using Hisat (v2.1.0)31, followed by assembly using Stringtie (v2.1.4)32. Following transcript assembly, we used GeneMarkS-T (v5.1)33 to perform gene prediction. Moreover, gene prediction based on unigenes and full-length transcripts (derived from PacBio sequencing) was performed using PASA (v2.4.1)34 software. These sequences were assembled using Trinity (v2.11)35. The gene models resulting from these varied methodologies, were integrated using the EVM (v1.1.1)36 software, with ensuing updates conducted via PASA. The assembly of the Xingan mandarin genome led to the prediction of 30,581 protein-coding genes, each averaging a length of 3,360.50 bp (see Table 3 for details).
Functional annotation of protein-coding genes
Gene functionality was deduced based on the highest alignment match to several protein databases, including NR, EggNOG37, KOG, TrEMBL38, InterPro, and Swiss-Prot38. This was accomplished using diamond blastp (v0.9.29.130)39 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database40, applying an E-value threshold of 1E−3. Protein domains were annotated using InterProScan (v5.34-73.0)41, derived from InterPro protein databases. The discernment of motifs and domains embedded within gene models was expedited using the Pfam42 database. The corresponding Gene Ontology (GO) IDs for each gene were systematically collated from a combination of databases, including TrEMBL, InterPro, and EggNOG. Altogether, functional annotation was possible for 89.08% (27,242) of the predicted protein-encoding genes with established genes, conserved domains, and GO terms (Table 4).
Annotation of non-coding RNA genes
Non-coding RNAs, which include a variety of functionally known RNAs such as miRNA, rRNA, and tRNA, do not encode proteins. To predict these non-coding RNAs, we adopted various strategies based on their structural characteristics. For tRNA identification, we utilized tRNAscan-SE (v1.3.1)43 with default parameters; barrnap (v0.9)44 was primarily employed for rRNA prediction with its default parameters. The prediction of miRNA, snoRNA, and snRNA was conducted using the Rfam (v14.5)45 database and Infernal (v1.1)46 software. Our exploration of the Xingan mandarin genome unveiled 2,293 ncRNAs, composed of 728 rRNAs, 415 tRNAs, 166 miRNAs, 325 snRNAs, and 659 snoRNAs.
Data Records
The raw data of Hi-C short reads, llumina DNA short reads, PacBio DNA long reads, RNA short reads, and have been deposited in the National Center for Biotechnology Information (NCBI Sequence Read Archive database with accession numbers SRR3182379947, SRR3182379848, SRR3182379749, SRR3182379650). The genome assembly has been deposited in NCBI under accession number JBKFGA00000000051. The annotation files have been uploaded in figshare52.
Technical Validation
To evaluate the accuracy of gene annotation, we compared the distributions of gene length, CDS length, exon length, and intron length in Xingan mandarin with those of Arabidopsis thaliana, pummelo (C. grandis), Mangshan mandarin, C. reticulata cv. Ponkan, and sweet orange (C. sinensis). The results showed that the gene structural features of Xingan mandarin are highly similar to those of other Citrus species, as illustrated in Fig. 4. Furthermore, we performed a BUSCO assessment on the predicted gene set using the embryophyta_odb10 database. The results showed that the predicted gene set has a completeness of 99.01%, including 97.71% complete and single-copy orthologs and 1.30% complete and duplicated orthologs, with only 0.37% fragmented orthologs and 0.62% missing orthologs. These BUSCO results indicate that our predicted gene set is highly complete, thus reflecting the reliability of the genome annotation.

Distribution of gene structural features across six species. Subpanels (a–d) show comparative analyses of (a) gene length, (b) CDS length, (c) exon length, and (d) intron length between Xingan mandarin (Citrus reticulata ‘Xingan’) and five other species: Arabidopsis thaliana, C. grandis, C. reticulata ‘Mangshan’, C. sinensis, and C. reticulata cv. Ponkan.
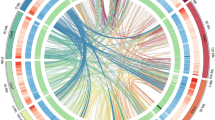
Chromosomal synteny analysis using MCScanX53 (with BLAST E-value ≤ 1 × 10−10) demonstrated strong collinearity among Xingan mandarin, Mangshan mandarin, and the mandarin haplotype of C. sinensis, characterized by nearly one-to-one chromosomal correspondence (Fig. 5). Notably, Xingan mandarin chromosomes exhibited a higher number of mapping fragments in Mangshan mandarin than in the mandarin haplotype of C. sinensis, providing empirical evidence for the reliability of the assembled chromosomal sequences.

Linear collinearity plot among Xingan mandarin, Mangshan mandarin, and Mandarin haplotype of C. sinensis.
To assess genome assembly quality, we employed BUSCO (v5.2.1)54 in conjunction with the embryophyta_odb10 database to curate a dataset of single-copy orthologs across major evolutionary lineages. This gene set was used for comparative analysis with the assembled genome, quantifying the proportion and completeness of orthologous genes. The final assembly achieved a BUSCO completeness score of 99.01%, reflecting exceptional gene space integrity. Additionally, our genome assembly demonstrated a robust long terminal repeat (LTR) assembly index (LAI)55 of 20.83, exceeding the threshold of 20 that defines a “golden reference” genome, indicating high structural integrity for LTR sequences. Using Merqury v1.356, we assessed the accuracy of the genome assembly with short-read sequencing data, obtaining a QV score of 46.65—a metric indicating an error rate below 0.0002%, which reflects near-ideal base-calling precision.
Finally, to gauge the completeness of the assembly and the uniformity of sequencing coverage, Illumina short reads and HiFi reads were mapped to the assembled genome using BWA17 and Minimap257 software, respectively. The completeness and coverage uniformity were evaluated based on alignment rates, the proportion of the genome covered, and the distribution of sequencing depths. The alignment results for the Illumina short reads revealed an alignment rate of 96.68%, a coverage of 99.81%, and an average sequencing depth of 50. On the other hand, the alignment results for the HiFi reads showcased an alignment rate of 99.61%, a coverage of 99.99%, and an average sequencing depth of 35 × . The collective results of the aforementioned analyses provide strong empirical evidence for the exceptional completeness and accuracy of the Xingan mandarin genome assembly, establishing a robust foundation for subsequent functional genomic studies and comparative analyses.
Code availability
If no detailed parameters were mentioned, all software and tools in this study were used with their default parameters. No specific code or script was used in this study.
Change history
12 December 2025
In this article the hyperlink provided for https://doi.org/10.6084/m9.figshare.29137547 in reference 52 was incorrect. The original article has been corrected.
References
Wu, G. A. et al. Genomics of the origin and evolution of Citrus. Nature 554, 311–316 (2018).
Spiegelroy, P. & Goldschmidt, E. E. The Biology of Citrus (2008).
Chapman, H. & Kelley, W. P. The mineral nutrition of Citrus. Ex The Citrus industry Volume I: history, botany, and breeding. First edition. Mineral Nutrition of Citrus Ex the Citrus Industry (1943).
Gu, Q. et al. Characterization of soluble dietary fiber from citrus peels (Citrus unshiu), and its antioxidant capacity and beneficial regulating effect on gut microbiota. Int J Biol Macromol 246, 125715 (2023).
Rao, M. J., Wu, S., Duan, M. & Wang, L. Antioxidant metabolites in primitive, wild, and cultivated citrus and their role in stress tolerance. Molecules 26, 5801 (2021).
Wang, X. et al. Targeted/untargeted metabolomics and antioxidant properties distinguish Citrus reticulata ‘Chachi’ from Citrus reticulata Blanco. Food Chem 462, 140806 (2025).
Wen, J. et al. An integrated multi-omics approach reveals polymethoxylated flavonoid biosynthesis in Citrus reticulata cv. Chachiensis. Nat Commun 15, 3991 (2024).
Wang, L. et al. Genome of Wild Mandarin and Domestication History of Mandarin. Mol Plant 11, 1024–1037 (2018).
Genfeng, L., Shanwen, H. & Wenbin, L. Two new species of Citrus in China. Acta Botanica Yunnanica (1990).
Deng, L. X. X. Citrus Breeding and Genetics in China (2007).
Wang, N. et al. Genomic conservation of crop wild relatives: A case study of citrus. PLoS Genet 19, e1010811 (2023).
Rao, M. J., Zuo, H. & Xu, Q. Genomic insights into citrus domestication and its important agronomic traits. Plant Commun 2, 100138 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–70 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–95 (2010).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–25 (2013).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12, 1269–76 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–8 (2005).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–8 (2007).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.1–4.10.14 (2009).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–80 (1999).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–44 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res 44, e89 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–60 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–5 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–66 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–52 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47, D309–D314 (2019).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–70 (2003).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 44, D457–62 (2016).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–40 (2014).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419 (2021).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–64 (1997).
Loman, T. A Novel Method for Predicting Ribosomal RNA Genes in Prokaryotic Genomes (2017).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33, D121–4 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–5 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31823799 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31823798 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31823797 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR31823796 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBKFGA000000000 (2025).
Deng, C. L. The genome assembly annotation for the Xingan mandarin, figshare. Dataset https://doi.org/10.6084/m9.figshare.29137547 (2025).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245 (2019).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Acknowledgements
This project was supported by the Science and Technology Major Project of Guangxi (Gui Ke AA22068092) and the National Natural Science Fund (U23A20198). We would like to thank TopEdit (www.topeditsci.com) for linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
The authors declare no competing interests.
Corresponding author
Ethics declarations
Competing interests
C.L.D. and X.X.W. conceived this project. C.L.D. and X.X.W. designed the experiments. C.L.D., X.X.W., H.M.F. and J.H. prepared the samples. C.L.D., Y.T. and C.C.W. investigated and collected wild citrus resources. C.L.D., X.X.W. and H.M.F. analysed the bioinformatics data. C.L.D. and X.X.W. wrote the article. P.L., S.Q.L. and X.Q.G. provided valuable suggestions on the research design and the improvement of the manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, C., Wu, X., Tang, Y. et al. A high-quality chromosome-scale genome assembly of Xingan mandarin (Citrus reticulata ‘Xingan’), a primitive Mandarin type. Sci Data 12, 1257 (2025). https://doi.org/10.1038/s41597-025-05577-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05577-6
