
Abstract
The genus Ormosia is in the Fabaceae family. China is one of its centers of origin, with almost all species being endemic, suggesting the need for genomic studies to understand its evolution, conservation, and use. Therefore, we generated a chromosome-scale genome assembly of O. boluoensis using Oxford Nanopore long reads combined with Illumina short reads. The initial genome assembly generated by NextDenovo had a total length of 1,638,935,015 bp and a contig N50 of 16,933,043 bp. The assembly after Hi-C reads scaffolding was 1,565,663,440 bp with the scaffold N50 of 201,058,012 bp, and 1,561,022,207 bp (99.70%) for sequences were anchored to 8 chromosomes. Repeats accounted for 75.06% of the assembly. There were 51,822 predicted genes. The BUSCO evaluation showed 98.2% completeness at genome assessment level. The high-quality assembly and annotation provide valuable insights into the genomic characteristics of O. boluoensis, providing resources for its effective conservation and future utilization.
Similar content being viewed by others

Background & Summary
The genus Ormosia (family Fabaceae) contains trees and shrubs and includes 130–150 species1,2. They are warm-adapted lowland plants, with current distributions including southeast Asia, north Australia, and South America1,2,3. China is one of its diversity centers, and its northernmost distribution extends to 23° north latitude4,5. Currently, there are approximately 37 Ormosia species in China, 34 of which are endemic2.
Ormosia species have hard, often red seeds used for jewelry and decorations1,4. They can also be used as landscape trees and harvested for timber4,6,7. Their seeds, roots, stems, bark, and leaves contain valuable secondary metabolites, such as alkaloids, flavonoids, isoflavones, terpenes, and lignin8,9,10. These substances have antioxidant, anticancer, insecticidal, bactericidal, and other pharmacological activities8. As legume species, Ormosia forms nitrogen-fixing nodules on its root system11,12,13.
Currently, four Ormosia nuclear genomes have been reported, including those of O. purpureiflora, O. emarginata, O. semicastrata14 and O. henryi15, all of which were assembled into the chromosome scales. Approximately 15 species, including O. boluoensis which we study here, have had their chloroplast genomes reported16,17. However, only one mitochondrial genome has been published, which is from O. boluoensis18. The nuclear genome sizes for Ormosia are generally around 1.5 Gb as shown in O. purpureiflora, O. emarginata and O. semicastrata, while the size is approximately 2.7 Gb in O. henryi. Regardless of genome size, nearly 70% or more of their sequences are detected as repetitive elements. Ormosia is in the Genistoid lineage within Fabaceae19, and its closest genera are Hovea and Poecilanthe. To date, no genomes have been reported for these two genera.
O. boluoensis is endemic to China, has only been found at two sites in southeast China: Guangdong Xiangtoushan (XTS) National Natural Reserve and Guangdong Nankunshan (NKS) Provincial Natural Reserve. Field investigations by Guo20 and the others in our group indicate that there are less than 1000 individuals in the reserves. Although both the chloroplast and mitochondrial genomes of O. boluoensis have been reported, its nuclear genome has not yet been assembled. Therefore, we present the chromosome-scale and fully annotated genome of O. boluoensis. Before genome assembly, chromosome number of O. boluoensis was observed using microscope and was determined to be 2n = 16. The assembly and annotation were then performed by using approximately 127.13 Gb Oxford Nanopore Technology (ONT) long reads, 122.07 Gb next-generation sequencing (NGS) short reads, 148.19 Gb high-throughput chromosome conformation capture (Hi-C) reads, and 22.29 Gb RNA-seq reads. The initial O. boluoensis genome assembly by Nextdenovo was 1,638,935,015 bp with the contig N50 of 16,933,043 bp. The assembly after Hi-C reads scaffolding by Juicer pipeline, 3d-dna and manually curated was 1,565,663,440 bp with the scaffold N50 of 201,058,012 bp, and 1,561,022,207 bp (99.70%) for sequences were anchored to 8 chromosomes. 75.06% of the assembled sequences were identified as repeats. A total of 51,822 genes were predicted. The BUSCO evaluation for the assembled genome showed 98.2% completeness. The high quality of O. boluoensis genome assembly provides valuable resources for its evolutionary and conservation studies.
Methods
Species description
O. boluoensis is a shade-tolerant small tree or shrub. It is distributed at altitudes between 640 and 840 m and generally found along streams. It has leathery leaves, which are simple and alternate (Fig. 1A). Its flowers are white (Fig. 1B), bisexual, mostly in panicles and rarely in racemes. It is outcrossing species and pollinated by insects. In 2018 and 2019, individuals with a diameter at breast height (DBH) larger than 5 cm were observed to flower and set seeds, but only a few actually flowered in these years20. In addition, because it suffers from severe pest/disease attacks on fruit/seeds (Fig. 1C,D), its seed yields were very low. No (healthy) seeds have been collected since 2020. Root-derived clonality was frequently observed in the field (Fig. 1E,F), indicating asexual reproduction. It has green bark (Fig. 1E).

Picture showing Ormosia boluoensis. (A) O. boluoensis leaves; (B) O. boluoensis flowers; (C) and (D) O. boluoensis seeds and fruit showing attack by worms/insects and/or disease; (E) and (F) O. boluoensis growth habitat and its clonal growth with root sprouting.
Chromosome number observation
The individual used for chromosome number observation in O. boluoensis was regenerated from seeds collected from the XTS Reserve. At room temperature, the root tips were pretreated with 0.002 M 8-hydroxyquinoline for 6 h and fixed in 3: 1 (v:v) absolute ethanol:glacial acetic acid for 24 h. The fixed root tips were hydrolyzed in 1:1 (v:v) 1 M absolute ethanol:hydrochloric acid for 7 min, washed by water, and stained with carbol fuchsin for 4 min. Meristems were subsequently excised, squashed and observed by microscope. Photographs were collected using an Olympus BX-43 microscope at 100x magnification with an Olympus DP26 camera. The chromosome number observation in O. boluoensis indicated 2n = 16 (Fig. 2A).

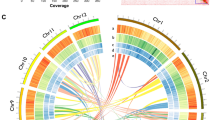
(A) Chromosome numbers observed in Ormosia boluoensis (scale bar: 10 μm); (B) Hi-C interaction heat maps (bin length 100,000 bp) for the O. boluoensis genome assembly; (C) Circos plot showing the genome features (chromosome, repeat density in length proportions, repeat density in numbers, gene density. All densities were estimated with a 1-Mbp sliding window.
Sample collection and sequencing
One O. boluoensis individual collected from the XTS Reserve was used for genome assembly. The collection was approved by the Management Office of Guangdong Xiangtoushan National Natural Reserve under the project of “Research on Manis pentadactyla and the other Rare and Endangered Animals and Plants in Guangdong Xiangtoushan National Nature Reserve — Genetic Diversity and Seedling Breeding of O. boluoensis (2024)”. For genome assembly and annotation, genomic DNA and RNA were isolated from its leaf tissues, and multiple libraries, including long- and short-read whole genome sequencing (WGS), Hi-C and RNA-seq libraries were constructed. Long-read WGS was applied using an Oxford Nanopore Technologies (ONT) PromethION sequencer. Short-read WGS, Hi-C, and RNA sequencing were performed using Illumina HiSeq X Ten platform sequencer with a 150-bp paired-end (insert size 300 bp) sequencing strategy.
Detailed libraries construction and sequencing information, including DNA/RNA preparation and library construction, has been reported in our previous studies21,22. Briefly, genomic DNA of O. boluoensis was extracted using the cetyltrimethylammonium bromide method. Extracted DNAs were used to build short-read and long-read WGS libraries. For the short-read WGS libraries, the DNAs were randomly fragmented to an average size of 200–400 bp for the final library construction. For the long-read WGS library, the extracted DNAs were size-selected and ligated with adapters to construct sequencing libraries. To perform Hi-C scaffolding, leaf tissues of O. boluoensis were immersed into nuclei isolation buffer with 2% formaldehyde for fixation. After fixation, nuclei were sheared into 300–600 bp fragments for the Hi-C library construction. After quality and integrity examination of extracted RNA, the RNA sequencing libraries were generated using RNA library preparing kit. After sequencing, these data were used for genome assembly, annotation, and comparative genomics analysis, with the default parameters in all programs unless otherwise specified.
Finally, the ONT sequencing platform generated approximately 127.13 Gb of WGS reads. The Illumina platform generated approximately 122.07 Gb of short WGS reads, 148.19 Gb of Hi-C reads, and 22.29 Gb of RNA-seq reads (Table 1).
Date pre-processing
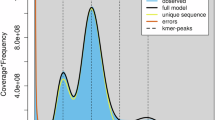
After sequencing, the adapters in long WGS reads were trimmed using Porchop v0.2.423 with the parameter “--check_reads 100000 --adapter_threshold 80”. Short WGS and Hi-C reads were quality trimmed using Sickle v1.3324 by removing the reads with base quality values less than 30 and lengths shorter than 80 bp. Short WGS reads were further error corrected using RECKONER v1.125, and duplicates were removed by FastUniq v1.126. Using the error-corrected reads of O. boluoensis, the genome size of O. boluoensis was estimated using Kmergenie v1.704427 with the parameters “–diploid–k 141”. The genome size of O. boluoensis estimated in Kmergenie was 1,467,753,196 bp.
Genome assembly
The O. boluoensis genome was assembled by Nextdenovo 2.3.128 using pre-processed Nanopore long reads with lengths larger than 1000 bp. The specific parameters for our Nanopore reads in Nextdenovo were set using “read_type = ont, input_type = raw” with the others at their default values. After assembly, the genomes were polished by Racon v1.4.2129 and Hapo-G v1.030 using long and short WGS reads, respectively, and each procedure was performed twice. After polishing, Pseudohaploid31 and Purge_Dups v1.2.532 were used to examine and remove duplications in the assembly.
Scaffhic v1.133 was subsequently used to identify misassembly and break the contigs with trimmed Hi-C reads. The Juicer pipeline 1.634 and 3d-dna v20100835 were then applied to perform scaffolding with Hi-C reads. The scaffolding results were visualized using Juicebox v2.04.0636, and the errors were manually corrected when necessary. The major corrections included chromosome boundary adjusting, misjoin and inversion error identifications. After scaffolding, TGS-GapCloser v1.0.137 was used to close the gaps with long WGS reads, and Racon and Hapo-G were used to polish the gap-closed genome. To detect potential chimeric assemblies, we used chimeric-contig-detector v1.0.238 to check them. The program contains two modes of chimera detection: one is “GC-Content Based Detection” mode, and the other is “Read-Pair Based Detection” mode. The latter mode uses short WGS reads.
The initial assembly in Nextdenovo was 1,638,935,015 bp with the contig N50 of 16,933,043 bp (Table 2). The assembly after Hi-C reads scaffolding was 1,565,663,440 bp with the scaffold N50 of 201,058,012 bp, and 1,561,022,207 bp (99.70%) for sequences assembled into 8 chromosomes (Fig. 2B). The largest chromosome size was 236,629,900 bp, and the shortest was 141,093,973 bp. No chimeras were detected under both “GC-Content Based Detection” and “Read-Pair Based Detection” mode by chimeric-contig-detector.
Repeat and gene prediction
Repeat and gene prediction in O. boluoensis followed the procedures previously described by Wang et al.21,22. Briefly, repeats were identified using EDTA v2.0.139 and RED v2.040, and their results were then combined and soft-masked in the assembled genome.
Based on the results of EDTA and RED, 70.35 and 63.04% of genomes, respectively, were identified as repetitive regions. According to EDTA (Table 3), the highest repetitive sequences were long terminal repeats (LTRs), accounting for 61.50% (962,829,486 bp) of the genome size, followed by terminal inverted repeats (TIRs) with 6.74% (105,292,142 bp) of the genome size. For LTRs, the large proportion of repetitive sequences were Gypsy-like, with a sequence size of 555,597,256 bp (35.49%).
Gene prediction was performed using BRAKER3 v.3.0.841 and Funannotate pipeline v1.8.1642. BRAKER3 is a gene annotation pipeline that integrates evidence from transcript reads and homologous proteins. For transcriptome-based gene annotation, RNA-seq reads were aligned to the soft-masked assembled O. boluoensis genome using Hisat2 v2.2.143 and then assembled with StringTie2 v2.2.144. For homology-based gene annotation, protein sequences from 13 reference genomes from Fabaceae (Table 4) were mapped to the assembled genome, generating hints on the gene structure using ProtHint v2.6.045. These results were used to train GeneMark-ETP v.1.046 and AUGUSTUS v3.5.047 to predict genes. The predictions were then combined and filtered by TSEBRA v1.1.2.548 to obtain non-redundant and consensus BRAKER3 gene sets, which were further input into Funannotate to generate the final gene annotation14. After gene prediction, a funannotate pipeline was used for gene functional annotation with different databases, including dbCAN v10.0, EggNOG v5.0.2, Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), InterPro v5.62–94.0, MEROPS v12.0, Pfam v35.0, SignalP 5.0b and UniProt v2023_0218,19. The reference proteins used for gene predictions are shown in Table 4.
By combining EDTA and RED results, 1,175,171,650 bp (75.06%) of the assembled genome was annotated and masked as repetitive components. The density of repeat sequences in the genome is shown in Fig. 2C. A total of 51,822 genes encoding 56,242 proteins were predicted. Functional annotation of these protein-coding genes showed that 40,750 genes (72.45%) were annotated to at least one database (Table 5).
Data Records
The assembled genome was submitted to GenBank under the accession number of JAIEWQ00000000049. The sequenced reads were deposited to NCBI Sequence Read Archive under the accession number SRR14802411 for the Nanopore reads50, SRR14725128 for Illumina WGS reads51, SRR14800493 for the Illumina Hi-C reads52, SRR14710764 for the Illumina RNA-seq reads53. O. boluoensis genome assembly and gene annotation files are submitted to figshare https://doi.org/10.6084/m9.figshare.29109572.v154.
Technical Validation
To evaluate the completeness of the assembly, Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.5.055 was used with the eudicots_odb10.2020-09-10 database. The database contains 2326 conserved core genes of eudicots. The completeness of the predicted genes with their longest transcripts was also evaluated using BUSCO with the eudicots_odb10.2020-09-10 database.
The completeness of the genome assessed by BUSCO at the genome assessment level with the parameter of “-m geno” was 98.2% complete, including 90.2% complete single-copy genes, 8% complete and duplicated genes, 1.4% fragmented genes, and 0.4% missing genes. Gene prediction completeness, as assessed using BUSCO with the parameter of “-m prot”, indicated a completeness score of 96.7% in O. boluoensis (89.9% complete and single-copy, 6.8% complete but duplicated), with 1.5% fragmented and 1.8% missing genes. We discovered that the BUSCO completeness score at the gene level was lower than that at the genome level, possibly due to gene prediction incompleteness. Therefore, future improvements to gene prediction are required.
The assembly quality was also examined by AssemblyQC v. 2.1.156 and sequencing read mapping ratios. The former helps estimate the Long Terminal Repeat (LTR) Assembly Index (LAI)57, which evaluates assembly contiguity using repetitive sequences, as well as k-mer-based accuracy and completeness by Merqury58. While for various types of long WGS, short WGS and RNA-seq reads, they were mapped to the assembly with Minimap2 v2.2959, BWA v0.7.1960 and Hisat2, respectively. Their mapping ratio were then calculated by Samtools v1.2161.
The LAI values for the O. boluoensis assembly was 13.23, meeting the “Reference genome” standard. Merqury indicated the quality value (QV) score was 30.01. Mapping-based evaluations indicated 98.52%, 94.99% and 94.82% mapping ratio for long WGS, short WGS and RNA-seq reads, respectively.
Data availability
The assembled genome can be downloaded on the website at https://identifiers.org/ncbi/insdc:JAIEWQ000000000. The raw Nanopore reads can be downloaded on the website at https://identifiers.org/ncbi/insdc.sra:SRR14802411, Illumina WGS reads at https://identifiers.org/ncbi/insdc.sra:SRR14725128, Hi-C reads at https://identifiers.org/ncbi/insdc.sra:SRR14800493, RNA-seq reads at https://identifiers.org/ncbi/insdc.sra:SRR14710764. The website of https://doi.org/10.6084/m9.figshare.29109572.v1 includes two compressed files. One is assembled genome under the file name of “blhd_change_chr_softmask.rar”. In the file, the repetitive sequences are softmasked (showing as lowercase letters) and non-repetitive sequences are shown as uppercase letters. The other is gene prediction and annotation file under the file name of “Ormosia_boluoensis-gene_annotation.rar. After decompression, six files will be generated. These are 1) all gene file under the name of “Ormosia_boluoensis_all_transcription.gff3” including all predicted genes and their transcripts information, 2) only longest transcripts file under the name of “Ormosia_boluoensis_only_longest_transcription.gff3” including all predicted genes and their longest transcripts information, 3) nucleotide sequences (all isoforms) of predicted genes under the name of “Ormosia_boluoensis_all_transcription-CDS.fa”, 4) translated sequences (all isoforms) of predicted genes under the name of “Ormosia_boluoensis_all_transcription-protein.fa”, 5) nucleotide sequences (only longest transcripts) of predicted genes under the name of “Ormosia_boluoensis_only_longest_transcription-CDS.fa”, 6) translated sequences (only longest transcripts) of predicted genes under the name of “Ormosia_boluoensis_only_longest_transcription-protein.fa”.
Code availability
All software and pipelines utilized in this study were performed following the guidelines of the published tools. The parameters and version numbers of software and databases are detailed in the Methods section. Any elements not specified in the Methods were executed using default parameters. No custom scripts were employed.
References
Torke, B. M. et al. A dated molecular phylogeny and biogeographical analysis reveals the evolutionary history of the trans-pacifically disjunct tropical tree genus Ormosia (Fabaceae). Mol. Phylogenet. Evol. 166, 107329, https://doi.org/10.1016/j.ympev.2021.107329 (2022).
Niu, M. et al. Two new synonyms of Ormosia semicastrata (Fabaceae, Papilionoideae, Ormosieae). Phytotaxa 613(2), 140–152, https://doi.org/10.11646/phytotaxa.613.2.3 (2023).
Wang, Z., Shi, G., Sun, B. & Yin, S. A new species of Ormosia (Leguminosae) from the middle Miocene of Fujian, Southeast China and its biogeography. Rev. Palaeobot. Palyno. 270, 40–47, https://doi.org/10.1016/j.revpalbo.2019.07.003 (2019).
Zhou, C. et al. Genetic structure of an endangered species Ormosia henryi in southern China, and implications for conservation. BMC Plant Biol. 23, 220, https://doi.org/10.1186/s12870-023-04231-w (2023).
Jiang, L. et al. Study on the potential suitable area of Ormosia in Guangdong Province based on the MaxEnt Model. Guangxi Sciences 31(1), 149–166, https://doi.org/10.13656/j.cnki.gxkx.20230719.001 (2024).
Li, L. et al. First report of dieback caused by Lasiodiplodia pseudotheobromae on Ormosia pinnata in China. Plant Dis. 104, 2551–2555, https://doi.org/10.1094/PDIS-03-20-0647-RE (2020).
Wei, L., Wang, G. & Xie, C. Predicting suitable habitat for the endangered tree Ormosia microphylla in China. Sci. Rep. 14, 10330, https://doi.org/10.1038/s41598-024-61200-5 (2024).
Zhang, L.-J. et al. A review on chemical constituents and pharmacological activities of Ormosia. Chin. Tradit. Herbal Drugs. 52(14), 4433–4442, https://doi.org/10.7501/j.issn.0253-2670.2021.14.035 (2021).
Zhou, Q.-Q. et al. Hosimosines A-E, structurally diverse cytisine derivatives from the seeds of Ormosia hosiei Hemsl. et Wils. Fitoterapia 170, 105661, https://doi.org/10.1016/j.fitote.2023.105661 (2023).
Zhou, W. et al. A new lignan from leaves of Ormosia xylocarpa. Rec. Nat. Prod. 17(1), 189–194, https://doi.org/10.25135/rnp.338.2203.2386 (2023).
Domínguez-Núñez, J. A. & Berrocal-Lobo, M. in Biofertilizers (eds Rakshit, A., Meena, V. S., Parihar, M., Singh, H. B. & Singh, A. K.) Chapter 20 - Application of microorganisms in forest plant. (Woodhead Publishing, Cambridge, United Kingdom, pp. 265–287, https://doi.org/10.1016/B978-0-12-821667-5.00026-9. 2021).
Ye, N. et al. Different leaf nutrient use strategies of nitrogen-fixing and non-nitrogen-fixing leguminous trees in South China. J. Trop. Subtrop. Bot. 31(3), 334–340, https://doi.org/10.11926/jtsb.4583 (2023).
Zhu, T. et al. Screening and identification of plant growth-promoting rhizobacteria (PGPR) in the rhizosphere of Ormosia henryi and their growth-promoting characteristics. J. Cent. South Univ. Forest. & Tech. 43(1), 43–49, https://doi.org/10.14067/j.cnki.1673-923x.2023.01.004 (2023).
Wang, Z.-F. et al. Chromosome-scale assemblies of three Ormosia species: repetitive sequences distribution and structural rearrangement. GigaScience 14, giaf047, https://doi.org/10.1093/gigascience/giaf047 (2025).
Zhou, C. et al. Chromosome-level genome assembly of the endangered tree species Ormosia henryi Prain. Sci. Data 12, 1065, https://doi.org/10.1038/s41597-025-05402-0 (2025).
Tang, J., Zou, R., Wei, X. & Li, D. Complete chloroplast genome sequences of five Ormosia species: Molecular structure, comparative analysis, and phylogenetic analysis. Horticulturae 9(7), 796, https://doi.org/10.3390/horticulturae9070796 (2023).
Wang, Z.-F. et al. The complete chloroplast genome of Ormosia purpureiflora (Fabaceae). Mitochondrial DNA B 6(12), 3327–3328, https://doi.org/10.1080/23802359.2021.1994901 (2021).
Wang, Z.-F., Zhang, Y., Zhong, X.-J., Kang, N. & Cao, H.-L. The complete mitochondrial genome of Ormosia boluoensis. Mitochondrial DNA B 6(8), 2109–2111, https://doi.org/10.1080/23802359.2021.1920503 (2021).
Zhao, Y. et al. Nuclear phylotranscriptomics and phylogenomics support numerous polyploidization events and hypotheses for the evolution of rhizobial nitrogen-fixing symbiosis in Fabaceae. Mol. Plant 14(5), 748–773, https://doi.org/10.1016/j.molp.2021.02.006 (2021).
Guo, Y. Preliminary study on endangered mechanism of Ormosia boluoensis with extremely small population (Master’s thesis. University of Chinese Academy of Sciences, 2020).
Wang, Z.-F., Rouard, M., Droc, G., Heslop-Harrison, P. & Ge, X.-J. Genome assembly of Musa beccarii shows extensive chromosomal rearrangements and genome expansion during evolution of Musaceae genomes. GigaScience 12, giad005, https://doi.org/10.1093/gigascience/giad005 (2023).
Wang, Z.-F. et al. Chromosome-level genome assembly and demographic history of Euryodendron excelsum in monotypic genus endemic to China. DNA Res. 31(1), dsad028, https://doi.org/10.1093/dnares/dsad028 (2024).
Porchop v0.2.4, https://github.com/rrwick/Porechop (Accessed 8 January 2019).
Joshi, N. A. & Fass, J. N. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33). 2011, https://github.com/najoshi/sickle (Accessed 28 August 2020).
Długosz, M. & Deorowicz, S. RECKONER: read error corrector based on KMC. Bioinformatics 33, 1086–1089, https://doi.org/10.1093/bioinformatics/btw746 (2017).
Xu, H. et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7(12), e52249, https://doi.org/10.1371/journal.pone.0052249 (2012).
Chikhi, R. & Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 30, 31–37, https://doi.org/10.1093/bioinformatics/btt310 (2014).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107, https://doi.org/10.1186/s13059-024-03252-4 (2024).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27(5), 737–746, https://doi.org/10.1101/gr.214270.116 (2017).
Aury, J. M. & Istace, B. Hapo-G, haplotype-aware polishing of genome assemblies with accurate reads. NAR Genom. Bioinform. 3(2), lqab034, https://doi.org/10.1093/nargab/lqab034 (2021).
Pseudohaploid, https://github.com/schatzlab/pseudohaploid (Accessed 28 August 2020).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
https://github.com/wtsi-hpag/scaffHiC (2022). Scaffhic v1.1Accessed 7 December.
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3(1), 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356(6333), 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6(2), 256–258.e1, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Xu, M. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9(9), giaa094, https://doi.org/10.1093/gigascience/giaa094 (2020).
Chimeric-contig-detector v1.0.2, https://github.com/megjohnson1999/chimeric-contig-detector (Accessed 27 August 2025).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275, https://doi.org/10.1186/s13059-019-1905-y (2019).
Girgis, H. Z. Red: an intelligent, rapid, accurate tool for detecting repeats de-novo on the genomic scale. BMC Bioinform 16(1), 227, https://doi.org/10.1186/s12859-015-0654-5 (2015).
Gabriel, L., et al BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 34(5), 769-777, https://doi.org/10.1101/gr.278090.123 (2024).
Funannotate v1.8.16, https://github.com/nextgenusfs/funannotate (Accessed 12 March 2023).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genom Bioinform. 2(2), lqaa026 (2020).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 34(5), 757–768, https://doi.org/10.1101/gr.278373.123 (2024).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34(suppl_2), W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Gabriel, L., Bruna, T., Hoff, K. J., Borodovsky, M. & Stanke, M. TSEBRA: transcript selector for BRAKER. BMC Bioinformatics 22, 1–12, https://doi.org/10.1186/s12859-021-04482-0 (2021).
Zhu, J.-P. et al. NCBI GenBank https://identifiers.org/ncbi/insdc:JAIEWQ000000000 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR14802411 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR14725128 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR14800493 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR14710764 (2025).
Zhu, J.-P. et al. A chromosome-scale assembly of Ormosia boluoensis (Fabaceae). Figshare https://doi.org/10.6084/m9.figshare.29109572.v1 (2025).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245, https://doi.org/10.1093/bioinformatics/btv351 (2019).
Rashid, U. et al. AssemblyQC: a Nextflow pipeline for reproducible reporting of assembly quality. Bioinformatics 40(8), btae477, https://doi.org/10.1093/bioinformatics/btae477 (2024).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46(21), e126, https://doi.org/10.1093/nar/gky730 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. & 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Acknowledgements
The study is supported by Guangdong Science and Technology Plan Project (2023A1111110001); Project of Research on Manis pentadactyla and the other Rare and Endangered Animals and Plants in Guangdong Xiangtoushan National Nature Reserve — Genetic Diversity and Seedling Breeding of O. boluoensis. Guangdong Provincial Forestry Bureau Project — Planning of the Provincial Plant Ex Situ Protection System and National Key Protected Plant Ex Situ Protection and Propagation; The National Natural Science Foundation of China (No. 32370406, 31970188).
Author information
Authors and Affiliations
Contributions
J.-P.Z., Z.-F.W., F.C. and H.-L.C. conceived and supervised the study. J.-P.Z., Z.-F.W., F.C., L.F. and H.-L.C. designed the methodology; J.-P.Z., Z.-F.W., F.C., L.F. and H.-L.C. performed data analysis and validation. J.-P.Z., Z.-F.W., F.C., L.F. and H.-L.C.; Z.-F.W., F.C., E.-P.Y., Y.-N.Z., Y.-M.H., X.-G.C., S.-W.D., Y.Z., J.-M.D., H.-H.L., W.-G.Z., F.-X.X. collected the samples; J.-P.Z., Z.-F.W., F.C., L.F. and H.-L.C. prepared original draft. J.-P.Z., Z.-F.W., F.C. and H.-L.C. obtained fundings. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, JP., Wang, ZF., Cheng, F. et al. A chromosome-scale assembly of Ormosia boluoensis (Fabaceae). Sci Data 12, 1659 (2025). https://doi.org/10.1038/s41597-025-05953-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-05953-2
