
Abstract
‘Huizao’ is a leading jujube (Ziziphus jujuba Mill.) variety valued for its high-quality dry fruit. Using PacBio HiFi long reads and Hi-C data, we generated a high-quality, chromosome-level, haplotype-resolved genome assembly for this cultivar, with genome sizes of 371.22 Mb and 385.42 Mb for the two haplotypes, and corresponding N50 values of 30.69 Mb and 31.26 Mb. Over 99.9% of the assembled sequences were anchored to 12 chromosomes. Genome annotation identified 32,065 protein-coding genes in Hap1 and 33,004 in Hap2, with 29,874 allelic gene pairs supported by collinearity and sequence similarity. Comparative analyses revealed extensive structural variants and allelic differences between the two haplotypes. This high-quality assembly addresses a critical gap in genomic resources for the ‘Huizao’ cultivar and provides a valuable foundation for allele-aware analyses, molecular breeding, and genetic diversity research in jujube.
Similar content being viewed by others
Background & Summary
Jujube (Ziziphus jujuba Mill.), the most important cultivated species of both genus Ziziphus Mill and family Rhamnaceae, is a major fruit tree native to China, renowned for its tolerance to drought, poor soil, salinity, and alkalinity. These tolerances make it increasingly important globally1. Jujube fruit is rich in sugars and vitamins and can be consumed fresh, dried, or processed into various products2,3. Additionally, jujube fruit has significant medicinal value, with polysaccharides, cyclic nucleotides, and flavones exhibiting antioxidant, anti-tumor, and immunomodulatory properties4,5,6,7. ‘Huizao’, a leading variety of jujube for dry fruit with excellent fruit quality, covers approximately 210,000 hectares and produces over 3 million tons annually, accounting for nearly 30% of global jujube production. Originating from the lower reaches of the Yellow River, the mother river of China, ‘Huizao’ is now predominantly cultivated in the oases surrounding the Taklamakan Desert, the second-largest desert in the world8,9.
In 2014 and 2023, our group published the first genome sequence and the first telomere-to-telomere (T2T) genome of jujube, using second- and third-generation sequencing technology, respectively, based on the cultivar ‘Dongzao’ (Z. jujuba Mill. ‘Dongzao’)10,11. In addition, chromosome-level genome assemblies have also been reported for the multi use jujube cultivar ‘Junzao’ (Z. jujuba Mill. ‘Junzao’)12, the wild sour jujube (Z. jujuba var. spinosa)13, and the table cultivar ‘Lingwuchangzao’ (Z. jujuba Mill. ‘Lingwuchangzao’) and ‘Shiguang’ (Z. jujuba Mill. ‘Shiguang’)14. However, a haplotype-resolved, chromosome-level genome assembly for dried jujube ‘Huizao’ is still lacking.
In this study, we report a high-quality, haplotype-resolved genome of ‘Huizao’, the leading jujube cultivar for dry fruit. The genome consists of two haplotypes: Hap1 (371,219,385 bp) and Hap2 (385,424,944 bp), with contig N50 values of 12.70 Mb and 10.68 Mb, and scaffold N50 values of 30.69 Mb and 31.26 Mb, respectively. This genome provides a valuable resource for studying functional genes related to key economic traits in jujube, accelerates the application of genomics in jujube molecular breeding, and facilitates studies on genomic diversity, allele-specific expression and the evolution of the Ziziphus genus.
Methods & Results
Sample preparation
Young leaves were collected from ‘Huizao’ jujube grown at the experimental base of Hebei Agricultural University (115.43°E, 38.83°N, 79.8 m altitude). A total of 15 g of healthy young leaf tissues was sampled. The leaves were immediately frozen in liquid nitrogen for subsequent PacBio HiFi and Hi-C library preparation and sequencing (Fig. 1a).

Overview of the ‘Huizao’ plant and genome estimation using PacBio Hifi reads. (a) Leaves, flowers and fruits of ‘Huizao’ jujube. (b) Estimation of genome ploidy, size, and heterozygosity using GenomeScope2.
HiFi SMRTbell library construction and sequencing
High-quality DNA was extracted using the SDS method and purified with the QIAGEN® Genomic Kit (Cat# 13343, QIAGEN). DNA purity was assessed using a NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and integrity was verified via agarose gel electrophoresis. The PacBio HiFi SMRTbell library was prepared using the SMRTbell Express Template Prep Kit 2.0 (PacBio, CA, USA). Long DNA fragments were sheared to 15–18 kb using a g-TUBE (Covaris, MA, USA), then concentrated and purified with AMPure PB beads (PacBio, CA, USA). Size selection for SMRTbell templates greater than 15 kb was performed using BluePippin (SageScience, MA, USA) to obtain large-insert SMRTbell libraries for sequencing. After data download, MD5 checksums were generated for the files to ensure data integrity.
Hi-C library construction and sequencing
For Hi-C library construction, approximately 2 grams of fresh leaves from the ‘Huizao’ jujube cultivar were used. Sample cells were fixed with formaldehyde to crosslink DNA with proteins, as well as proteins with each other. After crosslinking, the cells were lysed, and DNA quality was evaluated through sampling. Upon confirmation of sufficient quality, Hi-C fragment preparation was initiated.
Chromatin was digested using the restriction enzyme DpnII, which recognizes the GATC motif. The primer index used was CGCTCATT. The efficiency of enzymatic digestion was assessed by sampling. Following digestion, the DNA underwent biotin labeling, blunt-end ligation, and purification. DNA quality was re-evaluated at this stage, and upon meeting quality requirements, standard library construction proceeded.
Library construction included the removal of biotin from unligated DNA ends, ultrasonic fragmentation, end repair, A-tailing, and adapter ligation to generate sequencing-ready fragments. PCR amplification was then optimized and performed. The amplified products underwent quality control to assess enrichment for Hi-C junctions. Libraries that passed QC were sequenced on the Illumina NovaSeq platform using a paired-end 150 bp (PE150) sequencing strategy.
In total, Hi-C sequencing generated approximately 54.3 Gb of data, consisting of 181 million paired-end reads, which were used for chromosome-level genome scaffolding.
Genome size and ploidy estimation
The genome size and ploidy of the ‘Huizao’ jujube were estimated using 4.8 Gb of high-quality PacBio HiFi sequencing data (Table 1). To accurately assess genome size and heterozygosity, we performed GenomeScope modeling based on a series of odd-numbered k-mer sizes (k = 17 to 31). Among these, the 17-mer model yielded the best performance for our dataset, showing the lowest model error (0.116%), clear separation between homozygous and heterozygous peaks, and a more consistent estimation of repetitive content. Consequently, k = 17 was selected as the optimal parameter for k-mer analysis in this study, using K-Mer Counter (KMC, v3.0.0)15 (Fig. S1). The resulting k-mer frequency distribution was further analyzed with GenomeScope (v2.0)16 to estimate genome size, ploidy, and heterozygosity, with the parameters “-m64 -ci1 -cs10000 -cx10000 -p 2”. The analysis indicated that ‘Huizao’ jujube is diploid, with an estimated haploid genome size of approximately 361.46 Mb and a heterozygosity rate of 1.54% (Fig. 1b).
Genome assembly
De novo assembly of PacBio HiFi reads was performed using Hifiasm (v0.19.6-r595)17, with the following parameters: -o 04-HZ -t 80–ul-cut 20000 -D10–hom-cov 20. Both PacBio HiFi reads and Hi-C paired-end sequencing data were used to generate the initial assembly, resulting in two haplotype-resolved contig sequences.
The preliminary assemblies of Hap1 and Hap2 were 389.01 Mb and 393.82 Mb in size, containing 161 and 123 contigs, with contig N50 values of 11.77 Mb and 10.45 Mb, respectively. To eliminate haplotypic duplications and enhance assembly quality, we applied Purge_dups (v1.2.6) (https://github.com/dfguan/purge_dups). This refinement step produced final assemblies with improved contiguity: Hap1 was 371.65 Mb in size with 47 contigs and a contig N50 of 12.70 Mb, while Hap2 measured 385.33 Mb with 49 contigs and a contig N50 of 10.68 Mb.
Chromosome anchoring by Hi-C
To evaluate the quality of the Hi-C libraries, we conducted alignment and statistical analysis for both haplotypes (Hap1 and Hap2) using Hicup (v0.9.2)18 with the parameter “--re1 ^GATC,DpnII”. The results demonstrated high valid-pair percentages and reasonable ratios of intra- and inter-chromosomal interactions in both datasets (Table S1), indicating that the Hi-C libraries were of high quality and suitable for downstream chromosome-level genome assembly and analysis (Fig. S2). Raw Hi-C reads were first quality-filtered using fastp (v0.21.0)19 with default parameters, resulting 54.3 Gb of clean data, comprising 181 million paired-end reads. These reads were then aligned to the preliminary genome assembly using BWA (v0.7.19-r1273)20 with the -5SP parameter to accommodate Hi-C-specific split reads. The alignment output was processed with samblaster (v0.1.26)21 using default parameters to remove PCR duplicates. Low-quality and invalid alignments were filtered using samtools (v1.21)22 with the -F 3340 parameter. To further refine the data, we applied the filter_bam script from the HapHiC toolkit (v1.0.5)23, using the –nm 3 parameter to allow a maximum of three mismatches. The resulting filtered alignments were used for subsequent scaffolding analysis.
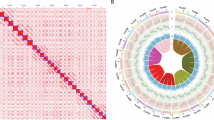
Scaffolding was performed using the HapHiC pipeline, with the restriction enzyme set to DpnII (recognition sequence: GATC), the chromosome number specified as 12, and the –processes 5 parameter enabled. The resulting scaffold structures were manually curated and refined using JuiceBox (v1.11.08)24 to adjust chromosome boundaries, resolve misjoins, and correct structural variations such as inversions and translocations (Fig. 2a). Subsequently, the juicer post tool was used to generate the final chromosome sequences and the corresponding agp file. To assess the quality of the chromosome-level assembly, the Hi-C contact matrix was visualized using the HapHiC plot tool.

Interaction heatmap of the two haplotype genomes and synteny between haplotypes. (a) Hi-C interaction heatmaps of the two haplotypes. (b) Collinearity relationship between the two haplotypes.
Both haplotypes were successfully clustered into 12 groups and ordered according to the reference genome11. The final assemblies anchored 371.65 Mb of contigs in Hap1 and 385.33 Mb in Hap2 to the chromosomes, achieving scaffold N50 values of 30.69 Mb and 31.26 Mb, respectively, with L50 values of 6 (Table 2). The completeness of single-copy genes was assessed using BUSCO (v5.8.2)25 with the embryophyta_odb10 database using default parameters. In Hap1, 2,326 genes were identified, of which 97.6% were complete and 0.5% were partial. Similarly, Hap2 also contained 2,326 genes, with 98.4% complete and 0.6% partial (Fig. 2b). These results demonstrate the successful assembly of a high-quality, haplotype-resolved, chromosome-scale genome for the ‘Huizao’ jujube cultivar (Fig. 3).

Circular maps of the two haplotypes of ‘Huizao’ jujube. (a) Chromosome name and size (b) Gene density. (c) GC skew. (d) GC content. (e) Repeat sequence density. (f) Collinearity of CDS genes.
PacBio HiFi reads were mapped to the genome, achieving coverage of 99.90% for Hap1 and 99.98% for Hap2. The BUSCO scores and mapping statistics confirmed the high completeness and accuracy of the assemblies (Table 2).
Genome annotation
Repetitive sequences in the ‘Huizao’ genome were annotated using both de novo and homology-based methods. A custom repeat library was built with RepeatModeler (v2.0.2a)26, RepeatScout (v1.0.6)27, and LTR_retriever (v2.9.0)28 and used by RepeatMasker (v4.1.2-p1)29 to annotate repeats in GFF format. Repetitive sequences at both the DNA and protein levels were identified by mapping to the Repbase database30 using RepeatMasker and RepeatProteinMask. Tandem repeats were annotated de novo with TRF (v4.10.0)31. In total, repetitive elements spanned 203.4 Mb (54.79%) of Hap1 and 215.3 Mb (55.87%) of Hap2, with LTRs being predominant (26.01% in Hap1, 26.77% in Hap2) (Table 3).
Protein-coding gene prediction was performed through a combination of de novo, homology-based, and transcriptome-based approaches. RNA-seq reads from leaf tissue were quality controlled and aligned to the assembled genome using STAR (v2.7.9a)32, followed by transcript assembly with StringTie (v2.1.7b)33 and structural annotation via PASA (v2.5.3)34. Protein sequences from six representative species35 (Malus domestica, Arabidopsis thaliana, Ziziphus jujuba, Prunus armeniaca, Populus, and Prunus persica) were retrieved from public NCBI databases and annotated with GeMoMa (v1.9)36. De novo gene prediction was performed using Augustus (v3.5.0)37.
The results were integrated using EVM (v2.1.0)38 with the parameters “–segmentSize 100000 –overlapSize 10000”, resulting in 32,065 protein-coding genes in Hap1 and 33,004 in Hap2. Functional annotation was carried out using InterProScan (v5.57–90.0)39 and eggNOG-mapper (v2.1.8)40, with data from TrEMBL, Swiss-Prot, InterPro, the NCBI Non-Redundant Protein Database (nr), eukaryotic orthologous groups, and Gene Ontology for comprehensive functional classification (Table 4). Except for EVM (v2.1.0), all other software were used with their default parameters.
Genome collinearity analysis
MCScan (v1.0)41 was used with default parameters to examine the collinearity between the two haplotype genomes of ‘Huizao’ jujube, with plots generated using the option ‘–minspan = 30’. A total of 50 collinear blocks were identified, encompassing 25,826 gene pairs. Of these, 78.67% of the genes were from Hap1 and 76.65% from Hap2. The genome collinearity analysis demonstrated a high degree of synteny between the two haplotype genomes (Fig. 2b).
Structural variation detection
Intra-species structural variations between the two haplotype genomes were identified using the SyRI (v1.7.0)42 pipeline with default parameters. Minimap2 (v2.28)43 was used to align the two haplotype genomes with the parameters “–eqx -ax asm5 -c –secondary=no.” The resulting SAM files were converted to BAM format, sorted, and analyzed for structural variations using the SyRI pipeline with default settings. The identified variations were classified into two categories: genomic rearrangements and sequence variations. Seven types of structural variation sites were detected, including 329 collinear regions, 48 inversions, 333 translocations, 182,766 insertions, and 182,368 deletions (Fig. 4a).

Comparative analysis. (a) Structural variations between the two haplotype genomes of ‘Huizao’. (b) Collinearity and structural variations between the two haplotypes of ‘Huizao’ and the reference genome of ‘Dongzao’.
Data Records
The genome assembly and associated raw sequencing data are available at the National Genomics Data Center (NGDC) under GSA accession numbers CRA02191344 and CRA02194745, with BioProject number PRJCA036471. The haplotype genomes of ‘Huizao’ jujube have been uploaded to the GWH database, with the assembly number GWHFIKR00000000.1 for Hap1 and GWHFIKS00000000.146 for Hap2. The annotation files have been deposited in Figshare47. In addition, the raw data have also been deposited in the National Center for Biotechnology Information (NCBI) under BioProject accession number PRJCA036471, with the sequencing data available in the SRA48 and the genome assembly in GenBank49,50.
Technical Validation
The completeness of the genome was assessed from both the assembled genome sequence and the annotated protein sequence perspectives. For genome sequence validation, we compared the two haplotype assemblies with the published T2T genome assembly of ‘Dongzao’ jujube using MUMMER (v4.0.0beta2)51 to evaluate collinearity and identify differences (Fig. 4b). Coverage was calculated using a custom Python script, yielding 99.0% for haplotype 1 and 99.8% for haplotype 2 (Table 2). Various assembly metrics, including contig N50, scaffold N50, and GC content, were also computed to assess the quality of the assembled genomes. Combined with the BUSCO results, both haplotype genomes exhibited high completeness.
Additionally, MUMMER (v4.0.0beta2) was used to compare the ‘Huizao’ haplotypes with the T2T genome assemblies of ‘Junzao’52 and ‘Dongzao’ jujube as reference genomes. The alignment was performed using nucmer with parameters (-l 100 -c 100). The resulting files were processed with delta-filter using parameters (−1 -i 98 -l 500), and the plots were generated with mummerplot (Fig. S3). These comparisons confirmed the high quality and completeness of the ‘Huizao’ genome assemblies.
Data availability
All data generated in this study, including the haplotype-resolved genome assembly, annotations, and raw sequencing reads, have been deposited in public repositories. The genome assembly and associated raw sequencing data are available at the National Genomics Data Center (NGDC) under BioProject number PRJCA036471, with GSA accession numbers CRA021913 and CRA021947. The haplotype genomes of ‘Huizao’ jujube have been deposited in the Genome Warehouse (GWH) with assembly numbers GWHFIKR00000000.1 (Hap1) and GWHFIKS00000000.1 (Hap2). The annotation files are available in Figshare (https://doi.org/10.6084/m9.figshare.29617400). In addition, the raw sequencing data have also been deposited in the National Center for Biotechnology Information (NCBI) under BioProject accession number PRJCA036471, with sequencing data available in the SRA, and the genome assemblies available in GenBank under accession numbers GCA_052692825.1 and GCA_052692835.1.
Code availability
No unpublished code was used in this study. All data processing commands were executed following the respective software manuals for the bioinformatics tools utilized.
References
Liu, M. et al. The historical and current research progress on jujube–a superfruit for the future. Hortic. Res. 7, 119, https://doi.org/10.1038/s41438-020-00346-5 (2020).
Feng, T. Functional nutrients and jujube-based processed products in Ziziphus jujuba. Molecules 29, 1234, https://doi.org/10.3390/molecules29031234 (2024).
Popstoyanova, D., Gerasimova, A., Gentscheva, G., Nikolova, S., Gavrilova, A. & Nikolova, K. Ziziphus jujuba: applications in the pharmacy and food industry. Plants 13, 2724, https://doi.org/10.3390/plants13192724 (2024).
Gao, Q. H., Wu, C. S. & Wang, M. The jujube (Ziziphus jujuba Mill.) fruit: a review of current knowledge of fruit composition and health benefits. J. Agric. Food Chem. 61, 3351–3363, https://doi.org/10.1021/jf4007032 (2013).
Lu, Y., Bao, T., Mo, J., Ni, J. & Chen, W. Research advances in bioactive components and health benefits of jujube (Ziziphus jujuba Mill.) fruit. J. Zhejiang Univ. Sci. B 22, 431–449, https://doi.org/10.1631/jzus.B2000594 (2021).
Ji, X. et al. Isolation, structures and bioactivities of the polysaccharides from jujube fruit (Ziziphus jujuba Mill.): A review. Food Chem 227, 349–357, https://doi.org/10.1016/j.foodchem.2017.01.074 (2017).
Liu, M. Chinese Jujube: Botany and Horticulture. (John Wiley & Sons, Ltd, 2010).
Liu, Y. et al. Analysis of volatility characteristics of five jujube varieties in Xinjiang Province, China, by HS-SPME-GC/MS and E-nose. Food Sci. Nutr. 9, 6617–6626, https://doi.org/10.1002/fsn3.2607 (2021).
Guo, M. et al. Analyzes of pan-genome and resequencing atlas unveil the genetic basis of jujube domestication. Nat. Commun. 15, 1, https://doi.org/10.1038/s41467-024-53718-z (2024).
Liu, M. J. et al. The complex jujube genome provides insights into fruit tree biology. Nat. Commun. 5, 5315, https://doi.org/10.1038/ncomms6315 (2014).
Yang, M. et al. Insights into the evolution and spatial chromosome architecture of jujube from an updated gapless genome assembly. Plant Commun 4, 100662, https://doi.org/10.1016/j.xplc.2023.100662 (2023).
Huang, J. et al. The jujube genome provides insights into genome evolution and the domestication of sweetness/acidity taste in fruit trees. PLoS Genet 12, e1006433, https://doi.org/10.1371/journal.pgen.1006433 (2016).
Shen, L. Y. et al. Chromosome-scale genome assembly for Chinese sour jujube and insights into its genome evolution and domestication signature. Front. Plant Sci. 12, 773090, https://doi.org/10.3389/fpls.2021.773090 (2021).
Wei, T., Li, H., Huang, X. & Yang, P. Chromosome-level genome assembly of two cultivated jujubes. Sci. Data 11, 1144, https://doi.org/10.1038/s41597-024-03992-9 (2024).
Deorowicz, S., Kokot, M., Grabowski, S. & Debudaj-Grabysz, A. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics 31, 1569–1576, https://doi.org/10.1093/bioinformatics/btv022 (2015).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research 4, 1310, https://doi.org/10.12688/f1000research.7334.1 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 https://doi.org/10.48550/arXiv.1303.3997 (2013).
Faust, G. G. & Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505, https://doi.org/10.1093/bioinformatics/btu314 (2014).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Zeng, X. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 10, 1184–1200, https://doi.org/10.1038/s41477-024-01755-3 (2024).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst 6, 256–258.e1, https://doi.org/10.1016/j.cels.2018.01.001 (2018).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245, https://doi.org/10.1007/978-1-4939-9173-0_14 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–i358, https://doi.org/10.1093/bioinformatics/bti1018 (2005).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Kohany, O., Gentles, A. J., Hankus, L. & Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7, 474, https://doi.org/10.1186/1471-2105-7-474 (2006).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467, https://doi.org/10.1159/000084979 (2005).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Jia, H. et al. PASA: identifying more credible structural variants of Hedou12. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1493–1503, https://doi.org/10.1109/TCBB.2019.2934463 (2020).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Methods Mol. Biol. 1962, 161–177, https://doi.org/10.1007/978-1-4939-9173-0_9 (2019).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Zdobnov, E. M. & Apweiler, R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848, https://doi.org/10.1093/bioinformatics/17.9.847 (2001).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122, https://doi.org/10.1093/molbev/msx148 (2017).
Tang, H. et al. JCVI: a versatile toolkit for comparative genomics analysis. Imeta 3, e211, https://doi.org/10.1002/imt2.211 (2024).
Goel, M., Sun, H., Jiao, W. B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol 20, 277, https://doi.org/10.1186/s13059-019-1911-0 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA021947 (2025).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA021913 (2025).
NGDC Genome Warehouse. https://ngdc.cncb.ac.cn/gwh/Assembly/88097/show (2025).
Yang, Y. et al. Haplotype-resolved genome assembly of the leading cultivar of jujube (Ziziphus jujuba Mill. ‘Huizao’). Figshare https://doi.org/10.6084/m9.figshare.29617400 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP614374 (2025).
Yang, Y. et al. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_052692825.1 (2025).
Yang, Y. et al. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_052692835.1 (2025).
Delcher, A. L., Salzberg, S. L. & Phillippy, A. M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10, Unit 10.13, https://doi.org/10.1002/0471250953.bi1003s00 (2003).
Li, K. et al. Haplotype-resolved T2T reference genomes for wild and domesticated accessions shed new insights into the domestication of jujube. Hortic. Res. 11, uhae071, https://doi.org/10.1093/hr/uhae071 (2024).
Acknowledgements
This work was supported by the general program from the National Natural Science Foundation of China (NO.32171817); the general program from the Natural Science Foundation of Hebei Province, China (NO. C2022204030); High-level talent project of Hebei Province.
Author information
Authors and Affiliations
Contributions
M.L. and M.Y. designed and managed the project; M.Y. and Y.Y. assembled the genome and annotated the genes; Y.Y., S.Z., Y.L., Z.Z., D.L., J.L., J.G. and J.S. performed the other bioinformatics analyses; S.Z., Y.L., Z.Z., D.L., J.L., J.G., J.S. and Q.H. collected and prepared the materials; Y.Y. drafted the manuscript; M.L. and M.Y. revised manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, Y., Zhang, S., Lan, Y. et al. Haplotype-resolved genome assembly of the leading cultivar of jujube (Ziziphus jujuba Mill. ‘Huizao’). Sci Data 12, 1800 (2025). https://doi.org/10.1038/s41597-025-06096-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06096-0
