
Abstract
Agroathelia rolfsii (A. rolfsii) is a globally distributed soilborne fungal pathogen that infects a wide range of crops, causing basal stem rot and yield losses. In this study, we report a high-quality genome assembly of A. rolfsii strain LC1 (43.3 Mb, N50 = 3.3 Mb, GC content = 46.3%) using integrated PacBio long-read and Illumina short-read sequencing technologies. Genome annotation identified 8,826 protein-coding genes, with 30.93% comprising repetitive sequences. Benchmarking Universal Single-Copy Ortholog (BUSCO) analysis demonstrated 97.7% genome completeness, supported by a high QV value of 44, with functional annotations covering 91.1% of the genes. A total of 1,260 carbohydrate-active enzyme (CAZyme) genes, predominantly glycoside hydrolases (37.7%), and 22 secondary metabolite biosynthetic gene clusters were identified, indicating strong host tissue degradation capability and diverse virulence factors. Phylogenetic analysis supported genetic diversity within the Agroathelia genus. This study provides key genomic resources for understanding the pathogenic mechanisms, host interactions, and resistance gene discovery of A. rolfsii, laying the foundation for the development of virulence-targeted control strategies.
Similar content being viewed by others
Background & Summary
The fungal pathogen Agroathelia rolfsii poses a significant threat to global agriculture, particularly in tropical and subtropical regions1. It affects over 500 plant species, including economically important crops such as peanut, tomato, and cotton2,3,4. Its asexual stage, Sclerotium rolfsii, is characterized by the production of highly resilient sclerotia, which can survive under adverse environmental conditions5. Once infected, plants exhibit basal stem and root rot, leading to wilting and, in severe cases, total crop failure. Yield losses can reach to 30% ~50%, making A. rolfsii a critical target for plant disease management research6,7.
While chemical fungicides, such as tebuconazole and flutolanil, have been used to control A. rolfsii, their prolonged application has raised concerns over environmental pollution and the development of fungicide resistance8,9. Biological control strategies, including the use of antagonistic microorganisms like Trichoderma spp., have shown potential but may lack consistency and universal applicability in field conditions10. Furthermore, the pathogen’s broad host range and strong ecological adaptability complicate efforts to develop effective and sustainable control measures.
In recent years, the rapid advancement of high-throughput sequencing technologies has provided powerful tools for studying fungal pathogens. Genomic studies have revealed virulence-related gene families in many plant-pathogenic fungi, including glycoside hydrolases, peptidoglycanases, and secondary metabolite biosynthetic gene clusters, which play key roles in host infection and tissue colonization11,12. Comparative genomic analyses have also provided insights into the genetic basis of pathogenicity, host adaptation, and environmental resilience, helping to identify key genes involved in fungal survival and infection strategies13. The emergence of genome-editing technologies, such as CRISPR-Cas9, has opened new possibilities for enhancing plant resistance by modifying key susceptibility genes14,15. Additionally, genomic information has facilitated the identification of novel targets for disease control, including genes essential for fungal virulence and survival16.
Studies on closely related pathogens, such as Sclerotium delphinii, which causes southern blight in Dendrobium officinale, provide additional context17. Genomic analyses of S. delphinii have revealed a significantly higher abundance of glycoside hydrolases (GHs) compared to A. rolfsii, which may explain its enhanced virulence. Furthermore, the identification of secondary metabolite gene clusters and pathogen-host interaction (PHI) factors in S. delphinii highlights the diverse strategies employed by fungal pathogens during infection. These findings emphasize the importance of comparative studies in understanding the molecular basis of fungal pathogenicity.
Here, we assembled a high-quality genome of A. rolfsii using an integrated approach that combined PacBio Single Molecule Real-Time (SMRT) long-read sequencing and Illumina short-read sequencing technologies. The quality of the genome assembly and annotation was evaluated based on the contiguity of assembled sequences (N50) and the completeness of conserved protein-coding genes. Protein functional annotations were performed by aligning the sequences against multiple databases, including Nr, SwissProt, KEGG, and KOG, ensuring comprehensive coverage. Furthermore, evolutionary insights were gained through comparative analyses with genomes of other Agroathelia species. This study provides a valuable genomic resource for advancing research on the pathogenicity, genetic diversity, and evolutionary dynamics of the Agroathelia genus.
Methods
Fungal strains
The A. rolfsii strain LC1 was isolated from the leaf of Coptis chinensis, and is stored at the Institute of Chinese Herbal Medicines, Hubei Academy of Agricultural Sciences, China. For fugal growth, the strain was incubated on solid potato dextrose agar (PDA) at 28 °C in the dark (Fig. 1).

Disease symptoms caused by Agroathelia rolfsii strain LC1 on field and potted Coptis chinensis, and colony morphology of LC1 on PDA. (a) Southern blight symptoms on field-grown C. chinensis infected by strain LC1. Dense white mycelia and sclerotia are observed on the left, accompanied by plant death, while healthy leaves appear on the right. (b, c) Pathogenicity assay on potted C. chinensis. (b) Mock-inoculated control plants at 10 days post-inoculation (dpi), showing no symptoms. (c) Inoculated plants at 10 dpi showing severe wilting and death. (d–f) Colony morphology of A. rolfsii strain LC1 on PDA at different time points: (d) 4 dpi, (e) 8 dpi, and (f) 10 dpi, with visible sclerotia formation.
DNA extraction and genome sequencing
Total genomic DNA was extracted using the GP1 method (Novogene, Beijing, China) following the manufacturer’s instructions. The purity of the DNA was assessed using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), and DNA concentration was quantified using the Qubit® DNA Assay Kit with a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). For Illumina short-read sequencing, a DNA library with a 350 bp insert size was prepared using the TruSeq Nano DNA HT Sample Preparation Kit (Illumina, USA) in accordance with the manufacturer’s recommendations. Sequencing was performed on the Illumina HiSeq X Ten platform with a paired-end read length of 2 × 150 bp, generating 2.3 Gb of Illumina data, corresponding to approximately 53 × genome coverage. For PacBio long-read sequencing, a sequencing library was constructed and sequenced on the Sequel II System platform. High-quality long reads were obtained using the Guppy basecalling software with default parameters, yielding 8.06 Gb of PacBio sequencing data, which represents approximately 186 × genome coverage.
Genome assembly
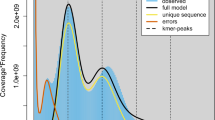
The genome size of A. rolfsii was first inferred from a 15-mer distribution using jellyfish software (v2.3.0) based on Illumina paired-end reads18, and the genome size was estimated to be 41.18 Mb (Fig. 2a; Table 1). For de novo genome assembly, PacBio long reads corrected with Falcon (v1.8.7)19 were assembled to generate an initial assembly by Wtdbg2 with the following parameters: -x sq and -g 44 m. The PacBio sequencing data provides significant advantages for genome assembly due to its long-read lengths, which simplify the complexity of assembly algorithms by enabling overlap-based assembly. To further enhance the accuracy and completeness of the assembly, Pilon (v1.23)20 was utilized to perform three iterative rounds of base correction using high-quality Illumina short reads with default settings. To the end, the assembled genome of A. rolfsii was found to be 43.3 Mb in length, with a Contig N50 of 3.3 Mb and a GC content of 46.3% (Fig. 2b; Table 1). The assembled genome consists of 50 Contigs (Table 1).

Genomic features of the A. Agroathelia isolate. (a) Genome survey based on 21-mer frequency distribution of base error-corrected reads. (b) Circos plot of the high-quality genome assembly. Tracks from outer to inner circle represent: contigs (n = 50), gene density (20-kb window), transposon density, simple repeat density, GC content (red above and green below the genome average), and syntenic blocks among contigs.
Genome annotation
Using RepeatModeler v1.0.8 (http://www.repeatmasker.org/RepeatModeler/), a de novo repeat library was generated and subsequently combined with the Repbase library v23.09 (http://www.girinst.org/repbase/). The merged library was then used for repeat identification with RepeatMasker v4.1.2-p1. This analysis identified 13.38 Mb of repetitive sequences, which accounted for 30.93% of the genome. Among these, long terminal repeats (LTRs; 27.56%) and DNA elements (1.6%) were the most abundant repetitive components, with unclassified repeats also making a significant contribution.
The repeat-masked A. rolfsii genome was annotated using Funannotate (http://github.com/nextgenusfs/funannotate), an automated pipeline designed for fungi but also applicable to other eukaryotes. Protein evidence was incorporated from curated UniProtKB/SwissProt datasets included in Funannotate. Gene prediction employed multiple tools integrated within the pipeline, including including Augustus v3.3.321, GeneMark-ES v4.3222, SNAP v2006-07-2823, and GlimmerHMM v3.0.424 (all ab initio predictors), as well as CodingQuarry v2.025, which utilizes transcriptome data for gene model inference. The combined results yielded a consensus gene model comprising 8,826 protein-coding genes, with an average length of 2,504 bp and a maximum length of 15,609 bp. A total of 7,907 (89.6%) of which were successfully annotated in at least one database (Table 2). Similarly, we utilized several complementary software tools to predict non-coding RNAs in the A. rolfsii genome, including RNAmmer for rRNA prediction, tRNAscan-SE for identifying tRNA regions and secondary structures, and cmscan (with default parameters) for detecting sRNAs and miRNAs by aligning sequences to the Rfam database. A total of 209 ncRNAs were identified in the A. rolfsii genome, including 160 tRNAs, 41 rRNAs (13 5S, 12 28S, and 16 18S), 1 sRNA, and 7 miRNAs.
Identification of CAZymes and biosynthetic gene cluster
The putative carbohydrate-active enzymes (CAZymes) in A. rolfsii were annotated using the dbCAN server (dbCAN HMMs 5.0)26, applying thresholds of an e-value lower than 1e-5 and at least 70% sequence coverage. CAZymes were categorized based on the modules outlined in the CAZyme database classification (http://www.cazy.org). The A. rolfsii genome encodes a total of 1,260 genes for putative CAZymes, which are distributed across six functional categories. Among these, glycoside hydrolases (475 genes, 37.7%) were the most prevalent, followed by glycosyltransferases (331 genes, 26.3%) (Table 3). Polysaccharide lyases were the least represented, with only one gene identified (Table 3). To explore secondary metabolism, the antiSMASH 4.0 tool was employed for identifying gene clusters within the A. rolfsii genome. A total of 22 clusters associated with secondary metabolites were predicted, including four terpene clusters, three type I polyketide synthase clusters, and one non-ribosomal peptide synthase cluster (Fig. 3a).

Identification of carbohydrate-active enzymes (CAZymes) and biosynthetic gene clusters (BGCs), and phylogenetic analysis. (a) Distribution of CAZyme and BGC families. (b) Phylogenetic analysis of nine species based on single-copy orthologous genes.
Data Record
All sequencing data, including the raw PacBio long reads and Illumina short reads, have been deposited in the Genome Sequence Archive (GSA27) in National Genomics Data Center28 under BioProject PRJCA042508. The PacBio raw data are available under accession CRA02743029, and the Illumina raw data under CRA02741730. The assembled genome has been made available in GenBank with JBQWDA00000000031. Additionally, the annotation data has been deposited at the Figshare repository32.
Data Overview
Protein sequences of 9 genomes were used for comparative genomics and phylogenomic analyses: A. rolfsii (this study), A. delphinii SD strain (GCA_041475395.1), A. rolfsii GP3 isolate (GCA_018343915.1), A. rolfsii Sr-RCBB strain (GCA_002940785.1), A. rolfsii CCF1 strain (GCA_036172465.1), A. rolfsii ZY isolate (GCA_018343895.1), A. rolfsii MR10 strain (GCA_000961905.2), Fusarium oxysporum Fo5176 strain (GCA_030345115.2), Fusarium solani F421 strain (GCA_040436825.1). The OrthoFinder (v2.5.1)33 with default settings was used to identify orthologous and paralogous genes. All protein sequences were aligned with MAFFT (v7.123b)34 and trimmed by trimAl (v1.4.rev22)35. The sequences were concatenated into a gene matrix and used for phylogenetic analysis (Fig. 3b). A phylogenetic tree was constructed by maximum likelihood (ML) method using IQ-TREE (v2.1.2)36, with 1,000 ultrafast bootstrap replicates. The best model (JTT + F + R6) was determined by ModelFinder37 and two Fusarium species were used as outgroups. A total of 758 single-copy orthogroups were identified and extracted for phylogenetic analysis (Fig. 3b). Among the analyzed strains, F. oxysporum and F. solani exhibit the highest gene counts, with 11,419 and 10,495 genes, respectively, forming an outgroup relative to the Agroathelia species. The Agroathelia rolfsii strain analyzed in this study contains 6,921 genes and clusters closely with A. delphinii (SD strain, 9,877 genes). Within A. rolfsii, notable gene count variations are observed, ranging from 4,051 genes in the GP3 isolate to 10,816 genes in the ZY isolate.These results highlight substantial genomic diversity within A. rolfsii, which may be attributed to differences in ecological adaptation, host specificity, or pathogenicity potential.
Technical Validation
Quality control of PacBio long reads was performed using Chopperv1.5.0 (http://gitcode.com/gh_mirrors/cho/chopper), resulting in 606,486 reads with an N50 length of 26 kb. Illumina short reads were processed using Fastp38 to remove low-quality reads, generating a total of 15,321,002 high-quality reads for subsequent analysis. The completeness of the genome was assessed using BUSCO (Benchmarking Universal Single-Copy Orthologs) v5.5.0 with the basidiomycota_odb10 database (genome mode)39. The evaluation yielded the following BUSCO statistics: 97.7% complete, 96.8% complete single-copy, 0.3% duplicated, 0.6% fragmented, and 2.3% missing (Table 4). The annotated and integrated proteins were further evaluated using BUSCO v5.5.0 with the basidiomycota_odb10 database (protein mode), which showed a complete BUSCO score of 88.6% (Table 5). Additionally, 89.6% of the genes were functionally annotated across four major databases (Nr, SwissProt, KEGG, and KOG) (Table 2), and future improvements will involve refining the genome annotation with full-length transcriptome data using the cDNA_Cupcake toolkit (https://github.com/Magdoll/cDNA_Cupcake). The genome assembly quality value (QV value) was assessed using Merqury (http://github.com/marbl/merqury), a k-mer-based tool designed for evaluating genome assembly accuracy and completeness without a reference genome. The process involved comparing the k-mers (specifically 21-mers) from short-read sequencing data with those from the assembled genome. The assessment yielded a high QV value of 44, reflecting the high base accuracy of the assembly.
Data availability
All sequencing data generated in this study have been deposited in public repositories. The raw PacBio long reads are available in the Genome Sequence Archive (GSA)27 at the National Genomics Data Center28 under accession number CRA027430 (BioProject PRJCA042508)29. The raw Illumina short reads are available under accession number CRA02741730. The assembled genome has been deposited in GenBank under accession number JBQWDA00000000031. The annotation data are accessible at the Figshare repository32.
Code availability
This study did not involve the use of custom scripts or customized command lines. All comparative analyses were conducted using publicly available software. Detailed information on the software versions and parameters can be found in the Methods section. For any software tools where specific parameters are not specified, default settings were applied.
References
Patra, G. K., Acharya, G. K., Panigrahi, J., Mukherjee, A. K. & Rout, G. R. The soil-borne fungal pathogen Athelia rolfsii: past, present, and future concern in legumes. Folia Microbiol (Praha) 68, 677–690, https://doi.org/10.1007/s12223-023-01086-4 (2023).
Shukla, V., Kumar, S., Tripathi, Y. N. & Upadhyay, R. S. Bacillus subtilis- and Pseudomonas fluorescens-Mediated Systemic Resistance in Tomato Against Sclerotium rolfsii and Study of Physio-Chemical Alterations. Front Fungal Biol 3, 851002, https://doi.org/10.3389/ffunb.2022.851002 (2022).
Wei, X., Langston, D. B. Jr. & Mehl, H. L. Comparison of Current Peanut Fungicides Against Athelia rolfsii Through a Laboratory Bioassay of Detached Plant Tissues. Plant Dis 106, 2046–2052, https://doi.org/10.1094/pdis-12-21-2789-re (2022).
Yi, R. H., Liao, H. Z., Li, K. Y. & Feng, F. Fruit rot causing by Athelia rolfsii (Sclerotium rolfsii) on jackfruit (Artocarpus heterophyllus) in China. Plant Dis, https://doi.org/10.1094/pdis-09-22-2250-pdn (2023).
Yu, D. et al. Progress on pathogenicity differentiation in Sclerotium rolfsii isolates from peanut. Chinese Journal of Oil Crop Sciences 44, 930–936 (2022).
Kumar, V. & Thirumalaisamy, P. Diseases of groundnut. Disease of field crops and their management. Indian Phytopathological Society, Today and Tomorrow’s Printers and Publishers, New Delhi, 459-487 (2016).
Chen, K. et al. Research progress on peanut southern stem rot caused by Sclerotium rolfsii. Chin J Oil Crop Sci 40, 302–308 (2018).
Shim, M. Y., Starr, J. L., Keller, N. P., Woodard, K. E. & Lee, T. A. Jr. Distribution of Isolates of Sclerotium rolfsii Tolerant to Pentachloronitrobenzene in Texas Peanut Fields. Plant Dis 82, 103–106, https://doi.org/10.1094/pdis.1998.82.1.103 (1998).
Franke, M. D., Brenneman, T. B., Stevenson, K. L. & Padgett, G. B. Sensitivity of Isolates of Sclerotium rolfsii from Peanut in Georgia to Selected Fungicides. Plant Dis 82, 578–583, https://doi.org/10.1094/pdis.1998.82.5.578 (1998).
Singh, S. et al. Harnessing Trichoderma Mycoparasitism as a Tool in the Management of Soil Dwelling Plant Pathogens. Microb Ecol 87, 158, https://doi.org/10.1007/s00248-024-02472-2 (2024).
Li, Y. et al. Fungal acetylome comparative analysis identifies an essential role of acetylation in human fungal pathogen virulence. Commun Biol 2, 154, https://doi.org/10.1038/s42003-019-0419-1 (2019).
Deng, K., Zhang, Y., Lv, S., Zhang, C. & Xiao, L. Decoding Pecan’s Fungal Foe: A Genomic Insight into Colletotrichum plurivorum Isolate W-6. Journal of Fungi 11, 203 (2025).
Xia, C. et al. Current Status and Future Perspectives of Genomics Research in the Rust Fungi. Int J Mol Sci 23, https://doi.org/10.3390/ijms23179629 (2022).
Song, R. et al. CRISPR/Cas9 genome editing technology in filamentous fungi: progress and perspective. Appl Microbiol Biotechnol 103, 6919–6932, https://doi.org/10.1007/s00253-019-10007-w (2019).
Xie, Z. et al. Genome editing in the edible fungus Poria cocos using CRISPR-Cas9 system integrating genome-wide off-target prediction and detection. Front Microbiol 13, 966231, https://doi.org/10.3389/fmicb.2022.966231 (2022).
Case, N. T. et al. Fungal impacts on Earth’s ecosystems. Nature 638, 49–57, https://doi.org/10.1038/s41586-024-08419-4 (2025).
Chen, Y. J. et al. Whole-genome sequence of Sclerotium delphinii, a pathogenic fungus of Dendrobium officinale southern blight. Genomics 116, 110932, https://doi.org/10.1016/j.ygeno.2024.110932 (2024).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054, https://doi.org/10.1038/nmeth.4035 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res 42, e119, https://doi.org/10.1093/nar/gku557 (2014).
Korf, I. Gene finding in novel genomes. BMC bioinformatics 5, 59 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879, https://doi.org/10.1093/bioinformatics/bth315 (2004).
Testa, A. C., Hane, J. K., Ellwood, S. R. & Oliver, R. P. CodingQuarry: highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genomics 16, 170, https://doi.org/10.1186/s12864-015-1344-4 (2015).
Zheng, J. et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res 51, W115–w121, https://doi.org/10.1093/nar/gkad328 (2023).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Members, C.-N. & Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res 51, D18–D28, https://doi.org/10.1093/nar/gkac1073 (2023).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA027430 (2025).
National Genomics Data Center https://ngdc.cncb.ac.cn/gsa/browse/CRA027417 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_052928445.1 (2025).
You, J. et al. High-Quality Genome Assembly and Annotation of Athelia rolfsii LC-1. figshare https://doi.org/10.6084/m9.figshare.30018793.v1 (2025).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20, 238, https://doi.org/10.1186/s13059-019-1832-y (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780, https://doi.org/10.1093/molbev/mst010 (2013).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973, https://doi.org/10.1093/bioinformatics/btp348 (2009).
Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37, 1530–1534, https://doi.org/10.1093/molbev/msaa015 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14, 587–589, https://doi.org/10.1038/nmeth.4285 (2017).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This study was supported by the Hubei Key Research and Development Program [2024BBB090]; the Science Research Program of Key Laboratory of Biology and Cultivation of Chinese Herbal Medicines, Ministry of Agriculture and Rural Affair [BZ2024001]; the Advantages Discipline Group (Biology and Medicine) Project in Higher Education of Hubei Province (2021–2025) [2022BMXKQT4] at Hubei University of Medicine; the National Nature Science Foundation of China [32200680]; the Science Research Program of Hubei Provincial Department of Education [T2023016]; the Joint supported by Hubei Provincial Natural Science Foundation and Shiyan-of China [2025AFD179, 2025AFD205, 2025AFD172] and the Young Top-notch Talent Cultivation Program of Hubei Province.
Author information
Authors and Affiliations
Contributions
J.Y. and Y.Z. designed the study; L.Y., T.T., F.W. and H.H. assembled, annotated and analyzed the genome; J.Y., Y.D., X.W. and W.Z. drafted the manuscript; Y.Z. and L. Z. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
You, J., You, L., Tang, T. et al. The high-quality genome sequence of Agroathelia rolfsii (Syn. Sclerotium rolfsii), the causal agent of southern blight in Coptis chinensis. Sci Data 12, 1833 (2025). https://doi.org/10.1038/s41597-025-06137-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-06137-8
