
Abstract
Research on CAR-T cell states is crucial for understanding the mechanisms of immunotherapy. Previous studies in live cells have been primarily limited by phototoxicity, resolution, and throughput, making it difficult to conduct further research and observations on cell states. To enable more detailed studies of cell states, we developed a microscopy imaging system with subcellular resolution, low phototoxicity, high imaging throughput, and automated data reconstruction. Using this system, we have generated and shared over 400 image sets that capture the cytotoxic effects of CAR-T cells on tumor cells. The data provide an isotropic spatial resolution of 320 nm, a temporal resolution of up to 2.5 seconds per volume, and long-term observations spanning up to 5 hours. This study reports an imaging system that fills an essential gap in the field, offers valuable insights into the cytotoxic processes of CAR-T cells, and significantly advances research in this area.
Similar content being viewed by others


Background
The efficacy of T-cell-based immunotherapies, including CAR-T-cell therapy, and the progression of immune diseases are closely dictated by the functional state of T cells1,2,3. Monitoring the dynamic interactions between CAR-T cells and tumor cells, particularly the formation and modulation of immune synapses, provides critical insights for elucidating T-cell physiology and optimizing clinical outcomes4.
The rapid dynamics of CAR-T cell synapses demand imaging techniques that combine high temporal resolution with minimal phototoxicity to avoid perturbing normal cellular physiology, which could otherwise lead to the misinterpretation of biological phenomena5. Despite recent advancements, the field still lacks high-resolution, long-term, and large-scale cellular imaging datasets necessary for deeper mechanistic insights. While traditional optical microscopy, such as confocal microscopy, enables the observation of subcellular structures4, it is often limited by low temporal resolution, slow volumetric acquisition, and significant phototoxicity that compromises cell viability during long-term experiments.
To overcome these limitations, light-sheet fluorescence microscopy (LSFM)4,6 has emerged as a preferred method due to its low phototoxicity and rapid 3D imaging capabilities6,7,8. However, because the primary subjects are often suspended cells, sample mounting remains a significant challenge for dual-objective LSFM designs, frequently restricting studies to individual cells and hindering long-term observations of large-scale CAR-T and target cell interactions9.
To address these challenges, Oblique Plane Microscopy (OPM)10,11,12,13,14 utilizes a single objective for both illumination and detection, allowing for a traditional and user-friendly sample mounting format. In this study, we developed a high-throughput Bessel Oblique Plane Microscopy (HBOPM) platform. By combining the OPM configuration with dual-ring Bessel light-sheet illumination, we extended the effective Rayleigh range and integrated microfluidic technology. The resulting system retains the low-phototoxicity advantages of LSFM while delivering high-throughput volumetric imaging with an isotropic subcellular resolution of ~320 nm. This approach enables the continuous, large-scale observation of over 400 interacting CAR-T and target cell pairs for durations exceeding 5 hours, providing a robust, high-resolution dataset for the study of cellular dynamics.
Methods
Cell lines and cell culture
The acute B-lymphocytic leukemia cell line Nalm6 (catalog No. CRL-3273) was cultured in RPMI 1640 medium (Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA). The lentivirus packaging cell line Lenti-X™293 T (catalog No. CRL-3216) was cultured in Dulbecco’s modified Eagle medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% FBS. Both cell lines were obtained from the American Type Culture Collection (ATCC) and were authenticated by short tandem repeat (STR) analysis before use. Furthermore, all cells were tested and confirmed to be negative for mycoplasma.
Recombinant plasmid construction and lentivirus packaging
The anti-CD19 CAR gene was constructed by a series of connected FMC 63 single-chain variable fragments (scFvs), a CD8a hinge/transmembrane region, a CD28 costimulatory domain, and an intracellular CD3ζ chain, which was followed by a T2A sequence and a Lifeact-EGFP gene in sequence. mTagRFP-Membrane-1 was purchased from Addgene (# 57992), and we replaced mTagRFP with mApple to generate the mApple-Mem plasmid for marking Nalm6 cells. Recombinant plasmids were extracted via the EndoFree Plasmid Maxi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. A lentivirus was generated via Lipo3000 (Invitrogen, Waltham, MA, USA) through transient transfection of Lenti-X293T cells with expression vectors (CD19 CAR-Lifeact-EGFP or mApple-Mem) and psPAX2 as well as PMD2.G packaging plasmids. The viral supernatants were collected 48 hours after transfection, filtered through 0.45 μm filters, concentrated via ultracentrifugation (Avanti J-26S XPI), aliquoted and stored at −80 °C.
Isolation, activation, transduction and culture of T cells
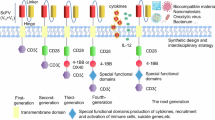
Peripheral blood mononuclear cells (PBMCs) from healthy donors were purchased from Milestone Biotechnologies, China. The cells were further isolated via density gradient centrifugation over Ficoll-Paque (GE Healthcare, Boston, MA, USA). Then, bulk CD3 + T cells (Fig. 1a) were separated from PBMCs via CD3 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions and subsequently stimulated with Dynabeads™ Human T-Activator CD3/CD28 (Gibco, Grand Island, NY, USA) at a 1:1 ratio in CTS™ OpTmizer™ medium (Gibco, Grand Island, NY, USA) containing 2 mM L-glutamine (Gibco, Grand Island, NY, USA), 5% human AB serum and 200 IU/ml rhIL-2 (PeproTech, Rocky Hill, NJ, USA). Within 24 h, the T cells were transduced with concentrated lentivirus at a multiplicity of infection (MOI) ranging from 2 to 5. Twenty-four hours later, the T cells were centrifuged and resuspended in fresh culture medium at a density of 1–2 × 106 cells/mL and then maintained at 37 °C and 5% CO2. The CAR transduction efficacy was tested by flow cytometry according to the percentage of GFP-positive cells 5 days after the addition of virus. Imaging and functional assays were performed after 12 days of culture in vitro.

System flow. (a) Acquire CAR-T cells. CAR-T cells (green) and Nalm6 cells (red) were then mixed at a 1:1 ratio on the chip. (b) Microchip fabrication process. After obtaining the silicon-based mask through photolithography, the microchamber chip is created by UV exposure of Bio-133 resin. (c)Using high-throughput Bessel oblique plane microscopy, we obtained 60x high-throughput 3-dimensional temporal sequence light sheet imaging. (d–g) The 3D sequence images acquired by OPM were subsequently reconstructed to reveal normal cell volume morphology.
Construction of mApple-Mem-expressing Nalm6 cell lines
Nalm6 cells were transduced with concentrated mApple-Mem lentivirus at an MOI of 30 to 50 for 4 hours. mApple-positive cells were sorted 5 days after infection via a Moflo XDP flow cytometer (Beckman Coulter) to obtain mApple-Mem-expressing Nalm6 cell lines.
Dasatinib treatment
For experiments on the effects of dasatinib, a drug reported to induce a function-off state in CAR-T cells, on key subcellular events during CAR-T cell-mediated killing of target cells, CAR-T cells were pretreated with 50 nM dasatinib (Selleck, Shanghai, China) for 24 hours15. Owing to the reversible effect of dasatinib, 50 nM dasatinib was also added to all subsequent staining, imaging, and other experimental solutions.
CAR-T-cell labeling
For labeling of the microtubules, CAR-T cells were stained with a SiR-tubulin probe (SpiroChrome, Switzerland) at a final concentration of 2 μM, incubated for 1 hour in a humidified 5% CO2 incubator at 37 °C, washed twice with warm phosphate buffer (PBS) and resuspended in an imaging solution consisting of phenol red-free 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% FBS, 25 mM N-2-hydroxy-ethylpiperazine-N′-2-ethanesulfonic acid (HEPES, Gibco, Grand Island, NY, USA), 100 U/mL penicillin and streptomycin (Gibco, Grand Island, NY, USA), and 1 μM SYTOX™ Blue stain (Invitrogen, Waltham, MA, USA).
Microchip preparation
Rationale and material selection
To facilitate parallel observation, repetitive positioning, and continuous tracking of large-scale cell populations, we developed a specialized open-top microchamber array chip. Unlike tissues, organoids, or adherent cells, suspended cells such as CAR-T and Nalm6 cells are prone to drifting or aggregating. Previous OPM imaging methods, such as those utilizing 96-well plates or fixed samples, struggle to prevent cell drift while maintaining high axial resolution. Furthermore, while PDMS is a common material for microfluidics, the refractive index mismatch between PDMS and the cell culture medium leads to significant aberrations and reflections at the interface. To overcome these challenges, we fabricated the microchambers using Bio-13316, a UV-curable resin with a refractive index (RI \(\approx \) 1.33) that matches water. This index-matching compatibility effectively eliminates spherical aberrations during light-sheet acquisition.
Design parameters
The chip consists of a 45 \(\times \) 45 array (totaling 2,025 units) of cylindrical microchambers (Fig. 1b). Each chamber features a diameter and depth of 50 µm. To ensure optimal light-sheet penetration and high-quality imaging, the thickness of the chamber floor was strictly controlled to be less than 10 µm.
Fabrication process
A 50 µm layer of SU-8 photoresist was spin-coated onto a 2-inch silicon wafer and patterned via UV exposure to create a master mold. Bio-133 resin was then applied to the mold, leveled with a coverslip, and cured under UV light. Following curing, the chip was carefully demolded and soaked in anhydrous ethanol for over 24 hours to remove residual adhesives and ensure biocompatibility17.
Mathematical modeling of cell distribution
The number of cells entering each microchamber follows a Poisson distribution, as the event of a cell falling into a chamber conforms to the probabilistic model of a low-probability event repeated across a massive population. The probability mass function is defined as:
where k is the actual number of cells in a microchamber and $\lambda$ is the expected number of cells per chamber.
Theoretically, when \(\lambda \) = 1, P (X = 1) reaches a maximum value of \(1/e\). When both CAR-T and target cells are seeded to satisfy a Poisson distribution with \(\lambda \) = 1, the probability of achieving an ideal 1:1 pairing reaches a maximum of \(1/{e}^{2}\approx 13.5 \% \) (or approximately 13.6% under optimal operation). Given the 2,025 chambers on a single chip, the expected yield of paired chambers is approximately 273. This mathematical framework allows for the calculation of appropriate cell concentrations required for experimental setup.
Automated identification and imaging
Because ideal pairings occur in only a fraction of the chambers, manual selection is labor-intensive and susceptible to subjective bias. Consequently, we developed an integrated intelligent recognition18 and imaging control algorithm. The system performs a low-magnification wide-field scan to automatically locate and score high-quality cell pairs, subsequently driving the motorized stage for high-throughput 3D volumetric imaging. This automated pipeline ensures objective data collection and maximizes the efficiency of the HBOPM system19.
High-throughput Bessel oblique plane microscopy
Oblique plane light-sheet microscope (OPM) setup
We constructed a single-objective system using an Olympus IX83 microscope, which facilitates various sample operations (Fig. 1c). The microscope frame was equipped with a motorized sample stage (U-780, Physik Instrumente) and a live cell workstation (STXG-WSKMX3WX-SET, TokaiHit). For light sheet illumination, a 10X (0.3 NA) objective collimated the four-color laser sources (Cobolt, 405/488/561/633 nm). The laser beam was expanded by a pair of beam expanders (AC254-050-A-ML, Thorlabs; MAD1-A, Lbtek) and then shaped by a cylindrical lens (ACY254-100-A, Thorlabs) to form a Gaussian light sheet. The light sheet passed through a custom-designed dual-ring mask, generating a Bessel beam with low side lobes and a long Rayleigh range. The beam was then reflected by a dichroic mirror (Di03-R405/488/561/635-t1-25 × 36, Semrock) and guided through lens combinations (TTL200MP, SM250*2, AD529-A). The light exits through a scan lens (CLS-SL, Thorlabs) and a galvanometer (GVS012, Thorlabs), where it enters the IX83 frame. The light sheet illumination was then directed onto the sample through the primary objective (O1, 60X, UPLSAPO60XS2, Olympus). The sample fluorescence was collected through the same objective and passed through a set of relay lenses and an additional objective (O2, UPLXAPO40X 0.95, Olympus), creating a 1.05x imaging system. An objective (O3, 60X, custom-designed, Special Optics) corrected the 30° inclined plane between O2 and O3, conjugating the sample plane to the focal plane of O2. The light then passed through O3 and a tube lens (Tl400, Thorlabs) and was split by dichroic mirrors to simultaneously image on the Kinetic camera and the Hamamatsu camera connected via a W-view adapter, capturing three fluorescence channels simultaneously.
Using the light sheet imaging method, we performed long-term, nanoscale observations of live cells. The 10X wide-field images captured are used in an algorithm we designed to automatically score and record cell coordinates, prioritizing high-scoring cells for 60X light sheet imaging. Wide-field imaging uses the native wide-field optical pathway of the microscope, employing a 10x objective to image larger fields of view for quickly locating regions of interest19.
Design and optimization of the double-ring mask
To overcome the inherent trade-off between axial resolution and the Rayleigh range (effective FOV) in conventional Gaussian light-sheets, we implemented a non-diffractive Bessel beam approach20. While standard single-ring masks produce significant side-lobes that increase phototoxicity, our double-ring optical intensity modulation strategy utilizes destructive interference between two coaxial Bessel beams to suppress these secondary structures21,22.
The mask’s transmittance function, \({T}_{{mask}}\), is defined by four radial parameters: outer radii (\({r}_{{omax}},{r}_{{omin}}\)) and inner radii (\({r}_{{imax}},{r}_{{imin}}\)). The modulated field \({U}_{1}\) is given by:
where \({U}_{0}\) represents the incident Gaussian field. Using MATLAB simulations based on scalar diffraction theory, we optimized these parameters to achieve ultrathin light-sheets with a tunable axial extent of 0.5–1.5 μm and a suppressed side-lobe ratio below 40%. For CAR-T and target cell interactions (approx. 20 μm height), the configuration ensures a robust Rayleigh range exceeding 40 μm at a 30° illumination angle.
Sample loading and microchamber seeding
Medium preparation
The sterile water within the microfluidic chip was replaced with pre-warmed T-cell culture medium, and 500 µL of medium was retained in the confocal dish to ensure the chip remained fully immersed.
Cell seeding
A 60 µL aliquot of CAR-T cell suspension at a density of \(2\times {10}^{6}/m{\rm{L}}\) was introduced into the dish and allowed to settle into the microchambers via gravitational sedimentation for 10 minutes. An equivalent volume of target cells was subsequently added following the same protocol.
Density conversion and monitoring
To account for the transduction efficiency inherent in CAR-T cell preparation, cell counts were adjusted based on the transduction rate to ensure that the number of functional cells within the microchambers met experimental requirements. Theoretically, the cellular distribution within the chambers conforms to the aforementioned Poisson distribution.
Multi-scale temporal strategy
CAR-T cell-mediated cytotoxicity manifests across three distinct temporal scales: Scale I (Seconds): Rapid dynamics such as retrograde actin flow. Scale II (Minutes): Global morphological transitions, including the formation of the immunological synapse (IS). Scale III (Hours): The complete immune cycle from recognition to target cell lysis. To observe these scales across a 5-hour duration while preserving cellular viability, we implemented a non-uniform temporal sampling strategy. For each ROI, we acquired 6 volumes at a high-speed interval of 2.5 s (covering 15 s of continuous observation to capture Scale I and II dynamics), followed by a 10-minute recovery period. This interval allows for the metabolic clearance of reactive oxygen species (ROS), preventing the cumulative phototoxic effects that typically limit T-cell observation windows to under 10 minutes in conventional OPM or confocal systems.
By integrating this protocol with an automated batch-imaging algorithm, we achieved sequential scanning of 40 cell pairs within each 10-minute cycle, effectively balancing high throughput with minimal physiological perturbation.
Phototoxicity mitigation and multichannel acquisition
Phototoxicity and bleaching control
To suppress photobleaching and maintain long-term cellular viability, specific hardware parameters and excitation protocols were optimized. The laser power, measured at the back aperture of the objective, was strictly limited to 40 µW (405 nm), 105 µW (488 nm), 46 µW (561 nm), and 240 µW (635 nm). To further minimize total radiant energy, the dead-cell marker SYTOX Blue was intentionally allocated to the short-wavelength channel and excited using ultra-low 405 nm laser power. Additionally, we utilized a high-speed camera exposure time of 19 ms per frame to significantly reduce the duration of individual light exposure per acquisition cycle, thereby limiting the cumulative dosage of photons received by the cells.
Synchronized multichannel detection
The system utilized a dual-camera architecture integrated with a W-view beam splitter to achieve efficient spectral separation. Short-wavelength channels (405/488 nm) were directed to the primary camera, while wavelengths exceeding 560 nm were routed to the secondary camera for synchronized detection. To maintain high-fidelity cytoarchitectural data while reducing specific wavelength-induced stress, we employed a time-division multiplexing strategy: the system acquired 5 consecutive volumes of the target cell membrane (561 nm laser excitation.), followed by 1 volume of the microtubule structure (637 nm laser excitation). This balanced approach ensures high-speed multi-channel coverage with minimal physiological perturbation over 5-hour longitudinal experiments. This interleaved sampling strategy enables comprehensive observation of cytoarchitectural changes across multiple channels. For researchers focusing on specific structural dynamics, the integrated dataset can be separated into individual channel records to facilitate independent analysis (Fig. 3e, f).
Principles of 3D volumetric reconstruction
We developed a MATLAB-based image processing and analysis system tailored for the reconstruction of 3D live-cell imaging data acquired via oblique plane microscopy (OPM). The primary objective of this system is to ensure high-fidelity restoration of raw data into its intrinsic orthogonal spatial coordinates. As illustrated in Figs. 2a, 3D scanning is performed using a Bessel light-sheet, where the z-axis represents the scanning direction and the xy-plane corresponds to the illumination plane. The raw image sequence (Fig. 2b) is captured at discrete z-positions by a detection arm that integrates an \({O}_{3}\) objective and a sCMOS camera (Fig. 2h).

Workflow for 3D volumetric reconstruction and the integrated GUI. (a–e) Principles of 3D volumetric reconstruction. (a) Schematic of the OPM scanning geometry using a light-sheet. (b,c) Results of direct volume reconstruction from raw stacks, showing characteristic geometric skewing. Blue-shaded regions delineate the physical space swept by the light-sheet illumination during the scan. (c,d) Results of the reconstruction algorithm based on affine transformations. Gray regions denote background noise padding applied to fill void volumes created during the geometric shearing correction. (f) The Matlab-based graphical user interface (GUI) for streamlined data reconstruction. (g) Example of the noise processing module within the GUI; black regions indicate areas filled with Gaussian noise to maintain background consistency in the reconstructed volume. (h) Simplified schematic of the single-objective optical path. (i) Illustration of the “Central crop” function for defining targeted reconstruction sub-regions.

Data structure. (a) Schematic overview of the reconstructed data directory organization and file structure. (b) MIP of the 488 nm and 561 nm channels at a single time point, alongside their merged composite image. (c) Visualization of a 4D volumetric dataset (x, y, z, channel) for a single time point. (d) Schematic of the 3D U-Net segmentation masks, identifying distinct cellular compartments. (e) Merged imaging data of four-color labeled samples. The green channel represents CAR-T cell actin (EGFP). The red channel shows Nalm6 cells (mApple-Mem plasmid transfection) for the first five volumes, and tubulin (SiR-tubulin) for the sixth volume. The blue channel indicates the nuclei of dead cells, stained with Sytox blue dye upon cell death. (f) Single-channel imaging data.
Generating a Maximum Intensity Projection (MIP) directly from the raw 3D stack results in significant geometric distortion (Fig. 2c, d). This is because the physical volume swept by the oblique illumination forms a skewed parallelepiped rather than a standard orthogonal volume. To rectify this spatial distortion, we implemented a reconstruction algorithm (RA) based on affine transformations, as illustrated in Fig. 2d, e.
Given an illumination angle \(\theta \), the forward imaging process is modeled as a composition of two linear transformations. First, a counter-clockwise rotation about the y-axis accounts for the tilted excitation plane, defined by the rotation matrix R:
Second, a shearing transformation along the x-axis compensates for the non-orthogonal scanning geometry inherent in OPM. This is defined by the shearing matrix S:
The total transformation of the forward imaging model is represented by the product SR. To restore the correct spatial relationships, the reconstruction matrix \({{\boldsymbol{T}}}_{r}\) is defined as the inverse of the imaging transformation:
By applying this inverse mapping, the RA calibrates the tilted coordinates via batch-processed affine transformations and voxel rearrangement, transforming skewed raw stacks into standard orthogonal volumes.
Data Records
The dataset is available at Science DB23. Key data types include: 10 sets of raw data on CAR-T cell-target cell interactions from healthy individuals, in.tif and.dcing formats; and 3 sets of raw data from drug-treated control groups. For data reconstruction software, please refer to the Code availability section.
GUI integration and parameter configuration
To facilitate data accessibility, the RA is integrated into a user-friendly Graphical User Interface (GUI) (Fig. 2f). The system utilizes a structured directory naming convention (prefixed with “v”) to encode essential acquisition metadata: “6” signifies the number of volumes acquired per sequence; “40” represents the number of fields of view (FOVs) or microchambers captured per cycle; “120” denotes the total light-sheet frames per volume, determined by the scan depth and interval; and “0–6” indicates the axial scanning step size (0.6 µm), optimized to satisfy the Nyquist-Shannon sampling criterion relative to the system’s axial resolution.
The GUI provides specialized modules for image optimization:
-
Noise: Minimizes background artifacts in reconstructed dark regions using Gaussian filtering and standard deviation normalization (Fig. 2g).
-
Pixel size: To ensure spatial fidelity, the pixel size is set based on the camera’s physical sensor specifications, allowing for the precise calculation of the effective lateral resolution in the reconstructed data.
-
Volume number: Users can select specific volumes (e.g., “vol1”) or initiate automated processing of the entire dataset by entering “Inf”.
-
Magnification: The system magnification is calibrated based on the optical configuration (set to 62.2 \(\times \)).
-
Digital binning: Digital binning options (2x and 4x) are available for pixel merging to reduce data volume and accelerate processing speeds.
-
Angle of light sheet: Critical OPM parameters, such as the tilt angles of the secondary and tertiary objective lenses, are accurately configured to ensure precise volumetric reconstruction (Fig. 2h).
-
Central crop: The “Central crop” function defines the reconstruction sub-region (Fig. 2i). An empirical value of “280” centers the reconstruction on specific microchamber coordinates, while a value of “−1” enables automated center localization.
Raw data are provided in two primary formats: Hamamatsu.dcimg files (labeled “Hd1_raw”) and multi-file Kinetic.tiff stacks (4 GB per file, prefixed “K_”). Reconstructed datasets are systematically organized within the MIP_ROI_0–39 directory (Fig. 3a).
Channel and temporal mapping
Each ROI contains 120 temporal volumes. Within the channel-specific folders, images correspond to the following biological markers: MIPxy_405: SYTOX Blue, used as a dead-cell marker to monitor target cell viability; MIPxy_488: Lifeact-EGFP, labeling the actin cytoskeleton of CAR-T cells; MIPxy_561: mApple and SiR-tubulin, labeling the tumor cell membrane and microtubules of CAR-T cells, respectively. MIPxy indicates a transverse (top-down) projection (Fig. 3b), while MIPxz provides a cross-sectional (side-view) projection.
3D analysis and segmentation data
For high-dimensional analysis, the ROIauto folder contains volumetric datasets in.h5 format. Each.h5 file (e.g., “00001.h5”) is a 4D record (x, y, z, channel) for a single time point (Fig. 3c). These are accompanied by 3D U-Net segmentation masks24,25,26 (Fig. 3d), stored as bitmaps where pixel integers identify cellular compartments:
0: Extracellular space; 1: CAR-T nucleus; 2: Target cell nucleus; 3: CAR-T cytoplasm/membrane; 4: Target cell cytoplasm/membrane.
For aggregate visualization, the Montage folder contains temporal MIP maps, while the Segmentation folder provides 3D segmented aggregate maps. Within these segmentation maps, specific grayscale values are assigned to identify cellular structures: CAR-T cell nuclei (200), target cell nuclei (150), CAR-T cell cytoplasm and membranes (100), and target cell cytoplasm and membranes (50). These records facilitate automated phenotyping and consistent data interpretation. All analyzed results are archived in Excel format to promote data reuse and transparency within the scientific community.
Technical Validation
To validate the low-phototoxicity performance of our system, we conducted a comparative analysis with conventional confocal imaging (Fig. 4a). While confocal microscopy was limited to capturing only 30 volumes within a 30-minute window, the HBOPM system successfully acquired 720 volumes in the same duration. Quantitative analysis of the photobleaching rates (Fig. 4b), modeled via exponential decay fitting, revealed that the half-decay point occurred at approximately 7 volumes for confocal microscopy, compared to 347 volumes for HBOPM. This demonstrates that for an equivalent level of signal attenuation, HBOPM can capture over 50 times more data, ensuring the maintenance of cell viability during prolonged longitudinal experiments.

Quantitative Analysis and Composition of Multi-Channel Imaging Data. (a) Comparison of photobleaching between confocal microscopy and our system. (b) Quantitative analysis of the photobleaching rate. (c) Comparison of morphology between normal cells and dasatinib-treated cells. (d) Graph of the IS area. We collected 419 biologically independent samples in 13 high-throughput experiments (n = 419). (e) Graph of actin flow speed. n = 419. (f) Graph of the mortality rate of Nalm6 cells labeled with SYTOX blue. Points represent individual CAR-T cells. #: dasatinib-treated group, *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar, 5 μm.
To verify the quantitative utility and biological sensitivity of the acquired data, we benchmarked the system’s performance using key metrics established in the literature19. These metrics include the immunological synapse (IS) area and retrograde actin flow speed, which characterize morphology and dynamics, as well as the tumor cell mortality rate for assessing cellular function.
To evaluate the dataset’s ability to resolve subtle physiological alterations, data from 10 healthy donors served as a baseline and were compared with a “positive control” group of 3 donors treated with dasatinib. Given its well-documented inhibitory effect on cytotoxicity, dasatinib was employed as a pharmacological benchmark to validate the system’s detection sensitivity. High-resolution 4D imaging successfully captured significant decreases in IS area and retrograde actin flow in the treated group (Fig. 4c-f), aligning closely with biological expectations. Furthermore, the correlation between attenuated actin dynamics and reduced Nalm6 cell mortality consistent with known mechanisms further confirms the high fidelity of the dataset at the physiological level.
Data availability
The datasets generated and analyzed during the current study are available in the Science DB repository under the https://doi.org/10.57760/sciencedb.19235. These data are publicly accessible.
Code availability
The associated codes are available on GitHub at https://github.com/feilab-hust/CAR-T-analysis for image analysis and https://github.com/feilab-hust/CAR-T-collection for data collection.
References
Baker, D. J., Arany, Z., Baur, J. A., Epstein, J. A. & June, C. H. CAR T therapy beyond cancer: the evolution of a living drug. Nature 619, 707–715 (2023).
June, C. H. & Sadelain, M. Chimeric antigen receptor therapy. New England Journal of Medicine 379, 64–73 (2018).
Majzner, R. G. & Mackall, C. L. Clinical lessons learned from the first leg of the CAR T cell journey. Nature medicine 25, 1341–1355 (2019).
Rochussen, A. M., Lippert, A. H. & Griffiths, G. M. Imaging the T-cell receptor: new approaches, new insights. Current Opinion in Immunology 82, 102309 (2023).
Mukherjee, M., Mace, E. M., Carisey, A. F., Ahmed, N. & Orange, J. S. Quantitative imaging approaches to study the CAR immunological synapse. Molecular Therapy 25, 1757–1768 (2017).
Hu, Y. S., Cang, H. & Lillemeier, B. F. Superresolution imaging reveals nanometer-and micrometer-scale spatial distributions of T-cell receptors in lymph nodes. Proceedings of the National Academy of Sciences 113, 7201–7206 (2016).
Ritter, A. T. et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 42, 864–876 (2015).
Ponjavic, A. et al. Single-molecule light-sheet imaging of suspended T cells. Biophysical Journal 114, 2200–2211 (2018).
Rosenberg, J., Cao, G., Borja-Prieto, F. & Huang, J. Lattice light-sheet microscopy multi-dimensional analyses (LaMDA) of T-cell receptor dynamics predict T-cell signaling states. Cell systems 10, 433–444. e435 (2020).
Sapoznik, E. et al. A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics. Elife 9, e57681 (2020).
Dunsby, C. Optically sectioned imaging by oblique plane microscopy. Optics express 16, 20306–20316 (2008).
Yang, B. et al. Epi-illumination SPIM for volumetric imaging with high spatial-temporal resolution. Nature methods 16, 501–504 (2019).
Bouchard, M. B. et al. Swept confocally-aligned planar excitation (SCAPE) microscopy for high-speed volumetric imaging of behaving organisms. Nature photonics 9, 113–119 (2015).
Yang, B. et al. DaXi—high-resolution, large imaging volume and multi-view single-objective light-sheet microscopy. Nature methods 19, 461–469 (2022).
Mestermann, K. et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Science translational medicine 11, eaau5907 (2019).
Han, X. et al. A polymer index-matched to water enables diverse applications in fluorescence microscopy. Lab on a Chip 21, 1549–1562 (2021).
Zhu, T. et al. Large-scale high-throughput 3D culture, imaging, and analysis of cell spheroids using microchip-enhanced light-sheet microscopy. Biomedical Optics Express 14, 1659–1669 (2023).
Shi, Y. et al. Smart lattice light-sheet microscopy for imaging rare and complex cellular events. Nature Methods 21, 301–310 (2024).
Wang, Z. et al. 3D live imaging and phenotyping of CAR-T cell mediated-cytotoxicity using high-throughput Bessel oblique plane microscopy. Nature Communications 15, 6677 (2024).
Zhao, Y. et al. Isotropic super-resolution light-sheet microscopy of dynamic intracellular structures at subsecond timescales. Nature Methods 19, 359–369 (2022).
Haouas, M., Chebbi, B. & Golub, I. Extension of the span and optimization of the optical “magic carpet”: generation of a wide quasi-nondiffracting light sheet. Journal of the Optical Society of America A 36, 124–131 (2018).
Antonacci, G., Domenico, G. D., Silvestri, S., DelRe, E. & Ruocco, G. Diffraction-free light droplets for axially-resolved volume imaging. Scientific Reports 7, 17 (2017).
Jie, W. ight sheet microscopy imaging dataset of CAR-T-cell-mediated cytotoxicity. Science Data Bank https://doi.org/10.57760/sciencedb.19235 (2025).
Lee, M. et al. Deep-learning-based three-dimensional label-free tracking and analysis of immunological synapses of CAR-T cells. Elife 9, e49023 (2020).
Wolny, A. et al. Accurate and versatile 3D segmentation of plant tissues at cellular resolution. Elife 9, e57613 (2020).
Arzt, M. et al. LABKIT: labeling and segmentation toolkit for big image data. Frontiers in computer science 4, 777728 (2022).
Acknowledgements
The authors would like to thank Dr. Zhaofei Wang,Dr. Yuxuan Zhao and Prof. Yuhui Zhang for providing us insightful biomedical supports, thank Optoelectronic Micro&Nano Fabrication and Characterizing Facility of Huazhong University of Science and Technology for providing technical support on photolithography technology. This work was supported by the National Natural Science Foundation of China (T2225014, T25B2008, 22534002, 82470239), National Key R&D Program of China (No. 2025YFF1500200), the National Research Center for Translational Medicine at Shanghai (NRCTM(SH)-2023-04), and the Shenzhen Science and Technology Program (GJHZ20220913143405011).
Author information
Authors and Affiliations
Contributions
J.W., L.C. and P.F. discussed the research and wrote the manuscript. J.W. was primarily responsible for system construction and data acquisition. J.J. and Y.F. performed the cell experiments. L.C. oversaw the overall design of the biological experiments, and P.F. supervised the overall design of the optical experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests exist.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Jin, J., Fang, Y. et al. Light sheet microscopy imaging dataset of CAR-T-cell-mediated cytotoxicity. Sci Data 13, 439 (2026). https://doi.org/10.1038/s41597-026-06829-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-06829-9
