
Abstract
Morus macroura with low chilling requirement is one of the primary cultivated mulberry varieties, demonstrates early and abundant fruiting characteristics with high yield and significant economic value, however, the genetic mechanisms of M. macroura are not well understood. In this study, we constructed a chromosome-level genome assembly and annotation of M. macroura. The contig-level genome was initially assembled in 322.62 Mb with a contig N50 of 17.98 Mb from PacBio HiFi reads. With Hi-C sequencing data scafolding, 99.34% of the initially assembled sequences were anchored and orientated onto 14 pseudo-chromosomes, generating a genome of 318.59 Mb with a contig N50 of 17.98 Mb. We identifed 173.34 Mb (54.41%) of repetitive sequences and 2,970 non-coding RNAs in the genome. A total of 21,824 protein-coding genes were predicted, with 21,181 (97.05%) functionally annotated genes. We found 97.21% and 97.46% complete BUSCO genes in the pseudo-chromosomes genome and predicted gene datasets. The high-quality assembly serves as a foundational resource for decoding regulatory networks of year-round fruiting and low-chilling adaptation in M. macroura.
Similar content being viewed by others
Background & Summary
Long-fruited mulberry (Morus macroura), belonging to the Moraceae family, is native to China, Malaysia, and India, with its primary distribution in South and Southwest China1,2. Preliminary surveys indicate that it grows at altitudes ranging from 100 to 1,800 meters. Notably, a rare variant with exceptionally long fruits (up to 18 cm) has been discovered, characterized by high yield and significant potential for development and utilization1,3 Generally prolific and easy to flower, the plant matures from late March to late May, coinciding with the off-season for fruit supply4. Its fresh berries can help alleviate seasonal shortages and, through cultivation practices, produce fruits year-round. The vibrant-colored berries, with their unique flavor, are widely favored and rich in fructose, glucose, seven types of vitamins, 21 amino acids, mineral salts, and trace elements5,6. As a “medicinal-edible dual-purpose” fruit in China, mulberries hold high medicinal value and considerable economic worth5,6. In China, economic development has shifted the sericulture industry northward and southward, with global warming making low chilling requirement germplasm key to industrial advancement. Recent studies have published chromosome-level genomes of Morus species, including M. notabilis, M. alba, M. atropurpurea, and M. yunnanensis7,8,9,10. Although low chilling requirement M. macroura is extensively used for fresh consumption and processing (Fig. 1), and substantial research has been conducted on its cultivation techniques and nutritional quality, its genetic foundations remain underexplored.

Morphological characteristics of M. macroura cv.‘Sijiguo 72 C’; (a) different developmental stages of mulberry fruit, (b) mulberry flowering branches, (c) mulberry fruit branches.
This study employed a combination of PacBio HiFi11 and Hi-C12 data to generate a high-quality chromosome-scale genome assembly of the cultivated long-fruit mulberry variety M. macroura cv. ‘Sijiguo 72 C’ (Fig. 2). The assembled genome spanned 318.59 Mb, with a contig N50 of 17.98 Mb and a scaffold N50 of 21.88 Mb (Table 1). Approximately 99.64% (316.47 Mb) of the contig sequences were anchored onto 14 pseudo-chromosomes, aligning with the known haploid chromosome count of M. alba (Table 1 and Fig. 2). Repetitive sequences accounted for 173.34 Mb of the genome, and 21,824 protein-coding genes were annotated (Table 6). These results demonstrate a contiguous and accurate genome assembly and annotation. Furthermore, comparative genomic analyses with six other Morus species revealed insights into their phylogenetic relationships, divergence times, and evolutionary history.

Chromosome-scale assembly genomic landscape of Morus macroura. Circos plot from the outer to the inner layers represents the following: (1) 14 pseudo-chromosomes length at the Mb scale; (2) GC content per Mb; (3) gene density per Mb repeat density per Mb; (4) Transposable element density; (5) Copia (blue) and Gypsy (purple) LTR retroelement density per Mb; and (6) center: intragenomic syntenic relationships.
This chromosome-scale genome assembly serves as a pivotal resource for characterizing agronomic traits in mulberry fruits and accelerating genetic breeding of Morus spp. It further establishes a foundation for elucidating regulatory mechanisms of winter bud dormancy and flowering, alongside comparative genomic analyses between M. macroura and other Morus species.
Methods
Sample collection and sequencing
For genome sequencing, fresh young leaves of M. macroura ‘sijiguo72c’ were collected from an adult individual at the Institute of Environment and Plant Protection, Chinese Academy of Tropical Agricultural Sciences, (Danzhou City, Hainan Province, N 19° 35′ E 109° 29′). High-molecular-weight genomic DNA was extracted using the CTAB method13 followed by purification with the Grandomics Genomic Kit according to the manufacturer’s protocol. DNA degradation and contamination were assessed via electrophoresis on 1% agarose gels. Purity was measured using a NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA), with acceptable ratios of 1.8–2.0 (OD₂₆₀/₂₈₀) and 2.0–2.2 (OD₂₆₀/₂₃₀). DNA concentration was quantified using a Qubit® 4.0 Fluorometer (Invitrogen, USA). Size selection and cleanup were performed using the SMRTbell Prep Kit 3.0. Sequencing was conducted on the PacBio Revio platform following the manufacturer’s manual. Raw reads were processed with CCS software (https://github.com/PacificBiosciences/ccs) under default parameters (min passes = 3, min RQ = 0.99), generating high-precision HiFi reads (Q > 20)14. This yielded 17.56 Gb of HiFi data, representing ~52.63 × genome coverage (Table 2).
Total RNA was extracted from four distinct tissues (leaf, stem, flower, and fruit) sampled from the same M. macroura plant. Subsequently, the mRNA was synthesized to cDNA, and four libraries were constructed with an insertion size of 350 bp using a MGIEasy Universal DNA Library Prep Kit V1.0 (CAT#1000005250, MGI) following the manufacturer’s instructions. The qualified libraries were sequenced on DNBSEQ-T7RS platform set in the PE150 program. Raw reads were filtered with fastp v0.23.4 Subsequently, the reads were filtered using fastp v0.23.4 with the following criteria: removal of reads containing adapters; remove reads with a proportion exceeding 10% N; remove reads where the proportion of low-quality (quality value < 20) bases exceeds 50%.with parameters.Generating an average of 8.97 GB of clean reads per sample (Table 2).
Contig-level genome assembly
HiFi reads were generated from filtered subreads using the CCS module in SMRT Link v12.015 with parameters:--maxLength = 50000, --minPasses = 3, and --min Predicted Accuracy = 0.99. After removing adapter sequences and discarding low-quality reads (average quality values rq < 0.99), the resulting HiFi dataset (reads with base quality ≥ Q20 and average rq > 0.99) yielded 17.56 Gb of CCS reads (52.63 × coverage), exhibiting an average length of 18.38 kb and an N50 of 18.96 kb (Table 2). These reads were converted from BAM to FASTQ format using bam2fastx v1.3.1.(https://github.com/ pacificbiosciences /bam-2fastx/). A pure third-generation assembly strategy was implemented with HiFi reads, employing Hifiasm v0.16.011 under default parameters to generate contigs. Post-assembly, redundant sequences were removed to produce a non-redundant preliminary assembly (322.62 Mb). Within this assembly, two contigs (104,112 bp; 0.03% of total length) were identified as bacterial contaminants and excised. Following the elimination of exogenous sequences—including non-target taxa, mitochondrial, and chloroplast DNA—the final genome assembly reached 320.58 Mb with a contig N50 of 17.98 Mb (Table 1).
Hi-C library construction and pseudo-chromosome anchoring
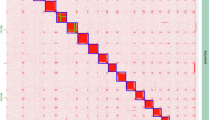
To anchor hybrid scaffolds to chromosomes, genomic DNA was isolated from tender leaves of M. macroura for Hi-C library construction. Leaves were sectioned into ~2 cm² pieces and cross-linked with 2% formaldehyde. Purified DNA was digested with DpnII restriction enzyme, biotinylated using biotin-14-dCTP, sheared to 300–400 bp fragments, and subjected to blunt-end repair. The Hi-C library was sequenced on the Illumina NovaSeq/MGI-2000 platform16. Raw reads were filtered with fastp v0.23.417, generating ~60.64 Gb (190.34 × coverage) of data for pseudomolecule assembly. Cleaned Hi-C data were aligned to contig assemblies using Bowtie2 v2.3.218 under parameters --very-sensitive -L 30. After merging paired-end reads, 480,988 uniquely aligned read pairs (45.64% of clean data) were obtained (Table S1). Hi-C scaffolding employed HiC-Pro v3.1.012, yielding 370,804 valid interaction pairs (77.09% of uniquely mapped reads; 35.18% of total clean data; Table S1). Quality-controlled data were aligned to the reference genome using BWA v0.7.1719 with mem -5SP, followed by filtering via filter_bam v2.0.0 (--nm 3) to retain reads with mapping quality ≥ 1 and edit distance < 3. Pseudomolecule construction utilized HapHiC v1.0.520 (command: HapHiC pipeline Genome bam Chrnumber) for clustering, reassignment, ordering, orientation, and assembly. Contigs totaling 316.47 Mb were anchored to 14 chromosomes (range: 14.19–58.92 Mb; Table 3), achieving a scaffolding rate of 99.34% (Table 1). The final pseudochromosome-level genome size was 318.59 Mb with 9 gaps (total gap length: 0.9 Mb) and a scaffold N50 of 21.88 Mb (Table 1). To validate anchoring accuracy, pseudochromosomes were partitioned into 100-kb bins for genome-wide interaction matrix construction, visualized as a heatmap using HiC Plotter v0.6.63621 (Fig. 3).

Heat map of genome-wide Hi-C intra-chromosome interactions in M. macroura. The interaction density is measured by the number of supporting Hi-C reads and illustrated by the color bar from dark red (high density) to light pink (low density).
Genome annotation and functional prediction
Identifying repeat sequences
The M. macroura genome assembly harbored abundant repetitive sequences, broadly classified as tandem repeats and interspersed repeats based on distribution patterns. 1. Microsatellite (SSR) analysis, GMATA v2.222 identified 117,316 SSR loci spanning 630,253 bp (0.20% of the genome). 2. Tandem repeat annotation, using Tandem Repeats Finder (TRF) v4.07b23 with parameters 2 7 7 80 10 50 500 -f -d -h -r, we detected 50,263 tandem repeats totaling 2,931,480 bp (0.92% genome length; Table 4). 3. Transposable element (TE) annotation pipeline, MITE-hunter24 (-n 20 -P 0.2 -c 3) generated a MITE library, and LTR_FINDER v1.0.725 and LTR_HARVEST v1.6.526 predicted LTR retrotransposons, and LTR_retriever v1.927 consolidated MITEs and LTRs into TE.lib and RepeatModeler v1.0.1128 produced RepMod.lib. Merged TE.lib, RepMod.lib, and RepBase database into a composite repeat library. RepeatMasker v4.1.2-p128 annotated repeats using this library. Combined annotations revealed 173.34 Mb repetitive sequences (54.41% genome coverage), dominated by: LTR retrotransposons: 34.92% (110.92 Mb), DNA transposons: 12.40% (39.40 Mb), LINEs: 2.48% (7.88 Mb), SINEs: 0.18% (0.57 Mb)(Complete classification in Table 4).
Identifying non-coding RNA (ncRNA) gene
Non-coding RNA (ncRNA) annotation was performed through an integrated approach: Rfam alignment: The genome was aligned against the Rfam database (release 14.9)29 using Infernal v1.130 with default E-value thresholds; tRNA prediction: tRNAscan-SE v1.3.131 was executed under standard parameters; rRNA identification: Homology-based detection of rRNA genes and subunits was conducted with RNAmmer v1.232. Integrated analysis revealed the following ncRNA repertoire (summarized in Table 5): Ribosomal RNA (rRNA), 1,170 genes (including subunits such as 5S/18S/28S); Small RNAs, 1,026 loci (e.g., snoRNAs, snRNAs, miRNAs); Regulatory RNAs, 7 elements (e.g., riboswitches or lncRNAs with regulatory roles); Transfer RNA(tRNA): 767 genes (covering all 20 standard amino acid-specific tRNAs).
Gene structure prediction
Gene structure prediction was performed by integrating three complementary approaches: homology-based prediction, transcriptome-based prediction, and ab initio prediction..1. Homology-based prediction: GeMoMa v1.6.133 was employed to infer gene models through cross-species comparison with six reference genomes: M. alba heyebai8, M. alba zhenzhubai10, M. notabilis7, Oryza sativa34], Arabidopsis thaliana35, and Prunus persica36; 2. Transcriptome-based prediction: quality-controlled RNA-seq reads were aligned to the reference genome using STAR 2.7.3a37; transcript coordinates were generated with StringTie v1.3.4 d38 under default parameters; trinity-assembled transcripts and full-length cDNAs were mapped to the soft-masked genome via GMAP v2014-10-239; PASA v2.3.340 integrated aligned data to assemble transcripts, with open reading frames predicted by GeneMark-ST v5.141; this pipeline yielded 24,189 transcriptome-supported genes (Table 6); 3. Ab initio prediction: Augustus v3.3.142 was trained on 3,000 high-confidence transcriptome-derived genes to generate a species-specific model. De novo prediction using this model identified 22,413 candidate genes (Table 7). Integration and refinement: Predictions from PASA, GeMoMa, and Augustus were consolidated using Evidence Modeler v1.1.1 (EVM)40 with weighted evidence priorities: PASA > GeMoMa > Augustus. The initial gene set was filtered through TransposonPSI43 to remove sequences containing potential transposable elements or coding errors. The final curated annotation contained 21,824 protein-coding genes with the following features (Table 6): average gene length of 4,034 bp, cds length of 1,305 bp, exon length of 230 bp, and intron length of 583 bp.
Gene function annotation
Functional annotation of the protein-coding genes was performed using Blastp v2.7.144 (-evalue 1e-5, -max_target_seqs. 1), against the following public databases: Non-redundant protein (NR; ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz), SwissProt, Kyoto Encyclopedia of Genes and Genomes (KEGG)45, Eukaryotic Orthologous Groups(KOG)46. Gene Ontology (GO)47 terms were assigned based on BLASTP matches through annotation transfer. Genome-wide functional profiling was conducted with InterProScan v5.32-71.048 with default parameters via Pfam database alignment. InterPro entries were mapped to GO annotations across three domains: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). This integrated approach annotated 21,181 genes (97.05% of the total gene set) with functional terms (Table 7).
Comparative genomic analysis
For comparative genomic analysis, we selected 9 species meeting three criteria: publicly available high-quality genome sequences with annotations, extensive prior research background, and documented medicinal or edible value.The taxa include 8 Moraceae species7,8,9,10,49 and P. persica (peach)35. Orthologs were identified using OrthoFinder based on the longest transcripts of protein-coding genes from 9 species. In the M. macroura genome, 17,326 gene families were identified,encompassing 20,959 genes (Fig. 4a). We also detected 387 expanded and 4,727 con-tracted gene families. The rooted tree generated by OrthoFinder v2.5.450. Ultrametric trees were constructed with r8s v1.81 using the OrthoFinder-rooted phylogeny Divergence between M. macroura and M. alba: ≈ 4.162 Mya (Fig. 4c), M. macroura and M. atropurpurea show minimal evolutionary distance (recent divergence), indicating close phylogenetic affinity.

Comparative genomic and evolution analysis of morus species. (a) A Venn diagram of specific and shared orthologs among 9 species; (b) Gene-based genome colinear comparison between M. macroura and M. alba. Conserved syntenic blocks are highlighted with grey color corresponding to the fourteen pseudo-chromosomes, indicating visible genome rearrangements occurred during evolution among morus species. (c) Phylogeny and divergence time analysis among 9 species. The divergence times among different plant species are labelled on the right (million years ago, Mya).To better visualize the branching patterns of Morus, the divergence distance between P. persica and F. chinensis was rescaled from 76 to 12. Similarly, the divergence time between F. chinensis and the common ancestor of Morus was adjusted from 31.218 to 9.218.
Whole-genome synteny
To understand the extend of genomic rearrangement of M. macroura during evolution, whole-genome synteny analysis was conducted between M. macroura and M. alba.The protein sequences of M. macroura and M. alba were blasted using blastp using an E-value cutoff of 1 × 10−5. The multiple alignments of syntenic blocks were identified by MCScanX51 with the parameter -s 15 (number of genes required to call a collinear block) and visualized by jcvi v1.2.87552 with a minimum span threshold of 30 genes (--minspan = 30). The analysis revealed interweaving conserved syntenic blocks across all fourteen M. macroura pseudochromosomes (Fig. 4b), indicating potential large-scale genomic rearrangements during Morus divergence.
Data Records
The raw sequencing data including the Hi-C sequencing, PacBio HiFi and Illumina NGS RNA-seq have been submitted to the NCBI Sequence Read Archive (SRA) under accession numbers SRR3502011253, SRR3502011354, and SRR34997372 to SRR3499737555,56,57,58, respectively. The final chromosome-level assembled genome sequences were deposited in the NCBI Assembly database under Accession Number PRJNA130563759. The genome annotation results, including repeated sequences, gene structure, and functional predictions were deposited in the Figshare database (https://doi.org/10.6084/m9.figshare.30143464)60.
Technical Validation
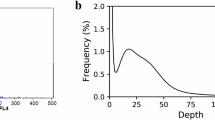
Genome assembly quality assessment(Integrating alignment statistics, ortholog completeness, and k-mer metrics). 1. HiFi read alignment, Minimap2 v2.2661 (-ax map-hifi) mapped 955,429 HiFi reads to the post-assembly error-corrected genome, achieving: Mapping rate, 99.64% (952,000 reads); Coverage, 99.9% of filtered short reads mapped; Depth analysis (Samtools v1.1862), the average depth of the third-generation data is 52.63x. When the statistical coverage depth is 1x, the coverage of the entire genome is 99.94%. 2. Evolutionary conserved element analysis, BUSCO v5.1.363 with embryophyte_odb10 database revealed high integrity:Genome-level completeness, 97.21% eudicot BUSCOs (Table 1); Gene model completeness, 97.46% eudicot BUSCOs (Fig. 5). 3. K-mer based quality metrics, using Merfin64 and Merqury v1.365 with 21-kmer profile: Consensus quality (QV), 62.55 (Q20≈99.999% accuracy); K-mer completeness, 80.90%. Interpretation: QV > 60 indicates fewer than 1 error per million bases.

Benchmarking of genome completeness of M. macroura genome assembly and annotation, evaluated by BUSCO based on embryophyta odb10 database which includes 1,614 genes.
Protein coding genes comparison with close species. To determine the prediction accuracy and reliability, the distribution of gene length, CDS length, exons number per gene, exon length, intron length, and exon number in M. macroura and other closely related species (M. alba8,M. alba zhenzhubai10, Morus notabilis7, O. sativa34, A. thaliana35, P. persica36) were determined. The consistent distribution tendency among all species further supported an ideal annotated gene dataset in M. macroura (Fig. 6).

Annotated genes comparison of the distribution of gene length (a), CDS length (b), C exon length (c), exon number (d), intron length (e) and intron number (f) in M. macroura with other closely related species. Te x-axis represents the length or number and the y-axis represents the density of genes.
In addition, the LTR Assembly Index (LAI) for the assembly was also calculated LTR_retriever v.2.9.067 to evaluate the contiguity of the assembly. The LAI score for the whole genome was estimated to be 26.45, surpassing the quality standard for reference genomes.
Hence, a high-quality completeness and accuracy M. macroura genome was assembled and annotated in the present study.
Data availability
The dataset has been deposited to GenBank [PRJNA1305637 and PRJNA1306310].
Code availability
All sofware and pipelines were executed according to the official instructions. No custom code was generated for this study.
References
Chu, Y. N. et al. A Survey on Leaf Quality and Partial Agronomic Traits of Wild Monus laevigata Wall Resources in Southem yunnan. Sci. Sericulture 40(5), 0928–0932 (2014).
Naik, M. V. G., Thumilan, B. M., Roy, B. & Dandin, S. B. Assessment of genetic diversity and interrelationship among wild mulberry (Morus macroura and M. Serrata) collections of India through DNA marker analysis. Indian Society of Plant Breeders 45(2), 169–175 (2006).
Yang, J. H. et al. Analysis and comprehensive evaluation of fuit quality of Mors macroura in Yumnan province. Southwest China Joural of Agricultural Scienees 37(5), 990–1000 (2024).
Yang, M. Study on cold-hardiness echanismof mulberry. Northwest A&F University Yanglin (2012).
Wu, H. Z. et al. Unveiling the unique aroma of Morus macroura through integrated volatile metabolome and transcriptome analysis. Sci. Hortic. 339, 113814 (2025).
Lou, D. Z. et al. Analysis of main components of five mulberry varieties in tropics. Plants. 13, 2763 (2024).
Ma, B. et al. The gap-free genome of mulberry elucidates the architecture and evolution of polycentric chromosomes. Hortic. Res. 10(7), uhad111 (2023).
Jiao, F. et al. Chromosome-level reference genome and population genomic analysis provide insight into the evolution and improvement of domesticated mulberry (Morus alba L). Mol. Plant. 13(7), 1001–1012 (2020).
Dai, F. W. et al. Genomic resequencing unravels the genetic basis of domestication, expansion, and trait improvement in Morus atropurpurea. Adv. Sci 10, 2300039 (2023).
Xia, Z. Q. et al. Haplotype-resolved chromosomal-level genome assembly reveals regulatory variations in mulberry fruit anthocyanin content. Hortic. Res. 11(6), uhae120 (2024).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C processing. Genome Biol. 16, 259 (2015).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325 (2006).
FastQC, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
SMRT Link, https://www.pacb.com/support/software-downloads/.
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7), 1665–80 (2014).
Chen, S. et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884–i890 (2018).
Langmead, B. & Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN] https://doi.org/10.48550/arXiv.1303.3997 (2013).
Zeng, X. F. et al. Chromosome-level scaffolding of haplotype-resolved assemblies using Hi-C data without reference genomes. Nat. Plants 10, 1184–1200 (2024).
Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome Biol. 16, 1–8 (2015).
Wang, X. & Wang, L. GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front Plant Sci. 7, 1350 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2), 573–80 (1999).
Han, Y. J. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids. Res. 38(22), e199 (2010).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids. Res. 35(Web Server issue), W265–8 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176(2), 1410–1422 (2018).
Bedell, J. A., Korf, I. & Gish, W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16(11), 1040–1 (2000).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids. Res. 33(Database issue), D121–4 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22), 2933–5 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids. Res. 25(5), 955–64 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids. Res. 35(9), 3100–8 (2007).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids. Res. 44(9), e89 (2016).
Shang, L. G. et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 16(8), 1232–1236 (2023).
Hou, X. R. et al. A near-complete assembly of an Arabidopsis thaliana genome. Mol. Plant 15(8), 1247–1250 (2022).
Zhou, P. et al. Genome sequencing revealed the red-flower trait candidate gene of a peach landrace. Hortic. Res. 10, uhad210 (2023).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open-source ab initio eukaryotic gene-finders. Bioinformatics 20(16), 2878–2879 (2004).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20(1), 278 (2019).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21(9), 1859–75 (2005).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids. Res. 43, e78 (2015).
Stanke, M. et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24(5), 637–44 (2008).
Urasaki, N. et al. Draft genome sequence of bitter gourd (Momordica charantia), a vegetable and medicinal plant in tropical and subtropical regions. DNA Res. 24(1), 51–58 (2017).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids. Res. 28(1), 27–30 (2000).
Galperin, M. Y. et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids. Res. 43, D261–9 (2015).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25(1), 25–9 (2000).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17(9), 847–8 (2001).
Zhang, X. T. et al. Genomes of the Banyan tree and pollinator wasp provide insights into fig-wasp coevolution. Cell 183(4), 875–889 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14 (2019).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids. Res. 40, e49–e49 (2012).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320(5875), 486–488 (2008).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR35020112 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR35020113 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34997372 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34997373 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34997374 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34997375 (2025).
Wu, H. Z. et al. Whole genome shotgun sequencing project. https://identifiers.org/ncbi/insdc.gca:GCA_055683485.1 (2025).
Wu, H. Z. et al. A high-quality chromosome-level genome assembly of the low chilling requirement mulberry, Morus macroura, Figshare, https://doi.org/10.6084/m9.figshare.30143464 (2025).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32(14), 2103–10 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16), 2078–9 (2009).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–2 (2015).
Formenti, G. et al. Merfin: improved variant filtering, assembly evaluation and polishing via k-mer validation. Nat. Methods 19(6), 696–704 (2022).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Acknowledgements
The work was supported by the Project of National Key Laboratory for Tropical Crop Breeding (NO.NKLTCBCXTD15) and China Agriculture Research System-Sericulture (CARS-18).
Author information
Authors and Affiliations
Contributions
S.C.W. and L.X. conceived and designed the study; T.G. and Y.L.Y. collected the samples and extracted the genomic DNA for sequencing; H.Z.W. and J.B.W. assembled the genome and performed Hi-C scaffolding and genome annotation; P.Q.L. and H.Z.W. performed technical validation; H.Z.W. wrote the draft manuscript. J.B.W. and L.X. modified the manuscript. All authors have read, revised, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, H., Wang, J., Geng, T. et al. A high-quality chromosome-level genome assembly of the low chilling requirement mulberry, Morus macroura. Sci Data 13, 458 (2026). https://doi.org/10.1038/s41597-026-07117-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-026-07117-2
