
Abstract
Global trade and human movements outspread animal species, for example ants, from their native habitats to new areas. This causes biosecurity concerns because an exotic ant might have adverse impacts on agriculture, the environment, or health; thus, incurring economic losses. The browsing ant, Lepisiota frauenfeldi, was first detected in 2013 at the Perth Airport. Since then, more discrete browsing ant infestations have been found in Perth and at the Ports of Darwin and Brisbane. This exotic ant has been deemed a significant pest in Australia and eradication efforts are underway. However, tackling this invasion requires an understanding of how these infestations are related. Are they same or separate or a combination of both? Here, we carried out a phylogenetic analysis using high-throughput sequencing data to determine their relatedness. Our results showed that each interstate incursion was separate. Furthermore, the Western Australian incursions might have two introductions. These findings are critical in devising effective biosecurity measures. However, we discovered that this information could only be revealed by analysing the whole mitochondrial genome; not by a single mitochondrial gene as typically done for species identification. Here, we sequenced 51 whole mitogenomes including three of its congener L. incisa for the first time, for tracing future infestations.
Similar content being viewed by others


Introduction
Global trade and human movements are influencing the geographic distribution of animal species1,2,3,4. Ants are the most studied terrestrial group for dispersal to new geographic locations2,5. When an ant species is introduced to a new area, it is often called an exotic or tramp ant6. An exotic or tramp ant is categorised as invasive7,8 when it spreads in the newly introduced site, dominates the local fauna, and exerts a negative impact on the surrounding ecosystem, biodiversity, environment, or public health2,9.
An invasive ant may cause significant economic losses to agriculture10,11,12,13,14,15, livestock16, and household properties2,17. For example, red imported fire ant (RIFA, Solenopsis invicta), a native of South America, caused an estimated US$5–6 billion losses in the USA2,17,18. The potential cost impacts in Australia would be in excess of $1.65 billion per year if RIFA were to establish permanently in Australia2,19. Furthermore, it adversely affects biota20,21, human health22,23,24,25, and wildlife26,27. These economic losses and damages warrant biosecurity measures put in place for early detection and eradication of any exotic pests28.
Lepisiota frauenfeldi, commonly known as browsing ant, was detected in April 2013 at the Perth Airport precinct, Western Australia. This was the first post border occurrence of the genus Lepisiota in Australia29. Since then, a further nine discrete browsing ant infestations have been found within the Perth metropolitan area, seven of which are believed to be linked to the original airport infestation. In June 2015 browsing ant was discovered at the Port of Darwin, Northern Territory and then in April 2019 at the Port of Brisbane, Queensland29. During surveillance activities for L. frauenfeldi, L. incisia has also been detected in 14 different locations in Western Australia since 2020, and 10 since 2022.
Browsing ant has been deemed a nationally significant pest and would likely become a considerable horticultural, environmental, and domestic pest if established in Australia. These aggressive ants are native to the Palearctic Bioregion30, and are ideally suited to Australian conditions. They form multi-queened super-colonies and monopolise food resources, quickly reaching high populations and displacing other invertebrates31.
Through tracing activities, authorities have uncovered 34 browsing ant infestations across Australia since 2013; 10 in Western Australia, 23 in the Northern Territory and one in Queensland29. These are all either eradicated or are under eradication, at a cost of many millions of dollars. This demonstrates browsing ants’ genuine capacity for spread and raises the question why they have suddenly emerged as such a prominent biosecurity risk across Australia, in such a relatively short period31.
When browsing ant was first discovered in Perth in 2013, very little in the way of written material or scientific literature was available to elucidate their biology, phenology, and pest characteristics such as population dynamics and dispersal mechanisms. And with so many infestations manifesting across Australia over just 6 years, we needed to understand the relatedness of the separate infestations to know if the incursions spread from one original incursion, were separate incursions, or a combination of both. DNA profiling using genetic markers such as ‘Sequence Tandem Repeats (STR) or microsatellites’32,33 could answer these questions by revealing any relatedness and even the origin of separate infestations, and also allow for advances in taxonomy and species identification31. However, there are currently no publicly available genetic markers or STR for browsing ants. Developing such markers from scratch would be time-consuming32 and delay the urgent biosecurity responses required to tackle this invasive pest incursion.
Instead, the relatedness of the incursions can be revealed by inferring the phylogeny of the ants using their mitochondrial genes34. Mitochondrial genes have widely been used to infer phylogeny in insects35,36. This is because mitochondrial genes are (i) high in copy number making them easier to amplify through the Polymerase Chain Reaction (PCR) assays37, (ii) maternally inherited38 thus lacking recombination39 presumably resulting in similar genealogical history for the entire molecule, (iii) high rate of nucleotide substitution allowing a chance to capture phylogeny signal without intensive sequencing40, and (iv) relatively conserved in size, content and synteny in animals41,42.
However, recent studies show that the whole mitochondrial genome can reveal a deeper-level phylogeny among the taxa than a single mitochondrial gene41,43,44. Furthermore, the recent advancement of sequencing technologies and decrease in sequencing costs make it cost and time effective to sequence the whole mitochondrial genome rather than partial, single or multiple genes41. Here, we sequence the whole mitochondrial genomes of 48 Lepisiota frauenfeldi individuals representing 10 incursions in Australia and four from overseas. We include Lepisiota incisa as an outgroup. Currently, there is no whole mitogenome data for L. incisa in Genbank45. We have constructed phylogenetic trees using the whole mitochondrial genomes as well as individual protein coding genes to reveal the relatedness of the L. frauenfeldi incursions in Australia. At the time of writing, all known infestations of L. frauenfeldi and L. incisa are either eradicated or under eradication.
Results
Phylogenetic analyses
Fifty-one complete mitochondrial genomes (mitogenomes) were sequenced using Oxford Nanopore Technologies (Table 1). Sequencing depth varied from 8 to 1785 × with a median value of 166 × (Table 2). The reconstructed mitogenomes were 16,455–17,430 bases in length. The average value for both N50 and NG50 was 17,116 bases. L50 and LG50 were 1 for all the constructed mitogenomes (Table 2). The reconstructed mitogenomes were subjected to Bayesian Inference and Maximum Likelihood based phylogenetic analysis.
The phylogenetic analysis data set contained whole mitogenome sequences of three replicates of the outgroup species (Lepisiota incisa) (Md1_1, W3_1 and W3_2), four from the Brisbane incursions, two from Northern Territory, 34 from Western Australia representing 10 browsing ant incursions across Australia (Table 1). The data set also included six browsing ants from overseas; one from Iran, two from Oman, two from Timor and one from Hawaii (Table 1).
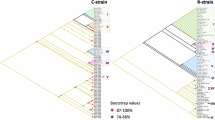
We constructed the phylogenetic tree using Bayesian and Maximum-Likelihood methods. Both methods recovered a similar topology (Figs. 1 and 2). The replicates from the same browsing ant incursions clustered together with strong posterior probability support ranging from 90 to 100 (Fig. 1) except for Br2_4. Br2_4 is a Brisbane incursion but grouped with Western Australian incursions in both Bayesian and Maximum-Likelihood based phylogenetic tress (Figs. 1 and 2). The outgroup species formed distinct clades (Figs. 1 and 2). Both methods revealed the incursions from Western Australia as two distinct taxa represented by red and pink colours (Figs. 1 and 2). The pink incursion fell as a sister taxon to the browsing ants from Oman with a support of 100% posterior probability (Fig. 1) and 88% bootstrapping (Fig. 2). The incursions from Northern Territory and Brisbane except Br2_4 appeared as separate clades. The incursions from Timor Leste and Hawaii grouped in one clade (Figs. 1 and 2). The browsing ants from Iran formed an individual clade on both trees (Figs. 1 and 2).

The Bayesian phylogenetic tree of the browsing ants inferred from mitochondrial genome sequence data. The tree was built using the MrBayes v3.256 program that implemented the ‘invgamma’ model and visualised using the FigTree v1.4.464 software. The numbers on the nodes represent the posterior probability percentages. The taxa are coloured to indicate potentially separate incursions. Red: Perth, WA; Pink: Fremantle, WA, and Oman; Dark blue: QLD; Light blue: NT; Light green: Timor Leste and Hawaii; Yellow: Iran; Black: outgroup (L. incisa). Taxa NC_061556.1 and OK569859 are retrieved from Tay et al. 202255.

The maximum-likelihood (ML) phylogenetic tree of the browsing ants inferred from mitochondrial genome sequence data. The tree was built using the RAxML v858 (Randomized Axelerated Maximum Likelihood) program that implemented the ‘GTRGAMMA’ model, and visualised using the FigTree64 software. The numbers on the nodes represent the percentages of 1000 bootstrap values. The taxa are coloured to indicate potentially separate incursions. Red: Perth, WA; Pink: Fremantle, WA, and Oman; Dark blue: QLD; Light blue: NT; Light green: Timor Leste and Hawaii; Yellow: Iran; Black: outgroup (L. incisa). Taxa NC_061556.1 and OK569859 are retrieved from Tay et al. 202255.
Mitogenome annotation
We annotated the mitogenomes of eight Lepisiota frauenfeldi individuals selected from the distinct clades of the Bayesian phylogenetic tree (Fig. 1). The Lepisiota frauenfeldi mitogenomes were a typical circular molecule with 17,090 base pairs (bp) in size (Fig. 3a–h) and consisted of 13 protein-coding genes (PCGs), 22 tRNA genes and two rRNA genes (Fig. 3a–h).

The mitochondrial genomes of eight Lepisiota frauenfeldi individuals collected from different locations. (a) specimen B1_5, collected from Belmont, Western Australia, Australia; (b) specimen Br1_4, collected from Brisbane, Queensland, Australia; (c) specimen Br2_1, collected from Brisbane, Queensland, Australia; (d) specimen NF1-1, collected from North Fremantle, Western Australia, Australia; (e) specimen NT1-1, collected from Northern Territory, Australia; (f) specimen O1-3, collected from Oman; (g) specimen PA2_5, collected from Perth Airport, Western Australia, Australia; and (h) specimen TL1, collected from Timor Leste. Protein coding genes, ribosomal RNAs (rRNA) and the transfer RNAs (tRNA) are represented by the green, red, and pink arrows, respectively.
Bayesian phylogenetic tree inferred from a single mitochondrial protein coding gene
We extracted 13 PCGs that were present in all the annotated mitogenomes and carried out Bayesian phylogenetic analysis using a single PCG (Fig. 4). Phylogenetic tree using cox1 gene (Fig. 4a) showed the outgroup (W3-1), Oman (O1-2), and North Fremantle (NF1-1) (Western Australia) incursions on distinctly different branches, instead of clustering Oman (O1-2) and North Fremantle (NF1-1) together as in the mitogenome-based tree (Fig. 1). cox2 gene-based tree showed that all incursions were separate i.e., no relatedness among the incursions (Fig. 4b). Likewise, all gene-based trees except nad5 showed that even the replicates of the same incursion sites such as Brisbane (Br1_4 and Br2_1) and Western Australia (B1_5 and PA2_5) were different (Fig. 4b–h,j–m). However, nad5 based tree grouped the Northern Territory (NT1-1) incursion together with Western Australian (NF1_1, B1_5 and PA2-5) and overseas incursions (O1_3 and TL1) (Fig. 4i) starkly contrasting to the topology of the mitogenome based trees (Figs. 1 and 2).

The Bayesian phylogenetic tree of the browsing ants inferred from a single mitochondrial protein coding gene (PCG) sequence data. The tree was built using the MrBayes v3.256 program that implemented the ‘invgamma’ model and visualised using the FigTree v1.4.464 software. The numbers on the nodes represent the posterior probability percentages. The tip labels are coloured to indicate that the same-coloured tips are grouped together in a clade when using the whole mitochondrial genome sequences for Bayesian phylogeny (Fig. 1). Red: Perth, WA; Pink: Fremantle, WA, and Oman; Dark blue: QLD; Light blue: NT; Light green: Timor Leste; Black: outgroup (L. incisa).
However, when the whole mitogenome sequences were used instead of just a single PCG, the Bayesian phylogenetic tree (Fig. 5) revealed the same topology as in Fig. 1. The relatedness among the incursions was revealed with a greater resolution, and 100% posterior probability support on the tree branches and 50–100% on the clades (Fig. 5). This mitogenome based tree separated the outgroup from all the other sequences, clustered the replicates of the same incursion together, and revealed that the browsing ant incursions in Western Australia (B1-5, PA2-5 and NF1-1), Northern Territory (NT1-1), Brisbane (Br1-4 and Br2-1) and Timor Leste (TL1) were all separate (Fig. 5). Furthermore, it showed that Western Australia had two separate incursions (B1-5, PA2-5 and NF1-1). Interestingly, the mitogenome based tree topology showed independence of sample sizes (Figs. 1 and 5), for example, 51 mitogenomes in Fig. 1 and nine mitogenomes in Fig. 5, but the topology demonstrated the same resolution.

The Bayesian phylogenetic tree of the browsing ants inferred from the whole mitochondrial genome sequences. The tree was built using the MrBayes v3.256 program that implemented the ‘invgamma’ model and visualised using the FigTree v1.4.464 software. The numbers on the nodes represent the posterior probability percentages. The tip labels are coloured to indicate that the same-coloured tips are grouped together in a clade when using the whole mitochondrial genome sequences for Bayesian phylogeny (Fig. 1). Red: Perth, WA; Pink: Fremantle, WA, and Oman; Dark blue: QLD; Light blue: NT; Light green: Timor Leste; Black: outgroup (L. incisa). Taxa NC_061556.1 and OK569859 are retrieved from Tay et al. 202255.
Mitogenome wide nucleotide identity
The nucleotide identity matrix extracted from the sequence alignment showed that L. frauenfeldi and L. incisa (outgroup) mitogenomes had about 20% (about 3500 bases out of 17,000) nucleotide variation (Fig. 6). The L. frauenfeldi mitogenomes varied between each other by only 2–650 bases (Fig. 6). However, none of the L. frauenfeldi mitogenomes had 100% nucleotide identity when compared to each other (Fig. 6).

Number of bases varying in the Browsing ant mitogenomes (17,090 bases) across different incursions. Taxa NC_061556.1 and OK569859 are retrieved from Tay et al. 202255.
Individual gene wide nucleotide identity
We extracted the nucleotide identity matrix from the individual gene sequence alignments that were used for constructing the Bayesian trees for each of the PCGs (Fig. 4). The matrices showed that there was always 100% nucleotide identity between some pairs of browsing ant individuals when considered only a single gene (Fig. 7).


Distance matrix of nucleotide alignment for individual Protein Coding Genes (PCGs). These alignments were used for constructing individual gene-wise Bayesian phylogenetic trees (Fig. 5).
Discussion
We sequenced and analysed the whole mitogenome to determine the relatedness of the browsing ant incursions from Australia and overseas.
Bayesian inference based phylogenetic analysis using the whole mitogenome sequences showed that Western Australian incursions were grouped into two clusters, one was in Fremantle (Fig. 1; pink cluster) and the other was across the rest of the state (Fig. 1; red cluster). The incursions within the red cluster might be a spread of a single incursion and/or might be multiple incursions from the same source/origin (Fig. 1; red cluster). It is interesting to note that one sample from Brisbane (Br2-4) also grouped with the main Western Australian cluster (red in colour), indicating a possible spread, or multiple inductions to Australia from a common origin. Furthermore, Western Australian clusters were separate from those in other Australian states and overseas with more than 90% posterior probability support on the clades (Fig. 1). Maximum-Likelihood method inferred the same topology (Fig. 2).
We further analysed the mitogenomes to determine if a single gene based phylogenetic tree can inform the relatedness of the incursions with a similar resolution like that with the whole mitogenomes. A single gene based phylogenetic tree will allow for developing a fast Polymerase Chain Reaction (PCR) based tool to diagnose future browsing ant incursions without needing to sequence the entire genome46. For this purpose, we annotated a subset of L. frauenfeldi mitogenomes comprising at least a representative from each distinct clade of the phylogenetic tree (Fig. 1). Annotation of the mitogenomes showed that all 13 typical protein coding genes (PCGs) in insect mitogenomes47,48,49 were present in L. frauenfeldi (Fig. 3). We also extracted the PCG sequences from one of our L. incisa samples to be included as outgroup in the gene-based phylogenetic analysis.
We continued our analysis with 13 PCGs and performed Bayesian phylogenetic analysis using a single PCG at a time. Our results demonstrated that none of the 13 PCGs could reveal the relatedness among the incursions unlike the mitogenome based trees (Figs. 1, 2 and 5). Our findings agree with the recent studies that the whole mitogenomes reveal phylogenetic relationships at a deeper level compared to those inferred from a single mitochondrial gene41,43,44,50,51. However, a single mitochondrial gene such as cox1 might still be an effective tool for barcoding animal species for species ID, though not likely to reveal effective relatedness within that species52,53,54.
The nucleotide identity analysis (Fig. 6) of our subset of mitogenomes offered an explanation as to why a single mitochondrial gene could not reveal the relatedness of the browsing ant incursions. This was because there were only 2- 62 base variations across the entire L. frauenfeldi mitogenomes except the OK569859 mitogenome (Fig. 6). OK569859 mitogenome was retrieved from NCBI55. It showed a 606–665 base variation when compared to the other mitogenomes. This high nucleotide variation accounts for the fact that the OK569859 mitogenome sequence itself has 141 N’s, and in alignment with most of the other genomes in Fig. 6, it has several big gaps. However, these gaps and missing columns in the alignment are ignored for the tree construction purposes in both Bayesian56,57 and Maximum-Likelihood58 algorithms as these algorithms use nucleotide substitution-based models. What this all boils down to is that there is a limited phylogenetic signal in the L. frauenfeldi mitogenome. These signals are further reduced in any fragment of the L. frauenfeldi mitogenome as demonstrated by the 100% nucleotide identity between genes in the nucleotide identity matrices (Fig. 7). The 100% nucleotide identity between any pair of genes meant that the corresponding gene had no phylogenetic signal for that pair of individuals (i.e., incursions) to separate them in the tree. And there was always 100% nucleotide identity between some pairs of individuals in any of the 13 PCGs (Fig. 7).
Tay et al. 202255 is an example of how a single mitochondrial gene can be limited for determining the relatedness of the browsing ant incursions. The study carried out a phylogenetic analysis of browsing ants using a partial cox1 gene (546 bp)55. The authors have claimed that the Brisbane and Perth browsing ant incursions might originate from India (Jodhpur) and Pakistan (Punjab), respectively, because of sharing 100% cox1 sequence. The maximum likelihood based phylogenetic tree has clustered them on the same clade with more than 90% bootstrap support55. However, our analysis based on the whole mitogenome shows that this may not be the case. While there are more cox1 genes than full mitogenomes available from NCBI, we have demonstrated here that this may not be enough information to accurately determine the relatedness between incursions. As well as using the partial cox1, Tay et al., 2022 did submit their draft mitogenomes of Perth (NC_061556.1) and Brisbane (OK569859) browsing ants55 to NCBI45 and we retrieved them and included in our phylogenetic analysis (Figs. 1, 2 and 5). The mitogenome based tree clustered Perth and Brisbane browsing ants on distinctly different clades (Figs. 1, 2 and 5). Based on this analysis, we can confidently conclude that the Brisbane and Perth browsing ant incursions are separate introductions. This is in contrast to the resolution obtained by partial cox1 gene analysis55.
The entirety of our results demonstrated that whole mitogenome based phylogenetic analysis was warranted to determine the phylogenetic relationship among the incursions. Using an individual PCG including the widely used cox1 could not reveal the relatedness of the browsing ant incursions in Australia.
Furthermore, to the best of our knowledge, our results provide a world first draft mitogenome of L. incisa, a closely related species to L. frauenfeldi, therefore a suitable outgroup for this browsing ant phylogenetic analysis. Taken together, our contribution of 48 L. frauenfeldi and three L. incisa mitogenomes will serve as an excellent resource for those wanting to trace any future browsing ant infestations in Australia. This will allow us to determine the relatedness of future browsing ant incursions. In other words, we will be able to address the biosecurity questions like “are the new browsing ant infestations in Australia a spread of the existing incursions, or separate introductions from the same or separate origins, or a combination of both?”. For this biosecurity surveillance purpose, our method i.e., whole mitochondrial genome-based phylogenetic analysis has proven to be affordable, scalable, and effective. Others who wish to pursue further into the population level analysis may wish to do so using the STR or microsatellite markers.
The information on the relatedness of browsing ant incursions in Australia is crucial in evaluating the risk of finding further infestations of browsing ant across Australia, and also in assessing the continued risk of future incursion. Currently, all known infestations in Australia are under eradication, and we expect that this process will be successful. Our findings also lay the foundation for further research to determine where from overseas the separate browsing ant incursions have hailed from. If we can determine the source of our incursions, we will stand better placed to implement mitigation procedures along the high-risk pathways, and also border and post-border biosecurity measures at home to reduce our risk.
Materials and methods
Materials
Browsing ants were collected from various incursions in Western Australia, Queensland and overseas complying with all relevant institutional, national, and international guidelines and legislation. Fifty-one individuals from these incursions were used for mitochondrial genome sequencing (Table 1).
DNA extraction and sequencing library preparation
Total DNA was extracted from a leg biopsy of the collected Browsing ants using Qiagen’s DNeasy Blood & Tissue Kits (Hilden, Germany) following the manufacturer’s instructions. The extracted DNA was quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific). DNA library for MinION sequencing was prepared using Oxford Nanopore Technologies’ PCR barcoding genomic DNA (SQK-LSK109) kit following the manufacturer’s instructions. The library was quantified using a Qubit 2.0 Fluorometer.
Loading the library for sequencing
A 75 µL loading reaction was made as follows: 37.5 μL Sequencing Buffer (SQB), 25.5 μL Loading Beads (LB) and 12 μL DNA library. A priming mixture was made by adding 30 μl of thawed and mixed Flush Tether (FLT) directly into a new thawed and mixed Flush Buffer (FB) tube. Then, the library reaction and the priming mix were loaded into the flow cell (FLO‐MIN106D, R9.5) of the MinION sequencer as per the manufacturer’s instructions.
Mitochondrial genome assembly
The nanopore electrical signals were basecalled using the guppy (version 6.0.1 + 652ffd179) (https://community.nanoporetech.com/downloads) ‘high-accuracy’ mode. The reads were trimmed for adapter sequences, demultiplexed and filtered out for low quality reads. Then, the high quality reads were mapped to NC_061556.1, a Lepisiota frauenfeldi mitochondrial genome55 (Fig. 8). For the low-quality mitochondrial genome assemblies, the nanopore electrical signals were basecalled again using the guppy ‘fast’ mode and the above quality control and mapping steps were repeated. For L. incisa, the first version of the mitochondrial genome was generated by mapping the reads of a L. incisa sample (W3_1) to L. frauenfeldi, NC_061556.155. The W3_1 mitochondrial genome assembly was improved by two rounds of mapping back the W3_1 reads to the W3_1 first-version mitochondrial genome. Then, the draft W3_1 mitochondrial genome was used as a reference to map W3_2 reads using minimap259 (v 2.24-r1122), and assembly Md1_1 reads using miniasm60 (v 0.3-r179) as miniasm produced better assembly than minimap2.

Computational workflow developed and implemented for determining the relatedness of the browsing ant incursions in Australia. BLAST basic local alignment search tool, ANI, average nucleotide identity.
Phylogenetic analysis
The final mitochondrial genomes of 48 L. frauenfeldi and three L. incisa were used for the phylogenetic analysis. The mitogenomes were aligned using Geneious61 and MUSCLE62 alignment (v3.8.425) under the default parameters. The alignment was exported out of Geneious and subjected to phylogenetic analysis using both Bayesian inference56,57 and Maximum-Likelihood58 methods. For Bayesian inference-based tree, the parameters were as follows: lset nst = 6 rates = invgamma; propset ExtTBR$prob = 0; mcmc ngen = 1,000,000 printfreq = 100 samplefreq = 1000 diagnfreq = 1000 nchains = 4 savebrlens = yes; sumt burnin = 12,500; sump burnin = 12,500. For Maximum-Likelihood based tree, the parameters were as follows: raxmlHPC -f a -m GTRGAMMA -p 12,345 −× 12,345 −# 1000.
Mitochondrial genome annotation
The assembled mitochondrial genome of the browsing ants was annotated using the MITOS63 web application (http://mitos.bioinf.uni-leipzig.de/index.py) and L. frauenfeldi, NC_061556.155 as a reference. In MITOS, ‘Invertebrate’ genetic code was applied. Thresholds for the MITOS protein search parameters were default (BLAST E-value Exponent = 2, Cutoff = 50, Maximum Overlap = 20, Clipping Factor = 10, Fragment Overlap = 20, Fragment Quality Factor = 10, Start/Stop Range = 6, Final Maximum Overlap = 35). The annotated mitochondrial genomes were circularised in Geneious61 using the assembled mitochondrial genome and the GFF (General Feature Format) file containing the annotation information.
Ethical approval
No ethical approval was required as both Lepisiota frauenfeldi and Lepisiota incisa are pest species.
Data availability
The mitochondrial genomes generated in the study have been deposited in the National Centre for Biotechnology Information (NCBI) and are publicly available at https://www.ncbi.nlm.nih.gov/ under the following GenBank accession numbers: B2_4 (OQ561465), B2_5 (OQ561466), Br1_2 (OQ561467), Br1_7 (OQ561468), Br2_4 (OQ561469), Bw1_1 (OQ561470), Bw1_2 (OQ561471), Bw2_1 (OQ561472), BW2_2 (OQ561473), I1_3 (OQ561474), Md1_1 (OQ561475), NF1_2 (OQ561476), NF1_3 (OQ561477), NT1_2 (OQ561478), O1_2 (OQ561479), PA1_6 (OQ561480), PA1_9 (OQ561481), PA2_3 (OQ561482), Rk1_1 (OQ561483), Rk1_2 (OQ561484), SI_H (OQ561485), T1_1 (OQ561486), T1_2 (OQ561487), T1_3 (OQ561488), T2_3 (OQ561489), TL2 (OQ561490), W1_1 (OQ561491), W1_2_3 (OQ561492), W1_2p_1 (OQ561493), W1_2p_2 (OQ561494), W1_2p_3 (OQ561495), W1_2p_5 (OQ561496), W1_2p_6 (OQ561497), W2_1 (OQ561498), W2_2 (OQ561499), W3_1 (OQ561500), W3_2 (OQ561501), W4_1 (OQ561502), W4_2 (OQ561503), W4_3 (OQ561504), W5_1 (OQ561505), W5_2 (OQ561506), W5_3 (OQ561507), B1_5 (OQ612679), Br1_4 (OQ612680), Br2_1 (OQ612681), NF1_1 (OQ612682), NT1_1 (OQ612683), O1_3 (OQ612684), PA2_5 (OQ612685), TL1 (OQ612686).
References
Capinha, C., Essl, F., Seebens, H., Moser, D. & Pereira, H. M. The dispersal of alien species redefines biogeography in the Anthropocene. Science 348, 1248–1251 (2015).
Xu, Y., Vargo, E. L., Tsuji, K. & Wylie, R. Exotic ants of the asia-pacific: Invasion, national response, and ongoing needs. Annu. Rev. Entomol. 67, 27–42 (2022).
Ingram, K. K., Bernardello, G., Cover, S. & Wilson, E. O. The ants of the Juan Fernández islands: Genesis of an invasive fauna. Biol. Invasions 8, 383–387 (2006).
Suarez, A. V., Holway, D. A. & Ward, P. S. The role of opportunity in the unintentional introduction of nonnative ants. Proc. Natl. Acad. Sci. 102, 17032–17035 (2005).
Siddiqui, J. A. et al. DNA barcoding of aphid-associated ants (Hymenoptera, Formicidae) in a subtropical area of southern China. ZooKeys 879, 117–136 (2019).
Ivanov, K. Exotic ants (Hymenoptera, Formicidae) of Ohio. J. Hymenopt. Res. 51, 203–226 (2016).
Holway, D. A., Suarez, A. V. & Case, T. J. Loss of intraspecific aggression in the success of a widespread invasive social insect. Science 282, 949–952 (1998).
Abbott, K. L. Spatial dynamics of supercolonies of the invasive yellow crazy ant, Anoplolepis gracilipes, on Christmas Island, Indian Ocean. Divers. Distrib. 12, 101–110 (2006).
Mack, R. N. et al. Biotic invasions: Causes, epidemiology, global consequences, and control. Ecol. Appl. 10, 689–710 (2000).
Cheng, S., Zeng, L. & Xu, Y. Mutualism between fire ants and mealybugs reduces lady beetle predation. J. Econ. Entomol. 108, 1560–1569 (2015).
Wu, D., Zeng, L., Lu, Y. & Xu, Y. Effects of Solenopsis invicta (hymenoptera: formicidae) and its interaction with aphids on the seed productions of mungbean and rapeseed plants. J. Econ. Entomol. 107, 1758–1764 (2014).
Morrison, J. E. Jr., Williams, D. F. & Oi, D. H. Effect of crop seed water content on the rate of seed damage by red imported fire ants (hymenoptera: formicidae). J. Econ. Entomol. 92, 215–219 (1999).
González-Hernández, H., Johnson, M. W. & Reimer, N. J. Impact of Pheidole megacephala (f.) (hymenoptera: formicidae) on the biological control of Dysmicoccus brevipes (cockerell) (homoptera: pseudococcidae). Biol. Control 15, 145–152 (1999).
Nelson, S. & Taniguchi, G. Ant Damage to Banana Fruits by Abdominal Secretions (University of Hawaii, 2012).
Sether, D. M., Ullman, D. E. & Hu, J. S. Transmission of pineapple mealybug wilt-associated virus by two species of mealybug (Dysmicoccus spp.). Phytopathology® 88, 1224–1230 (1998).
Teal, S., Segarra, E., Barr, C. & Drees, B. The cost of red imported fire ant infestation: the case of the texas cattle industry. Tex. J. Agric. Nat. Resour. 12, 86–95 (1999).
Lard, C. F., Hall, C. R. & Salin, V. The economic impact of the red import fire ant on the homescape, landscape, and the urbanscape of selected metroplexes of Texas: A part of the Texas fire ant initiative 1997–1999. Tex. AM Univ. Dep. Agric. Econ. Fac. Pap. Ser. https://doi.org/10.22004/ag.econ.23994 (2001).
Pereira, R. M., Williams, D. F., Becnel, J. J. & Oi, D. H. Yellow-head disease caused by a newly discovered Mattesia sp. in populations of the red imported fire ant Solenopsis invicta. J. Invertebr. Pathol. 81, 45–48 (2002).
Wylie, F. R. & Janssen-May, S. Red imported fire ant in Australia: What if we lose the war?. Ecol. Manag. Restor. 18, 32–44 (2017).
Gotelli, N. J. & Arnett, A. E. Biogeographic effects of red fire ant invasion. Ecol. Lett. 3, 257–261 (2000).
Porter, S. D. & Savignano, D. A. Invasion of polygyne fire ants decimates native ants and disrupts arthropod community. Ecology 71, 2095–2106 (1990).
Liu, Y.-S. et al. Establishment and social impacts of the red imported fire ant, Solenopsis invicta, (hymenoptera: formicidae) in Taiwan. Int. J. Environ. Res. Public. Health 18, 5055 (2021).
MacConnell, J. G., Blum, M. S. & Fales, H. M. The chemistry of fire ant venom. Tetrahedron 27, 1129–1139 (1971).
More, D., Kohlmeier, R. & Hoffman, D. Fatal anaphylaxis to indoor native fire ant stings in an infant. Am. J. Forensic Med. Pathol. 29, 62–63 (2008).
Wang, L. et al. Mental health effects caused by red imported fire ant attacks (Solenopsis invicta). PLoS ONE 13, e0199424 (2018).
Morrison, L. W. Long-term impacts of an arthropod-community invasion by the imported fire ant. Solenopsis invicta. Ecology 83, 2337–2345 (2002).
Dziadzio, M. C., Long, A. K., Smith, L. L., Chandler, R. B. & Castleberry, S. B. Presence of the red imported fire ant at gopher tortoise nests. Wildl. Soc. Bull. 40, 202–206 (2016).
Colunga-Garcia, M., Magarey, R. A., Haack, R. A., Gage, S. H. & Qi, J. Enhancing early detection of exotic pests in agricultural and forest ecosystems using an urban-gradient framework. Ecol. Appl. 20, 303–310 (2010).
Commonwealth of Australia. Browsing ant | National pest & disease outbreaks. https://www.outbreak.gov.au/current-responses-to-outbreaks/browsing-ant (2022).
Bolton, B. Antweb. https://www.antweb.org/page.do?name=world (2022).
Widmer, M. Browsing ants. https://www.agric.wa.gov.au/biosecurity/browsing-ants (2021).
Stanley, U. N., Khadija, A. M., Bukola, A. T., Precious, I. O. & Davidson, E. A. Forensic DNA profiling: Autosomal short tandem repeat as a prominent marker in crime investigation. Malays. J. Med. Sci. MJMS 27, 22–35 (2020).
Merheb, M. et al. Mitochondrial DNA, a powerful tool to decipher ancient human civilization from domestication to music, and to uncover historical murder cases. Cells 8, 433 (2019).
Behura, S. K. Insect phylogenomics. Insect Mol. Biol. 24, 403–411 (2015).
Cameron, S. L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Galtier, N., Nabholz, B., Glémin, S. & Hurst, G. D. D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 18, 4541–4550 (2009).
Rooney, J. et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. Clifton NJ 1241, 23–38 (2015).
Sato, M. & Sato, K. Maternal inheritance of mitochondrial DNA. Autophagy 8, 424–425 (2012).
Hagström, E., Freyer, C., Battersby, B. J., Stewart, J. B. & Larsson, N.-G. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 42, 1111–1116 (2014).
Xu, S. et al. High mutation rates in the mitochondrial genomes of Daphnia pulex. Mol. Biol. Evol. 29, 763–769 (2012).
Sullivan, K. A. M., Platt, R. N., Bradley, R. D. & Ray, D. A. Whole mitochondrial genomes provide increased resolution and indicate paraphyly in deer mice. BMC Zool. 2, 11 (2017).
Lavrov, D. V. & Pett, W. Animal mitochondrial DNA as we do not know it: mt-genome organization and evolution in nonbilaterian lineages. Genome Biol. Evol. 8, 2896–2913 (2016).
Chan, Y.-C. et al. Mitochondrial genome sequences effectively reveal the phylogeny of Hylobates gibbons. PLoS ONE 5, e14419 (2010).
Song, N., Cai, W. & Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 7, 10429 (2017).
NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46, D8–D13 (2018).
Dudas, G. & Bedford, T. The ability of single genes vs full genomes to resolve time and space in outbreak analysis. BMC Evol. Biol. 19, 232 (2019).
Lee, C.-C., Wang, J., Matsuura, K. & Yang, C.-C.S. The complete mitochondrial genome of yellow crazy ant, Anoplolepis gracilipes (hymenoptera: formicidae). Mitochondrial DNA B Resour. 3, 622–623 (2018).
Kumar, V. et al. The first complete mitochondrial genome of marigold pest thrips, Neohydatothrips samayunkur (Sericothripinae) and comparative analysis. Sci. Rep. 9, 191 (2019).
Hyde, J. et al. The first complete mitochondrial genomes of subterranean dytiscid diving beetles (Limbodessus and Paroster) from calcrete aquifers of Western Australia. Aust. J. Zool. 65, 283–291 (2018).
Duchêne, S., Archer, F. I., Vilstrup, J., Caballero, S. & Morin, P. A. Mitogenome phylogenetics: The impact of using single regions and partitioning schemes on topology, substitution rate and divergence time estimation. PLoS ONE 6, e27138 (2011).
Zhang, L. et al. Complete mitochondrial genomes reveal robust phylogenetic signals and evidence of positive selection in horseshoe bats. BMC Ecol. Evol. 21, 199 (2021).
Robba, L., Russell, S. J., Barker, G. L. & Brodie, J. Assessing the use of the mitochondrial cox1 marker for use in DNA barcoding of red algae (Rhodophyta). Am. J. Bot. 93, 1101–1108 (2006).
Goodall-Copestake, W. P., Tarling, G. A. & Murphy, E. J. On the comparison of population-level estimates of haplotype and nucleotide diversity: a case study using the gene cox1 in animals. Heredity 109, 50–56 (2012).
Ward, R. D. & Holmes, B. H. An analysis of nucleotide and amino acid variability in the barcode region of cytochrome c oxidase I (cox1) in fishes. Mol. Ecol. Notes 7, 899–907 (2007).
Tay, W. T., Court, L. N., Hoffmann, B. D. & Polaszek, A. Draft mitogenomes of the invasive ant Lepisiota frauenfeldi (Mayr 1855) (Hymenoptera: Formicidae). Mitochondrial DNA B 7, 1183–1185 (2022).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016).
Geneious Prime 2021.2.2 (https://www.geneious.com).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Rambaut, A. FigTree. http://tree.bio.ed.ac.uk/software/figtree/.
Acknowledgements
We thank Dr Ben Hoffmann (Commonwealth Scientific and Industrial Research Organisation, Northern Territory, Australia) and Riki Fulton (National Browsing Ant Eradication Program, Northern Territory, Australia) for suppling Browsing ant samples from Hawaii and the Northern Territory. We thank the Pawsey Supercomputer Centre (https://pawsey.org.au/) for the computing resources.
Author information
Authors and Affiliations
Contributions
M.A.P., T.K. and M.K. conceptualised the study. M.W. collected and supplied the ants. M.A.P. performed DNA extractions, prepared Oxford Nanopore sequencing libraries, generated sequencing data using MinION sequencer, developed, and carried out the bioinformatics workflow for reconstructing the Browsing ant mitochondrial genomes, performed mitochondrial genomic analysis, and interpreted the outputs. T.K. performed DNA extractions, prepared Oxford Nanopore sequencing libraries, and generated sequencing data using MinION sequencer. All authors critically discussed the analyses and results. M.A.P. wrote the manuscript. M.W. wrote part of the introduction section. M.K., T.K. and M.W. provided editorial comments. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prodhan, M.A., Widmer, M., Kinene, T. et al. Whole mitochondrial genomes reveal the relatedness of the browsing ant incursions in Australia. Sci Rep 13, 10273 (2023). https://doi.org/10.1038/s41598-023-37425-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-023-37425-1
This article is cited by
-
Mitochondrial genome of Bactrocera fruit flies (Tephritidae: Dacini): features, structure, and significance for diagnosis
BMC Genomics (2025)
-
Mitogenomic Architecture of Mycetophylax conformis (Hymenoptera: Formicidae): Evidence for Conserved Gene Order and Strand Asymmetry in Myrmicinae
Biochemical Genetics (2025)
