
Abstract
The whole genome sequence (WGS) of prevalent MRSA strains harboring mecA gene obtained from skin and soft tissue infections (SSTIs) in Nigerian hospitals were profiled for pathogenomic structure and evaluated for clonal diversity. The two MRSA strains identified among 66 isolated multi-drug resistant S. aureus from a collection of 256 clinical samples were phenotyped for antibiotic resistance and genotyped for mecA, SCCmec, and spa types. The mecA positive MRSA was analysed using whole-genome sequencing for resistomes, virulomes, phylogenomic profiles and clonal diversity. The identified MRSA-CC7-ST789-t091-SCCmecV strains from a female child (aged 1 year) with severe otorrhea and an adult male (aged 23) with purulent wound abscess showed high-level resistance to streptomycin, vancomycin, kanamycin, sulfamethoxazole and ciprofloxacin. Both strains harbored abundant resistomes, inherent plasmids, chromosomal replicons and typical seven housekeeping genes (arc3, aroE4, glpF1, gmk4, pta4, tpi6, yqiL3). The most abundant putative virulomes were pathogenesis-associated proteins (included hemolysin gamma, leucocidins, proteases, staphylococcal superantigen/enterotoxin-like genes (Set/Ssl), capsule- and biofilm-associated genes, and hyaluronate lyase). Comparative phylogenomic analysis revealed the relatedness of the two clonal strains with prevalent MRSA-CC7 pathotypes observed in Italy (2013 and 2014), Denmark (2014), Thailand (2015 and 2016), USA (2018), and Nigeria (2016 and 2020); and share high genetic similarities with livestock strains from cow milk and cattle. Identified MRSA-CC7-ST789-t091-SCCmecV pathotypes implicated in SSTIs from Nigeria harboring repertoires of antibiotic resistance and virulence genes, and genetic relatedness with livestock strains; show the possibility of gene transfer between animal and human. Adequate hospital MRSA infection control and geno-epidemiological surveillance for animal and human transfer is required.
Similar content being viewed by others


Introduction
In the last two decades, there have been several reports on the emerging clones of methicillin-resistant Staphylococcus aureus (MRSA) implicated in various systemic infections ranging from mild skin and soft tissue infections (SSTIs) to life-threatening septicemia, endocarditis, toxic shock, and necrotizing pneumonia1. Globally, MRSA was widely reported as a multidrug resistant pathogen to diverse narrow and extended-spectrum antibiotics2. The ability to adapt to various ecological niches enhance the continuous dissemination of MRSA and epidemiology in hospital- and community-acquired infections3,4.
Community-acquired MRSA (CA-MRSA) was reported in SSTIs with different severity in skin abscess, necrotizing fasciitis, and pneumonia associated with high morbidity5,6. Select CA-MRSA strains possess distinctive Panton-Valentine leukocidin (PVL) and other considerable virulence determinants associated with SCCmec type IV or type V with very low antibiotic susceptibility7,8,9. Global distribution of various human clonal complexes, particularly CC7 is not commonly reported in humans but is highly noted in livestock10. The emergence and evolution of MRSA in different ecological niches and host species, including the involvement of its associated mobile genetic elements (MGEs) for horizontal transfer of genetic determinants gave rise to the acquisition of antibiotic-resistance genes and new genotypes (such as livestock- and human-associated CC398)11,12.
Although CC9 is usually reported in Asia13, the United States14, Europe15, and Africa16, the carriage of CC7 frequently reported in animal populations is now recognized as newly emerging zoonotic strains in humans15,16. Phenotypic methods and PCR genotyping have not provided a robust clonal signature as whole genome sequencing (WGS) for the evaluation of CA-MRSA and Livestock-associated MRSA (LA-MRSA) infections or related zoonotic diseases. However, the current study on the genome structure of implicated strains from human source further reveals the evolution of virulent MRSA with high-level antibiotic resistance in SSTIs (wound and ear infections) in respect to niche adaptability.
Furthermore, comparative genomic analyses provide phylogenetic diversity of strains with zoonotic potential to establish infection, exhibiting evolutionary traits expressed through defined core genome composition for cell maintenance, survival, colonization, and infection processes through accessory species-specific surface and virulence regulatory proteins17. Recent findings of animal MRSA phylogeny from livestock, colonizing the human tissue to cause systemic infection16, portray a growing concern for the increasing human nosocomial MRSA-CC7 infections.
The influence of encoded plasmids on virulome and resistome transfer allows high dissemination of zoonotic MRSA pathotypes leading to high pathogenicity and disease severity. Despite many reports on the prevalence of MRSA strains in the last few years13,15,16, data on pathogenomic profile, phylogenetic relatedness and specific features relating to the transmission, infectivity and persistence of zoonotic MRSA-CC7 in SSTIs are lacking. Therefore, this study profiles the pathogenomic structure and clonal diversity of emerging human MRSA-CC7-ST789-t091-SCCmecV clonal traits with high zoonotic potential from SSTIs.
Methods
Isolates collection and ethical approval
A collection of 256 clinical samples from skin and soft tissue infections (SSTIs) (including purulent discharges, skin aspirates and effusions, wounds, otorrhea, eye infection, throat, and endocervical samples) were obtained from overall 12,654 patients attending Federal Medical Centre, Abeokuta Nigeria between June 2017 and August 2018. Two PCR-genotyped MRSA strains encoded with mecA gene from previous multi-drug resistant S. aureus (n = 66) isolates from clinical samples were selected for pathogenomic profiling17. These MRSA strains were re-phenotypically characterized on Baird-Parker agar and Mannitol salt agars, Gram stained for cellular morphology, and examined for catalase and coagulase production as previously discussed18. We confirm that all methods were carried out in accordance with relevant guidelines and regulations. Ethical permission for the study was granted by the Federal Medical Centre Abeokuta Health Research Ethics Committees with protocol approval: FMCA/470/HREC/09/2017; NHREC/08/10–2015 and informed consent of individual subjects and/or their legal guardian(s) was obtained with permission.
Strain genotyping and resistance phenotyping
The MRSA isolates were re-assessed for resistance to oxacillin (10 µg/disc) at an inhibition zone ≤ 21 mm18. The strains were further confirmed using real-time polymerase chain reaction (PCR) targeting the nuc and tuf (gene marker for Staphylococcus spp), mecA (marker for MRSA) and pvl (gene encoding Panton-Valentine leukocidin) (Table S1) as previously described19. SCCmec typing was based on a multiplex PCR that discriminates between Types I, II, III, IVa, IVb, IVc, IVd, and V. The amplification reaction was carried out in a volume of 25 µl containing 10 µl of 2 × MyTaq HS Mix, 9 µl SCCmec primer mix, 5 µl water, and 1 µl Template DNA (Table S2) as previously described17,19. Typing for S. aureus protein A (spa gene) was performed in constituted reaction mixture of 2 × MyTaq HSMix (10 µL), containing spa primers; spa1095F (5’-AGACGATCCTCCGGTGAGC-3’), spa1517R (5’ GCTTTTGCAATGTCATTTACTG-3’) of 5 µL each and 1 µL DNA. The reaction was followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, elongation at 72 °C for 30 s, and final extension at 72 °C for 5 min19. The obtained amplicon products were purified with GFX PCR DNA and Gel Band Purification Kit (GE Healthcare), sequenced with forward primer spa1095F using BigDye 3.1 terminator sequencing, and analyzed on ABI Genetic Analyzer 3500Dx (Applied Biosystems, CA, USA). The categorization of spa types was performed with the Based Upon Repeat Pattern (BURP) algorithm of the Ridom Staph Type software version 1.4 (RidomGmbH, Sedanstr, Germany)20. The antibiotic resistance profile of the confirmed MRSA isolates was determined by broth microdilution as previously described17 with 12-panel antibiotics consist of tetracycline (TE), ceftazidime (CAZ), ciprofloxacin (CIP), gentamycin (GEN), amoxycillin-clavulanic acid (AMC), cefuroxime (CRO), ofloxacin (OFX), sulfamethoxazole (SXT), erythromycin (E), fosfomycin (FOX), vancomycin (VA) and Linezolid (LZD). Methicillin-resistant S. aureus (ATCC 43,300) and Methicillin-sensitive strain S. aureus (ATCC 29,213) were used as controls. The phenotypic resistance was interpreted according to CLSI guidelines21.
DNA Extraction and whole genome sequencing
Characterized MRSA isolates were cultured on sheep blood agar (Oxoid GmbH, 46,483, Wesel, Germany), and DNA was extracted from a colony using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Germany) according to the manufacturer’s protocol modified by adding of 10 ml lysostaphin to the lysis buffer. DNA library was prepared using an Illumina Nextera DNA Flex kit (Illumina Inc., United States) and 150 bp paired-end sequencing run was done on an Illumina NextSeq 500 instrument (Illumina Inc., San Diego, CA, USA). The sequence reads were assessed for quality, trimmed, and de novo assembled with the in-house developed Aquamis pipeline (https://gitlab.com/bfr_bioinformatics/AQUAMIS/), which implements fastp for trimming and shovill (based on SPAdes) (https://github.com/tseemann/shovill) for assembly22. Reference search for assembly quality was performed with minimal coverage depth of > 80 using mashv2.123 and quast v 5.0.224. The quality of assemblies was checked for single-copy and duplicated ortholog analyses. The fraction majority species was > 0.97, and the total genome length was > 2.5 Mbp.
Bioinformatics analysis
Genome visualization, annotation, and antibiotic resistome profiling
The whole genomic sequences of S. aureus were annotated with the PATRIC v.3.5.392 tools for comparative genomic analysis (including specialty genes, functional subsystems categorization (Table S3), and Phylogenetic diversity)25. The assembled genome of the two MRSA strains as shown in Table S3 were annotated using RAST tool kit (RASTtk) available at http://rast.nmpdr.org/26, providing the size, GC content, number of contigs, N50, L50, average coverage and the number of RNAs and protein-coding sequences. A circular graphical display of the distribution of the genome annotations is provided (Fig. 1A). This includes outer to inner rings, the contigs, CDS with homology to known antimicrobial resistance genes, virulence factors, and other related genes belonging to several subsystems (Fig. 1B). The WGS-Based typing of the two MRSA for SCCmec types was determined in-silico using the web-based tool SCCmecFinder (https://cge.cbs.dtu.dk/services/SCCmecFinder)27 and Spa types were analysed in-silico using the assembled genomic sequences on the online platform tool Spa Typer 1.028. WGS data online platform tool MLST 1.829 was used to analyse for the Multilocus sequence typing (MLST) and eBURST30 for all sequence types (STs) in the MLST database (http://saureus.mlst.net) to assign clonal complexes (CC).

(A). Illustrated circular map of the MRSA-CC7-ST789-t091-SCCmecV (2,578,347 bp) (B).The CDS on the strain genome indicate the subsystem (set of proteins) that implement a specific biological process or structural complex8.
Antibiotic resistome profiling
For comprehensive antibiotic resistance profiling, the GoSeqIt via ResFinder pipeline, Antibiotic Resistance Gene-Annotation database (ARG-ANNOT)31, and comprehensive antibiotic resistance database (CARD)32 tools were used to annotate and identify antibiotic resistance genes. To determine the associated resistomes that were plasmid-borne or chromosomal-borne, the contigs of these resistance genes were predicted from the BLASTn analysis based on nucleotide homologies. Plasmid replicons were predicted through PlasmidFinder33.
Pathogenicity and virulome predictions
To predict the pathogenicity in human hosts, PathogenFinder available at https://cge.cbs.dtu.dk/services/PathogenFinder/34 was used, and annotated proteins with functional assignments were mapped to KEGG pathways35. PATRIC annotation includes two types of protein families25, and this genome has 2,613 proteins that belong to the genus-specific protein families (PLFams) and 2,648 proteins that belong to the cross-genus protein families (PGFams). Virulence determinants (sequences and functions) were analyzed with the pathogenic bacteria database using the NCBI BLASTN tool25,35.
Comparative phylogenomic analysis and metadata insights
The endemic clonal subtype genome with more than 90% homologous sequence types downloaded from the NCBI database (https://www.ncbi/) were analyzed with PATRIC for genomic comparison and phylogenomic analysis via CSI Phylogeny-1.436. The reference and representative genomes the PATRIC provided were included in the phylogenetic analysis and identified by Mash/MinHash24. PATRIC global protein families (PGFams)25 were selected from these genomes to determine the phylogenetic placement of this genome. The protein sequences from these families were aligned with MUSCLE37 and the nucleotides for each of those sequences were mapped to the protein alignment. The joint set of nucleotide alignments were concatenated into a data matrix, and RaxML38 was used to analyze this matrix, with fast bootstrapping of 100 replicates to generate phylogenetic tree38.
Data analysis and availability
Global prevalence and distribution of the strain subtype from 2015 to 2020 (Table S4) were analyzed and visualized in Bubble chart created with GraphPad Prism 8.0. The whole-genomes of the two strains were deposited in NCBI SRA GenBank with BioProject number PRJNA1073721 with accession no. SAMN39837611 and SAMN39837610 for the strains from wound abscess and ear infection respectively.
Ethics approval and consent to participate
Written ethical permission for the study was obtained from Federal Medical Centre, Abeokuta Nigeria which serves as a referral clinic for internal medicine in Southwest Nigeria with approval number: FMCA/470/HREC/09/2017 and NHREC/08/10-2015.
Result
Demographics, phenotypic resistance, and expressed gene characteristics
From a total of 66 S. aureus isolates from clinical samples of diagnosed skin and soft tissue infection (including purulent discharges, skin aspirates and effusions and wounds), two MRSA genotypes were detected. MRSA strain from a female child aged 1 year with severe otorrhea showed phenotypic resistance to streptomycin, vancomycin, kanamycin, sulfamethoxazole and ciprofloxacin with MIC ranging from 16 to 64 µg/mL. Other MRSA strain from a male adult aged 23 with purulent wound abscess showed similar resistance to gentamycin with MIC 16–128 µg/mL. Both isolates expressed similar gene markers (nuc, tuf, mecA genes) with the same clonal complex (CC7), spa type (t091) and harboured SCCmec type V referring to MRSA-CC7ST789-t091-SCCmecV (Table 1).
Strain genome annotations and resistance
Figure 1A revealed the WGS circular map of the MRSA-CC7-ST789-t091-SCCmecV annotations and differences in genome structure of the two strains including the size (ear MRSA, 2,815,094 bp and wound-MRSA, 2,790,159 bp), coding DNA sequences, RNA, antimicrobial resistance genes, virulence factors, and GC content (Table S3). The CDS on the strain genome indicates the subsystems (set of proteins) that implement a specific biological process or structural complex8. The CC7-ST789-t091-SCCmecV annotations revealed the proportion of genes belonging to subsystem that comprised metabolism (83, 540), protein processing (40, 210), stress response, defense, virulence (36, 128), energy (26, 178), DNA processing (20, 94), cellular processes (15, 88), RNA processing (13, 52), membrane transport (11, 72), and regulation and cell signaling (4, 16) (Fig. 1B). The antibiotic susceptibility patterns of the isolates were further confirmed, showing several putative acquired resistance genes and chromosomal mutations (Table 2). The clones exhibited resistomes of aminoglycoside modifying enzymes (aph(3')-III, aac(6')-aph(2'') and ant(6)-Ia)), methicillin (mecA) and beta-lactamase (blaZ) genes, protein synthesis inhibitor for tetracycline (tetK), dfrG mediating high-level resistance to trimethoprim, antibiotic resistance gene cluster, cassette, or operon including TcaA, TcaB, TcaB2, TcaR, gyrA and gyrB mutation enhancing resistance to fluoroquinolones, rifampicin resistance genes (rpoB, rpoC), gidB conferring Streptomycin resistance and efflux pump conferring antibiotic resistance (NorA). The strains harboured resistance determinants coding for protein altering cell wall charge, and gene regulators modulating the antibiotic resistance expression. Both MRSA harbour inherent plasmid and chromosomal replicons and revealed seven typical housekeeping genes (arc3, aroE4, glpF1, gmk4, pta4, tpi6, yqiL3).
Putative virulome Factor Predictions
Annotation of the whole-genome virulome analysis of pathogenesis-associated proteins included significant functional assignments for the catalytic enzyme (with Commission numbers based on oxidation, reduction, hydrolysis, group transfer), pathways (ppa), genus-specific family (PLfam) and cross-genus family (PGfam) (p < 0.05; Fig. 2A). Of these protein-coding sequences (CDS per 10000 bp), more than Log20.0620 were significantly expressed (p < 0.05; Fig. 2B). Highest level of aureolysin (toxin), Staphylokinase; (hostimm); Leukocidin D, Leukocidin E and.

(A).Protein annotation for hypothetical protein (hp), functional assignments (fp), catalytic enzyme commission numbers (EC), Gene Ontology (GO) assignments, proteins mapped with KEGG pathways assignments (ppa), genus-specific family (PLfam) assignments and cross-genus family (PGfam) assignments (*p < 0.05; Fig. 2A). (B). protein coding sequences (CDS) proportional to the detection frequency (C). Virulome detection rate (scn, Staphylococcal complement inhibitor; sak, Staphylokinase; splA, Serine protease A; splB, Serine protease B; splE, Serine protease E; aur, Aureolysin; sep, Enterotoxin; hlgA, Gamma-hemolysin chain II precursor; hlgB, Gamma-hemolysin component B precursor; hlgC, Gamma-hemolysin component C precursor; lukD, Leukocidin D; lukE, Leukocidin E).
Gamma-hemolysin component B precursors (exoenzymes) were predominant virulence-encoded pathogenesis-associated proteins (with functionalities for adherence, toxin regulation, motility, antiphagoctyosis, invasion, and biofilm formation (Fig. 2C).
The clonal strain obtained from the wound infection possessed immunodominant antigen, defensin resistance, transferrin binding protein, putative transporter, Type I restriction-modification system, single sequence specificity protein, and hyaluronate lyase (Table 3).
Comparative phylogenomic diversity
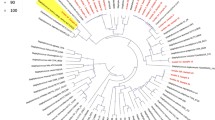
The phylogenomic analysis revealed related clonal complex CC7 with other global pathotypes. High prevalence trend of MRSA-CC7 clonal pathotypes was observed in the year 2010 to 2020 in Italy (2013, 2014), Denmark (2014), Thailand (2015 and 2016), USA (2018), and Nigeria (2016, 2020) (Table S3; Fig. 3A). The codon tree-based phylogenetic analysis of MRSA isolates (labeled red dots) showed genetically related MRSA clones built with the PATRIC pipeline, with a set of 1000 conserved core genes. The phylogenetic relationship and epidemiological distribution of all deposited MRSA-CC7 genomes provided the prevalent human clonal types from Nigeria with diverse human and animal strains (Fig. 3B). Further investigation of the clonal diversity revealed genetic similarity of the human strain MRSA-CC7 clustering with animal strains (Staphylococcus aureus subsp. aureus CP010526.1) in cow milk from Indian and Staphylococcus aureus subsp. aureus N315 158879.11 in cattle from the USA (Fig. 4).

(A). Bubble chart showing the related MRSA-CC7 strain occurrences and distribution over the years in various countries (B) Rooted phylogenetic tree of clonally related MRSA isolates (red dots indicate the studied strains). A graphical illustration of phylogenetic tree including global strains (n = 49) and their respective accession number with strains from the present study (red dots) clustering into different clades (A, B1, B2, C, D and E). The two MRSA strains from this study clustered with human and animal strains depicting a genetic relatedness (red rectangle).

Maximum likelihood phylogenetic tree of clonally related MRSA isolates. A phylogenetic tree reporting the clustering of MRSA genome from SSTIs with livestock-associated MRSA CC7 strains (red highlight) sharing similar genomic composition. Having different clusters further indicates an important genetic diversity with endemic clonal subtypes.
Discussion
In the last two decades, MRSA infection has gained enormous attention, particularly in hospital settings with increasing morbidity and persistent nosocomial infections, which has become a major public health issue. Wound infections (including abscesses, surgical sites, or burn wounds) are suitable niches for Staphylococci colonization due to the enormous repertoire of genes encoding for tissue tropism due to contaminated dressing materials, adulterated or substandard antiseptic agents and poor hygiene or hospital environment39. Recurring staphylococci ear infections in children are common phenomenon of the middle ear passage, facilitated by poor clinical management by quacks, unguided application of antibiotic ear drops for treatment, and unhygienic perforation of the ear lobe5. Similar multidrug resistance MRSA in ear infections were earlier reported in different communities having population with declining socio-economic status3,5.
The phenotypic resistance of the identified strains further suggests a possible non-inherited resistance associated with specific processes such as biofilm formation or stationary growth phase40. This resistance in aminoglycosides (streptomycin and kanamycin), glycopeptide (vancomycin) and fluoroquinolone (ciprofloxacin) is commonly reported41, rendering several classes of antibiotics as treatment option unachievable.
The SCCmec_V encoding mecA gene (mediating methicillin resistance) co-carried other resistance genes, further provided evidence for high–level resistance of these strains but WGS gave more insight into functional and wide arrays of virulomes enhancing the strain colonization, transmission, and pathogenesis7,8. The annotated genome of the MRSA-CC7-ST789-t091-SCCmecV provides arrays of coding DNA sequences (CDS) involved in various functional processes attributed to the conservation of the strain evolutionary trait, metabolic variability, and cellular processes17. The observed staphylococcal draft genomes of 2.8 Mbp with low GC content expressed numerous assigned CDS on the strain genome subsystems that regulate specific and functional biological processes (carbohydrate, protein, amino acid synthesis, regulatory and cell signaling systems) and structural complexes (cell wall and capsule formation, cell division) which are essential for survival, colonization, and pathogenesis34,42.
The present MRSA-CC7 strains carried a broad repertoire of antimicrobial resistance genes which agrees with the phenotypic antibiotic class resistance pattern. Antibiotic resistance gene cluster, cassette, or teicoplanin-associated operons (TcaA, TcaB, TcaB2, TcaR), functionally regulate protein pathways in response to the expression of resistance. The aph(3')-III and aac(6')-aph(2'') resistomes encode a bi-functional enzyme that confers resistance to a broad spectrum of aminoglycosides while mecA and blaZ mediate inactivation of beta-lactam antibiotics43. The identified classical gene clusters including protein synthesis inhibitors for tetracycline (tet(K)) and trimethoprim (dfrG), mutations in genes encoding DNA gyrA and gyrB conveying resistance to fluoroquinolone (particularly to ciprofloxacin with MIC 16 µg/ml and 32 µg/mL as shown in Table 1), inhA, rpoB and rpoC gene mutation causing resistance to isoniazid and rifampicin respectively31,43, further increase the resistance of these strains to many antibiotics. The efflux pumps activity controlled by the cascaded mechanism by BceA, BceB, and NorA enhance the inactivation of several antibiotics44. Expression of plasmid and chromosomal replicons indicate the longtime evolution of replicons gene sequences along with the current host with the capability for possible intergeneric transfer. WGS profiling has further aided classification of the strain as defined by the Multilocus sequence types based on the seven housekeeping genes (arc3, aroE4, glpF1, gmk4, pta4, tpi6, yqiL3), differentiation of the alleles for clonal complexes (CCs), spa types and SCCmec compares to phenotypic characterization and biotyping schemes29,45.
The annotated clonal strain CC7-ST789-t091-SCCmecV from wound and ear infection expressed hypothetical proteins with functional assignments covering the strain‐specific information on functional and molecular properties, enzyme functionality for differential catalysed reaction, kinetics, substrates/products, inhibitors, co-factors, activators, structure and stability (Fig. 1). Other proteins mapped to KEGG pathways provide insight into high-level functional virulence and other regulatory proteins belonging to cross-genus protein families35. Collections of putative virulence factors play significant roles in the pathogenesis and survival of these pathotypes. Significant expression of aureolysin suggests the ability of the strains to inhibit phagocytosis processes and the reduction or killing of neutrophils through the cleavage process of the central complement protein C37,46. The evidence of staphylokinase secretion predicted by the hostimm ensures persistent tissue tropism, promoting the colonization and penetration of the skin barriers by activating plasminogen7,47. However, the interaction of staphylokinase and plasminogen induce high rate of abscesses which could be seen in ear infection48. Moreover, the endemic clone encoded variants of PVL (Leukocidin D and Leukocidin E components) that are major constituents of a bi-component layer with gamma hemolysins induce cell activation, causing Ca2 + influx and apoptosis of the host cell13,17,49. Expression of LukED promotes S. aureus replication in vivo through direct killing of phagocytes, thereby promoting the survival of MRSA clones and further restrict neutrophil-mediated killing; and promote tissue pathogenesis17,49. Previous studies have not provided the linkage between SCCmec type V and PVL production50,51, but differential expression of PVL and other putative virulence determinants provide a remarkable contribution to the colonization, virulence, and adaptability of these pathotypes in hospital-acquired infections. Predominant virulomes encoding pathogenesis-associated proteins for adherence, toxin regulation, antiphagoctyosis, invasion, and biofilm formation indicate developed complex mechanisms for adaptability and survival during exposure to various environmental stressors in hostile biological niches42,49,52.
Moreover, the virulence-encoded pathogenesis-associated factors and encoded repertoire of virulence genes from diverse sources predominantly aid the regulation of toxin production which induces host cell membrane damage and triggers cytokine formation53,54. Detection of protease and capsule- and biofilm-associated genes further confirms the ability to intensify necrotization of epithelial mucosa and functional invasive mechanism and enhance attachment and colonization with biofilm production55,56.
Phylogenomic analysis with related clonal complex CC7 showed a clear distinction with other globally reported strains belonging to the same clonal complex. The prevalence trend of the clone in the last ten years indicates persistent and recurring distribution from 2013 to 2020 in Asia, Europe, America, and Africa. Over time, the prevalence of these clones increased steadily over the continents and became the predominant clone from 2015 to 2018 (Table S3; Fig. 3A), driving a wave of new evolutionary traits of possible epidemic potential. In this study, the obtained clusters of the global and two studied MRSA strains highlight related clonal clusters (Fig. 3B) while the studied MRSA strains were further found to cluster separately with human and animal strains suggesting unique clonal types with high clonal expansion with other MRSA-CC710,36,57.
Considering the potential of WGS to highlight and provide a tentative interpretation of the genetic differences with metadata, there is a high-level relatedness of these human clones with animal strains depicting a zoonotic structure having conserved virulence determinants associated with the clonal subtypes. The implication of differential resistome and virulome determinants separating these clones from other phylogenetic pathotypes should be explored to provide information on the dynamics of strain evolution. The clonal similarity of Nigeria strains with animal subtypes (Staphylococcus aureus subsp. aureus CP010526.1) from cow milk in India and Staphylococcus aureus subsp. aureus N315 158879.11 in cattle from the USA portrays a zoonotic transmission from livestock to humans due to predominant contact with livestock. Recording zoonotic MRSA-CC7 clonal lineages in hospital settings is of relevant importance to public health and its impact on epidemiology in Nigeria and Africa. Possible genetic re-assortment due to gene exchange within the entire segment could occur during co-infection of LA-MRSA and HA-MRSA, leading to the emergence of MRSA hybrid types, causing the evolution of new trait sharing both human and animal ancestral clonal diversity58,59. The present study could not confirm the hybrid types of the identified clones but the observed genetic variation can be ascertained in future studies in relation to the geographically-associated clusters prominent in Europe, Asia, and America. The genetic variation of SCCmec types could predict homoplastic loss of accessory components, giving rise to diverse phylogeny associated with HA-MRSA having a defined and unique epidemic clonal lineage60,61.
Conclusion
Although the two MRSA strains (MRSA-CC7-ST789-t091-SCCmecV) from ear and wound infections from Nigerian populace analysed here were relatively small, but the encoded repertoire of resistomes, virulomes, and highly conserved subsystem cell signaling and cellular processes enhance their survival, colonization, and pathogenesis. Phylogenomic analysis further reveals the genetic relatedness of these MRSA-CC7 isolates with globally reported livestock strains known to be prevalent and recurring in the last ten years. The genomic composition and myriads of subsystem proteins encoded by these strains are potential elements that could aid the prediction of possible hospital MRSA transmission. Effective monitoring of MRSA-CC7 dissemination and pragmatic surveillance programs are required to prevent the possible genetic transfer from animal sources to human and the evolution of pathogenic hybrid strains.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Reddy, P. N., Srirama, K. & Dirisala, V. R. An update on clinical burden, diagnostic tools, and therapeutic options of Staphylococcus aureus. Infect. Dis. Res. Treat. 10, 1179916117703999 (2017).
Sanders, T., Bentum, J., Fox, A., Egyir, B. & Watters, C. Characterization of MRSA and ESBL pathogens from patients with surgical-site infections in Accra Ghana. Antimicrob. Steward. Healthc. Epidemiol. 2(S1), s86–s86 (2022).
Ghaznavi-Rad, E. et al. Predominance and emergence of clones of hospital-acquired methicillin-resistant Staphylococcus aureus in Malaysia. J. Clin. Microbiol. 48(3), 867–872 (2010).
Abd El-Hamid, M. I. et al. Clonal diversity and epidemiological characteristics of ST239-MRSA strains. Front. Cell. Infect. Microbiol. 25(12), 782045 (2022).
Sartelli, M. et al. WSES/GAIS/WSIS/SIS-E/AAST global clinical pathways for patients with skin and soft tissue infections. World J. Emerg. Surg. 17(1), 1–23 (2022).
Moore-Lotridge, S.N., Bennett, M.R., Moran, C.P., Schoenecker, J.G., & Thomsen, I.P. (2022). MRSA and Virulent MSSA Infections. In Pediatric Musculoskeletal Infections (pp. 95–107).
Mohamadou, M. et al. High prevalence of Panton-Valentine leukocidin positive, multidrug resistant, Methicillin-resistant Staphylococcus aureus strains circulating among clinical setups in Adamawa and Far North regions of Cameroon. Plos one. 17(7), e0265118 (2022).
Chang, S. C., Lin, L. C. & Lu, J. J. An lnu (A)-carrying multi-resistance plasmid derived from sequence type 3 methicillin-resistant staphylococcus lugdunensis may contribute to antimicrobial resistance in staphylococci. Antimicrob. Agents Chemother. 66(8), e00197-e222 (2022).
El-Ashker, M. et al. Antimicrobial resistance pattern and virulence profile of S. aureus isolated from household cattle and buffalo with mastitis in Egypt. Vet. Microbiol. 240, 108535 (2020).
Sieber, R. N. et al. Phage-mediated immune evasion and transmission of livestock-associated methicillin-resistant Staphylococcus aureus in humans. Emerg. Infect. Dis. 26(11), 2578 (2020).
Huber, C. et al. How to survive pig farming: Mechanism of SCCmec element deletion and metabolic stress adaptation in livestock-associated MRSA. Front. Microbiol. 13, 969961 (2022).
Luzzago, C. et al. Survey of Staphylococcus aureus carriage by free-living red deer (Cervus elaphus): Evidence of human and domestic animal lineages. Transbound. Emerg. Dis. 69(5), e1659–e1669 (2022).
Chen, C. J. & Huang, Y. C. New epidemiology of staphylococcus aureus infection in Asia. Clin. Microbiol. Infect. 20(7), 605–623 (2014).
Belhout, C., Elgroud, R. & Butaye, P. Methicillin-resistant staphylococcus aureus (MRSA) and other methicillin-resistant staphylococci and mammaliicoccus (MRNaS) associated with animals and food products in Arab countries: A review. Vet. Sci. 9(7), 317 (2022).
Obasuyi, O. et al. Molecular characterization and pathogenicity of Staphylococcus aureus isolated from Benin-city Nigeria. Microorganisms 8(6), 912 (2020).
Abdulgader, S. M., Shittu, A. O., Nicol, M. P. & Kaba, M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: A systematic review. Front. Microbiol. 6, 348 (2015).
Akinduti, A. P. et al. Clonal diversity and spatial dissemination of multiantibiotics resistant Staphylococcus aureus pathotypes in Southwest Nigeria. PLoS ONE 16(2), e0247013 (2021).
Rabelo, M. A. et al. Phenotypic methods for determination of methicillin resistance in Staphylococcus spp from health care workers. J. Bras. Patol. Med. Lab. 49, 91–96 (2013).
Fosheim, G. E., Nicholson, A. C., Albrecht, V. S. & Limbago, B. M. Multiplex real-time PCR assay for detection of methicillin-resistant Staphylococcus aureus and associated toxin genes. J. Clin. Microbiol. 49(8), 3071–3073 (2011).
Mellmann, A. et al. Based upon repeat pattern (BURP): An algorithm to characterize the long-term evolution of Staphylococcus aureus populations based on spa polymorphisms. BMC Microbiol. 7, 98. https://doi.org/10.1186/1471-2180-7-98 (2007) (PMID: 17967176).
Clinical and Laboratory Standards Institute (2018). Performance standards for antimicrobial susceptibility testing, sixteenth informational supplement, document M100-S20. Wayne, PA, USA: CLSI, (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Ondov, B. D. et al. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D. & Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150 (2018).
Wattam, A. R. et al. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucl. Acids Res. 45, D535–D542 (2017).
Aziz, R. K. et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 9, 1–15 (2018).
Kaya, H. et al. SCC mec Finder a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. Msphere 3(1), e00612-e617 (2018).
Bartels, M. D. et al. Comparing whole-genome sequencing with sanger sequencing for spa typing of methicillin-resistant staphylococcus aureus. J. Clin. Microbiol 52, 4305–4308 (2014).
Larsen, M. V. et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol 50, 1355–1361 (2012).
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P. & Spratt, B. G. eBURST: Inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol 186, 1518–1530 (2004).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother 67, 2640–2644 (2012).
Alcock, B. P. et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucl. Acids Res. 48(D1), D517–D525 (2020).
Carattoli, A. et al. In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother 58, 3895–3903 (2014).
Cosentino, S., Voldby Larsen, M., Møller Aarestrup, F. & Lund, O. PathogenFinder—distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 8, e77302 (2013).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucl. Acids Res. 44, D457-462 (2016).
Ahrenfeldt, J. et al. Bacterial whole genome-based phylogeny: Construction of a new benchmarking dataset and assessment of some existing methods. BMC Genom. 18, 19 (2017).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 32(5), 1792–1797 (2004).
Lang, J. M., Darling, A. E. & Eisen, J. A. Phylogeny of bacterial and archaeal genomes using conserved genes: Supertrees and supermatrices. PloS one 8(4), e62510 (2013).
Tang, Q. et al. Role of wound microbiome, strategies of microbiota delivery system and clinical management. Adv. Drug Deliv. Rev. 1(192), 114671 (2023).
Sadiq, F. A. et al. Phenotypic and genetic heterogeneity within biofilms with particular emphasis on persistence and antimicrobial tolerance. Future Microbiol. 12(12), 1087–1107 (2017).
Ayepola, O. O., Olasupo, N. A., Egwari, L. O., Becker, K. & Schaumburg, F. Molecular characterization and antimicrobial susceptibility of Staphylococcus aureus isolates from clinical infection and asymptomatic carriers in Southwest Nigeria. Plos one 10(9), e0137531 (2015).
Amoako, D. G. et al. Genome mining and comparative pathogenomic analysis of an endemic methicillin-resistant Staphylococcus aureus (MRSA) clone, ST612-CC8-t1257-SCCmec_IVd (2B), isolated in South Africa. Pathogens 8(4), 166 (2019).
Vanhoof, R., Godard, C., Content, J., Nyssen, H. J. & Hannecart-Pokorni, E. Belgian Study Group of Hospital Infections (GDEPIH/GOSPIZ). Detection by polymerase chain reaction of genes encoding aminoglycoside-modifying enzymes in methicillin-resistant Staphylococcus aureus isolates of epidemic phage types. J. Med. Microbiol. 41(4), 282–290 (1994).
Monteiro, K. L., de Aquino, T. M. & Mendonça Junior, F. J. B. An update on Staphylococcus aureus NorA efflux pump inhibitors. Curr. Topics Med. Chem. 20(24), 2168–2185 (2020).
Vivas, M.C., & Gutierrez, A.D.C.M. (2018). Typification methods and molecular epidemiology of Staphylococcus aureus with methicillin resistance. In Staphylococcus Aureus. IntechOpen.
Laarman, A. J. et al. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 186(11), 6445–6453 (2011).
Kwiecinski, J. et al. Staphylokinase promotes the establishment of Staphylococcus aureus skin infections while decreasing disease severity. J. Infect. Dis. 208(6), 990–999 (2013).
Peetermans, M. et al. Plasminogen activation by staphylokinase enhances local spreading of S. aureus in skin infections. BMC Microbiol. 14(1), 1–12 (2014).
Monecke, S. et al. Microarray-based characterisation of a Panton-Valentine leukocidin-positive community-acquired strain of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 12(8), 718–728 (2006).
Hamilton, S. M. et al. In vitro production of Panton-Valentine leukocidin among strains of methicillin-resistant Staphylococcus aureus causing diverse infections. Clin. Infect. Dis. 45(12), 1550–1558 (2007).
Mimica, M. J., Berezin, E. N., Damaceno, N. & Carvalho, R. B. SCC mec type IV, PVL-negative, methicillin-resistant Staphylococcus aureus in cystic fibrosis patients from Brazil. Curr. Microbiol. 62, 388–390 (2011).
Shore, A. C. et al. Characterization of a novel arginine catabolic mobile element (ACME) and staphylococcal chromosomal cassette mec composite island with significant homology to Staphylococcus epidermidis ACME type II in methicillin-resistant Staphylococcus aureus genotype. Antimicrob. Agents Chemother. 55, 1896–1905 (2011).
Ramsamy, Y. et al. Pathogenomic analysis of a novel extensively drug-resistant Citrobacter freundii isolate carrying a blaNDM-1 Carbapenemase in South Africa. Pathogens 9(2), 89 (2020).
Kong, C., Neoh, H. M. & Nathan, S. Targeting Staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins 8(3), 72 (2016).
Campbell, M. J., Beenken, K. E., Ramirez, A. M. & Smeltzer, M. S. The major role of sarA in limiting Staphylococcus aureus extracellular protease production is correlated with decreased virulence in diverse clinical isolates in osteomyelitis. Virulence Just-accepted 8, 2022–2111 (2022).
Wang, L., Wang, H., Zhang, H. & Wu, H. Formation of a biofilm matrix network shapes polymicrobial interactions. ISME J. 17(3), 467–477 (2023).
Akinduti, P. A. et al. Emerging vancomycin-non susceptible coagulase negative Staphylococci associated with skin and soft tissue infections. Ann. Clin. Microbiol. Antimicrob. 21(1), 1–105 (2022).
Lakhundi, S. & Zhang, K. Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin. Microbiol. Rev. 31(4), e00020-e118 (2018).
John, J., Narendrakumar, L., Thomas, S. & Nelson-Sathi, S. Hybrid genome assembly and annotation of multidrug-resistant Staphylococcus aureus genotype ST672-SCCmec type IVd (2B). J. Glob. Antimicrob. Resist. 32, 74–77 (2023).
Bianco, C. M. et al. Pre-epidemic evolution of the MRSA USA300 clade and a molecular key for classification. Front. Cell. Infect. Microbiol. 13, 13 (2023).
Aloba, B. K. et al. An emerging Panton-Valentine leukocidin-positive CC5-meticillin-resistant Staphylococcus aureus-IVc clone recovered from hospital and community settings over a 17-year period from 12 countries investigated by whole-genome sequencing. J. Hospital Infect. 132, 8–19 (2023).
Acknowledgements
The authors acknowledge the management of Federal Medical Centre, Abeokuta for assisting data collection, Aboderin W. and Dr S.K Akinwande for their technical assistance.
Funding
Deutsche Forschungsgemeinschaft (DFG), Germany in cooperation with the World Academy of Science (TWAS), Italy for providing sponsorship for the genomic analysis at the Staphylococci Unit, BfR, Germany. The publication was partly supported by the management of Covenant University, Ota, Nigeria. The aforementioned support agents had no role in study design, data collection and analysis, interpretation of data, the decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Each author has made a substantial contribution as follows; P.A., B.O., and M.E. provide conception of the study; P.A., B.O. and P.O. analysed and interpreted the data; P.A. and P.O. performed genomic analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Akinduti, P.A., Motayo, B.O., Maged, EA. et al. Pathogenomic profile and clonal diversity of potential zoonotic MRSA-CC7-ST789-t091-SCCmecV from human skin and soft tissue infections. Sci Rep 14, 19326 (2024). https://doi.org/10.1038/s41598-024-67388-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-67388-w
