
Abstract
Endothelial cell dysfunction can lead to various vascular diseases. Blood flow disorder is a common symptom of vascular diseases. Regenerative angiogenesis, which involves transplanting vascular cells or stem cells into the body to shape new vasculature, can be a good therapeutic strategy. However, there are several limitations to using autologous cells from the patients themselves. We sought to investigate the new vascular cells that can play a role in the formation of angiogenesis in vivo using stem cells from alternative animals suitable for cellular therapy. Porcine is an optimal animal model for xenotransplantation owing to its physiological similarity to humans. We used differentiated porcine endothelial cells (pECs) as a therapeutic strategy to restore vessel function. Differentiated pECs formed vessel-like structures in mice, distinguishing them from stem cells. MMPs activity and migration assays indicated that differentiated pECs possessed angiogenic potential. Tube formation and 3D spheroid sprouting assays further confirmed the angiogenic phenotype of the differentiated pECs. Immunofluorescence and immunoprecipitation analyses revealed claudin-mediated tight junctions and connexin 43-mediated gap junctions between human ECs and differentiated pECs. Additionally, the movement of small RNA from human ECs to differentiated pECs was observed under co-culture conditions. Our findings demonstrated the in vivo viability and angiogenetic potential of differentiated pECs and highlighted the potential for intercellular communication between human and porcine ECs. These results suggest that transplanted cells in vascular regeneration completed after cell therapy have the potential to achieve intercellular communication within the body.
Similar content being viewed by others


Introduction
Endothelial cells (ECs) form the innermost layer of blood vessels and play a pivotal role in regulating vascular function1. They regulate blood flow, inflammation, immune response, and platelet deposition. Their dysfunction can lead to various vascular diseases, including hypertension, ischemic heart disease, atherosclerosis, and thrombosis2. Peripheral artery disease (PAD) is characterized by gradual narrowing of blood vessels, which hinders blood circulation. The incidence of PAD increases, particularly with advancing age3. Current treatments such as vascular bypass and stent surgeries address the symptoms but do not restore the underlying EC dysfunction. Dysfunctional ECs compromise vascular homeostasis, contributing to irregular plaque formation, increasing the risk of strokes and heart failures and impairing angiogenesis4,5,6,7. Implantation of healthy ECs into compromised blood vessels is a promising strategy to restore vascular homeostasis. Vascular regeneration, involving angiogenetic growth factors like vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF) and basic fibroblast growth factor (bFGF), is crucial for improving blood flow disorders in ischemic heart disease and PAD8. Recent research includes cellular therapy that induces angiogenesis by injecting stem cells, but this approach can be invasive and has ethical issues9,10.
Pluripotent stem cells (PSCs) found in multicellular organisms are undifferentiated or partially differentiated cells with the remarkable ability to differentiate into various specialized cell types. They also exhibit indefinite proliferation, which is crucial for the development and maintenance of tissues11. Examples of PSCs include embryonic stem cells (ESCs), epiblast stem cells (EpiSCs), induced pluripotent stem cells (iPSCs), and adult stem cells (ASCs)12,13. Pluripotent stem cells use various signaling pathways to maintain their pluripotent state depending on the species from which they originate. However, ethical issues remain an important topic in the case of cell therapy using human stem cells14,15,16.
Xenotransplantation is the transplantation of organs, cells, or tissues from one species to another. Various large animals were used for xenotransplantation, including pigs, monkeys, and chimpanzees17. Pigs closely resemble humans in terms of their organ size and physiological metabolism and are considered optimal candidates for organ xenograft donation18,19. Ongoing studies aim to modulate the porcine immune system for organ transplantation in humans. Strategies include creating pig organs with immune response factors removed to avoid rejection and conducting chimeric research, allowing human organs to grow in immunodeficient pigs20,21. Xenogeneic cellular therapy using porcine cells may emerge as a promising strategy to restore tissue function22.
Cell–cell junctions are specialized structures that facilitate communication, adhesion, and interaction between neighboring cells in multicellular organisms, include gap junctions that link intercellular channels for various molecules, such as ions, miRNAs, amino acids, and electronic signals23,24,25. Gap junctions are formed by connexin proteins (some of which are cell-specific) that play a role in immune responses, invasion, and cell proliferation, and endothelial cells express connexin 37, connexin 40, and connexin 4326,27,28.
In a previous study, we successfully differentiated ECs from porcine epiblast stem cells (pEpiSCs)29. Using magnetic-activated cell sorting (MACS) with CD31 antibody as an endothelial cell marker, we selected differentiated porcine ECs (pECs) with functional EC properties30. This study aimed to confirm whether the properties of differentiated pECs differ from those of healthy human ECs, and whether they function as ECs in vivo. Our findings demonstrated that differentiated pECs organize into tight and gap junctions. Moreover, differentiated pECs exhibited no discernible differences between porcine and human ECs. Organized gap junctions between these cells serve as intercellular channels. Additionally, we demonstrated that differentiated pECs contributed to the formation of a vascular network in vivo.
Materials and methods
Cell culture
Primary human umbilical vein endothelial cells (HUVECs) were obtained from PromoCell. Differentiated porcine endothelial cells (differentiated pECs) were established and sorted as previously described30. CD31 antibody and magnetic bead were mixed. ECs differentiated from porcine epiblast stem cells and magnetic bead labeled with CD31 antibody were suspended 0.1% BSA-DMEM medium for 1 h. Then, the mixtures were washed with 0.1% BSA/DMEM to removed unlabeled cells. Sorted ECs were seeded on coated with 0.2% gelatin culture plates. Swine umbilical vein endothelial cells (SUVECs) and differentiated pECs were plated on 0.2% gelatin-coated plates. HUVECs, SUVECs, and differentiated pECs were cultured in medium 199 (Sigma, M4530) with 20% fetal bovine serum (FBS) (GenDEPOT, F0900-050), 100 µg/ml heparin (Sigma, H3149), and 30 µg/ml endothelial cell growth supplements (ECGS) (Corning, 306006) at 37 °C in 5% CO2. HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (WELGENE, LM-001-05) with 10% FBS and 1X penicillin/streptomycin (Lonza, 17-602E) at 37 °C in 5% CO2.
Establishing SUVECs
The pig umbilical cord was placed on a clean pad, blood on the surface was wiped off, and both ends were cut. Porcine vein endothelial cells were isolated by extracting the veins from Wharton’s jelly. The inner part of the isolated vein underwent three washes with 37 °C phosphate buffered saline (PBS). One end of the washed vein was occluded, 1 mg/ml collagenase type 1 (Gibco, 17018–029) was injected, and the other end was also occluded. The vein was incubated three times for 5 min each in 37 °C PBS. The incubated vein was fixed to a 15 mL conical tube, and the contents were collected after centrifugation at 500 g for 5 min at 24 °C. The cell pellet was pre-incubated for 10 min using M199 medium (Sigma, M4530) supplemented with 20% FBS (Gibco, 16000–044), 50 µg/ml heparin (Sigma, H3149), and 100 µg/ml penicillin/streptomycin (Sigma, P0781) in a 0.2% gelatin-coated T-75 flask. The supernatant was transferred to a new 0.2% gelatin-coated T-75 flask and cultured using M199 medium at 37 °C in 5% CO2.
Plasmid constructs
HA-tagged Human connexin 43 (NM_000165.5) (HA-hCx43) was cloned into a pCMV-HA plasmid. Myc-tagged porcine connexin 43 (NM_001244212.1) (Myc-pCx43) was cloned into the pCMV-Myc plasmid. Briefly, to construct HA-human connexin 43 (hCx43), the coding sequence of hCx43 was amplified by PCR, and the PCR products were digested with SalI and KpnI enzymes. This PCR product was then cloned into the SalI and KpnI sites of pCMV-HA, resulting in the generation of HA-hCx43 vectors. The hCx43 sequence was derived from cDNA of HUVECs using the following primers: forward: 5′-CGAGTCGACAATGGGTGACTGG-3′ and reverse: 5′-CGAGGTACCCTAGATCTCCAGGTC-3′. To construct Myc-porcine connexin 43 (pCx43), the coding sequence of pCx43 was amplified by PCR, and the PCR products were digested with SalI and KpnI enzymes. The resulting PCR product was then cloned into the SalI and KpnI sites of pCMV-Myc, yielding Myc-pCx43 vectors. The pCx43 sequence was obtained from cDNA of pEpiSCs using the following primers: forward: 5′-CAAGTCGACAATGGGTGACTGGAGTGCC-3′ and reverse: 5′-CGCGGTACCCTAGATCTCCAGGTCATCA-3′.
Zymography assay for matrix metalloproteinase (MMP) activity
Cell lysates were harvested in lysis buffer (0.025 M Tris–HCl pH 7.5, 0.1 M NaCl, 1% NP-40, protease/phosphatase inhibitors cocktail, Cell Signaling, 5872S) and centrifuged at 15,000 rpm for 15 min at 4 °C. Proteins were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) containing 0.1% gelatin. After electrophoresis, gels were agitated in renaturing solution (2.5% Triton X-100) at 25 °C for overnight. After washing, gels were incubated in developing buffer (0.05 M Tris–HCl pH 7.8, 0.2 M NaCl, 0.005 M CaCl2, and 0.02% Tween-20) for 30 min. Gelatin gels were stained with Coomassie Blue staining solution (0.5% Coomassie Blue R-250, 5% methanol, and 10% acetic acid) and washed in destaining solution (5% methanol and 10% acetic acid) until gelatinolytic activity appeared as clear bands on a blue background.
Three-dimensional (3D) spheroid sprouting assay
The 3D spheroid sprouting assay was conducted as previously described30. Spheroids were formed using hanging drop cultures in 20% methocel solution. Cells were counted at 500 cells/spheroid and incubated to form a 3D spheroid structure at 37 °C overnight. Spheroids were collected in 10% FBS in PBS, centrifuged at 100 g for 5 min at 25 °C, and the supernatant was discarded. Collected spheroids were resuspended in 20% FBS in methocel. The collagen matrix was composed of medium 199, acetic acid, and 100 mg/mL collagen 1 (Corning, 354249) in a 1:4:4 ratio. The collagen solution and spheroids were mixed in a 1:1 ratio and the mixture was placed in 24-well culture plates. Collagen polymerization was performed at 37 °C for 1 h. Endothelial cell growth supplements in endothelial cell culture medium was added to the cells and cultured for 24 h. Sprouted spheroids were fixed in 4% formaldehyde (Sigma, 252549) for 10 min. After PBS washing, permeabilization was performed using 0.2% IGEPAL (Sigma, I3021) for 10 min, followed by washing with PBS. Blocking was performed with 3% bovine serum albumin (BSA) in PBS at room temperature for 1 h. Phalloidin (1:1000; Invitrogen, A12379) and 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, D1306) were used to stain the spheroids. Sprout length was measured using ImageJ 1.52 software.
Tube formation
Tube formation assays were conducted as previously described30. A total of 2 × 104 cells were seeded on Matrigel-coated (Corning, 354234) 96-well plates. Cells were cultured at 37 °C in 5% CO2. Images were captured using an Olympus fluorescence microscope (Olympus, DP70) with DP Manager software (Olympus, version 3.1.1.208).
Migration assay
Transwell chambers (Falcon, 353097) were coated with Fibronectin (Corning, 354008) at 0.1 mg/mL for the migration assay. A total of 2.5 × 104 cells were cultured in serum-free M199 medium in Transwell chambers. Complete M199 medium (500 µL) was added to the lower chamber. Fibronectin-coated Transwell plates were placed in 24-well plates. Cells were counted at 12.5 × 104 cells/mL, and 200 µL/well cell suspension was placed in the Transwell chambers. After 12 h of incubation at 37 °C, cells inside the Transwell were removed with a cotton swab. Migrated cells were fixed in 4% formaldehyde for 5 min, stained with hematoxylin for 6 min, and eosin for 2 min (Abcam, ab245880). Images were captured using an Olympus fluorescence microscope (Olympus, DP70) with DP Manager software.
Xenograft
The animal experiment was conducted in accordance with the Guide for the Care and Use of Laboratory Animals, as well as adhering to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2.0. 5-week-old male Balb/c nude mouse were purchased from ORIENT Bio. All mouse were maintained under strict SPF (specific pathogen free) conditions. The mouse were allocated randomly to three group. A total of 1 × 107 group 1: pEpiSCs, group 2: differentiated ECs, and group 3: purified differentiated ECs stained with PKH26 were mixed with growth factor-reduced Matrigel (Corning, 354230). Subsequently, after ketamine (100 mg/kg) anesthesia, 0.1 mL of Matrigel mixed with 1 × 106 cells was injected subcutaneously into the right and left posterior flanks of each mouse. pEpiSCs injection group was used as control. The sample group was established at n = 4 per group and a total of 12 mouse were used. The mice were sacrificed by cervical dislocation after 14 days and the tissues were separated. Tissues were fixed in 4% paraformaldehyde for 1 h and then incubated in 30% sucrose at 4 °C overnight. The tissues were embedded in optimal cutting temperature compound (OCT) compound for cryosectioning.
Tissue histochemistry
Tissues embedded in OCT compound were cut into 10 µm sections. These slides were incubated with CD31 antibody (1:100, NOVUS, NB100-2284) for 90 min, then incubated with goat anti-rabbit IgG FSD 488 (BioActs, RSA1245) for 90 min at room temperature. After PBS washing, samples were stained with 5 µg/mL of DAPI in PBS for 15 min at room temperature. Tissue sections were washed in distilled water for 30 min for hematoxylin and eosin (H&E) staining. The slides were stained using a H&E staining kit (Abcam, ab245880). Images were captured using an Olympus fluorescence microscope (Olympus, DP70) with DP Manager software (Olympus, version 3.1.1.208).
Immunofluorescence
SUVECs and sorted pECs stained with PKH26 were mixed with HUVECs. The mixed cells were plated on a 0.2% gelatin-coated slides and cultured at 37 °C for 8 h. After culture, the cells were fixed with 4% PFA (Sigma, P6148) for 20 min at 4 °C and permeabilized with 0.2% Triton-X (Sigma, T8532) for 10 min at room temperature. The cells were then washed three times with PBS-Tween 20 (0.1%) at 50 rpm for 5 min each at room temperature and blocked with blocking solution (5% bovine serum albumin (BSA) (Sigma, A3311) in PBS-T) for 1 h at room temperature. Subsequently, the coverslips were incubated overnight at 4 °C with primary antibodies: Connexin 43 (1:400, Abcam, ab11370), Claudin-5 (1:200, Affinity Bioscience, AF5216). Incubated coverslips were rinsed three times with PBS-T at 50 rpm for 5 min and incubated with the secondary antibody: Goat anti-rabbit IgG, FSD™ 488 (1:500, BioActs, RSA1245). Nuclei in cells were stained with 5 µg/mL of DAPI in PBS at room temperature for 15 min. Images were acquired using a LEICA fluorescence microscope (LEICA DM 2500) and analyzed using Leica Application Suite (LAS) software (LEICA, version 2.7).
Co-immunoprecipitation
HEK293T cells were cotransfected with HA-hCx43 and Myc-pCx43. After transfection, the cells were lysed in lysis buffer. The cell lysates were incubated with IgG (SC-2025, Santa Cruz) or Myc antibody (60003–2-Ig, Proteintech) or HA antibody (51064–2-AP, Proteintech) at 4 °C overnight. Agarose beads (SC-2003, Santa Cruz Biotechnology) were added to the protein and incubated at 4 °C for 2 h. Beads were washed three times with lysis buffer and boiled with 2X sample buffer for 10 min. Protein samples were investigated with Myc and HA antibodies by western blotting.
PKH26 staining
Cells were detached using 0.05% trypsin, and the supernatant was removed. The cell pellet was washed with PBS and centrifuged at 300 g for 3 min at room temperature. A dye mixture of 4 µM was prepared by mixing 2 µL of PKH26 with 0.5 mL of diluent C from the PKH26 mini kit (Sigma, MINI26-1KT). The dye mixture was then added to the pelleted cells and incubated at room temperature for 1 min. After incubation, 1 mL of heat-inactivated FBS was added to the cell mixture and incubated for another minute at room temperature. The cell mixture was centrifuged at 300 g for 3 min at 24 °C and the supernatant was removed. The cell pellet was washed with PBS and centrifuged to remove the supernatant.
Small nucleotide transport
HUVECs were transfected with Cy3-control siRNA (Ambion, AM4621) using RNAimax (Invitrogen, 13778–150) in serum free medium 199 (Sigma, M4530). Subsequent co-culture of HUVECs with differentiated pECs or SUVECs cultured using transwell for overnight at 37 °C in 5% CO2. After co-culture, HUVECs transfected with Cy3 were removed by swabbing before fluorescence imaging. The cells were fixed with 4% PFA (Sigma, P6148) for 10 min at room temperature. The cells were then washed three times with PBS. Nuclei in cells were stained with 5 µg/mL of DAPI in PBS at room temperature for 15 min. Images were captured using an Olympus fluorescence microscope (Olympus, DP70) with DP Manager software (Olympus, version 3.1.1.208).
Statistical analysis
Graph pad prism software v7.00 (GraphPad) was used to data analysis. For Migration cell number, branch points and sprouts length analysis, one-way ANOVA used statistical significance among groups. The data are presented as the means ± SEM. 0.05 > P values was considered statistically significant.
Results
Formation of vascular structures in vivo by differentiated pECs
We previously established a differentiation protocol to generate ECs from pEpiSCs29. Following differentiation, we isolated and purified endothelial cells using a MACS sorting assay with CD-31, a specific marker of endothelial cell membranes. Our previous study demonstrated the functionality of purified pECs as fully competent cells. We injected pEpiSCs, non-purified differentiated ECs and purified differentiated ECs into mice to assess the in vivo vascular-forming potential of differentiated pECs. Prior to injection, cells were labeled with PKH26 for subsequent identification (Fig. 1A). The tissue sections were then analyzed to determine the presence of CD31 (an endothelial cell marker) to assess the formation of vasculature by the injected cells. The pEpiSCs and non-purified differentiated ECs failed to organize into vascular structures. In contrast, only purified differentiated pECs exhibited the capability and formed well-defined vascular structures (Fig. 1B,D). Co-localization analysis of PKH26 and CD31 fluorescence further substantiated the vascular-forming ability of differentiated pECs in vivo (Fig. 1C). These findings strongly support the conclusion that differentiated pECs possess the capacity to form functional vascular structures in vivo.

Formation of vascular structures in vivo by differentiated pECs. (A) schematic outline of porcine cells transplantation in Balb/c nude mouse. Created with BioRender.com (B) Immunofluorescence images depicting CD-31 expression (green) in injected cells, including porcine epiblast stem cells (pEpiSCs), non-purified differentiated pECs (unsorted EC), and purified differentiated pECs (sorted EC). All cells were labeled with PKH 26 (red) prior to injection. Nuclei was counterstained with DAPI (blue). (C) Fluorescence intensity analysis of representative immunofluorescence images measured in RGB profile of ImageJ. (D) Hematoxylin and eosin (H&E) staining images illustrating the morphological characteristics of injected cells, including pEpiSCs, non-purified differentiated pECs, and purified differentiated pECs. Scale bar = 50 µm.
Matrix metalloproteinase activity and migration ability of differentiated pECs
Angiogenesis is a critical aspect of endothelial cell function that significantly relies on cellular migration. Matrix metalloproteinases are a family of enzymes responsible for extracellular matrix (ECM) degradation, play a pivotal role in facilitating angiogenesis31,32. Matrix metalloproteinase-mediated breakdown of the ECM is essential for the migration and alignment of endothelial cells, enabling the formation of new blood vessel structures. Among MMP family members, MMP-2 and MMP-9 are particularly crucial for angiogenesis33. To assess the migratory activity of ECs, we examined MMP activity using zymography. HUVECs, SUVECs, and differentiated pECs exhibited comparable MMP-2 activities (Fig. 2A). Furthermore, there was no discernible difference in the MMP-9 activity between SUVECs and differentiated pECs (Fig. 2A). Migration of ECs is a key component of angiogenesis. We evaluated EC migration using a Transwell migration assay (Fig. 2B). The number of migrated cells was consistent among HUVECs, SUVECs, and differentiated pECs (Fig. 2C). These findings indicate that differentiated pECs possess MMP activity and migration abilities, which are essential attributes of angiogenesis. Importantly, these capabilities are comparable to those observed in human ECs, suggesting the translational potential of differentiated pECs for therapeutic applications.

Differentiated pECs MMP activity and migration ability. (A) Zymography analysis illustrating MMP activity in HUVECs, SUVECs, and differentiated pECs. (B) Assessing migration ability through a transwell migration assay for HUVECs, SUVECs, and differentiated pECs. Scale bar = 20 µm. (C) Migration cell number was counted by using ImageJ.
Angiogenic function of differentiated pECs
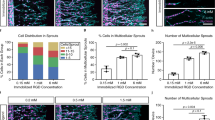
Having established the MMP activity and migration capacity of differentiated pECs, we further examined their angiogenic functions. A tube formation assay was performed to assess angiogenesis. Remarkably, HUVECs, SUVECs, and differentiated pECs exhibited the ability to form intricate tube structures (Fig. 3A,B). We used a spheroid sprouting assay to validate angiogenesis in a three-dimensional context. Spheroid-shaped HUVECs, SUVECs, and differentiated pECs were embedded in a collagen matrix, revealing their capacity for 3D angiogenesis (Fig. 3C,D). Notably, differentiated pECs demonstrated extended sprouting compared with HUVECs and SUVECs (Fig. 3C). Intriguingly, differentiated pECs exhibited superior angiogenic ability compared with SUVECs. These findings collectively demonstrate that HUVECs and differentiated pECs possess comparable angiogenic capabilities, highlighting the potential of differentiated pECs for application in angiogenesis-related therapeutic interventions.

Differentiated pECs have angiogenesis function. (A) Tube formation images in HUVECs, SUVECs, and differentiated pECs on Matrigel for 4 h. Scale bar = 500 µm. (B) Branch points of HUVECs, SUVECs, and differentiated pECs. (n = 3) (C) Spheroid sprouting images of HUVECs, SUVECs, and differentiated pECs embedded in collagen gels. Phalloidin (red) stained cytoskeleton for sprouting measurement. The nucleus was stained with DAPI (blue). Scale bar = 275 µm. (D) Sprouts length was measured by ImageJ. (n = 4) Values presented as mean SEM. * P < 0.05, ** P < 0.01.
Porcine ECs form a tight junction with human ECs
Tight junctions are crucial cell–cell adhesion protein complexes and prominently feature claudin proteins. We examined the protein interactions between HUVECs and pECs (SUVECs and differentiated pECs) to investigate whether porcine endothelial cells establish tight junctions with human endothelial cells. We utilized PKH 26 staining of porcine ECs (SUVECs and differentiated pECs) to distinguish between HUVECs and porcine ECs. Co-culture of HUVECs with stained porcine ECs allowed the monitoring of tight junctions. There was an intercellular interaction between the tight junction protein claudin in SUVECs and HUVECs, indicating the formation of tight junctions between these cells (Fig. 4A). Further investigation was aimed at ascertaining whether a similar junction exists between differentiated pECs and HUVECs. Claudins from differentiated pECs also interacted with HUVECs (Fig. 4B). Notably, no discernible differences were observed between SUVECs and differentiated pECs in their ability to form tight junctions with HUVECs. Collectively, these findings suggest that pECs (including SUVECs and differentiated pECs) can establish tight junction complexes with human ECs, emphasizing the potential for intercellular communication between these cell types.

Tight junction formation between porcine and human ECs. (A) Immunofluorescence image depicting claudin (green) in HUVECs and SUVECs stained with PKH 26 (red). Nuclei were stained with DAPI (blue). (B) Immunofluorescence image illustrating claudin (green) in HUVECs and differentiated pECs stained with PKH 26 (red). Nuclei was stained with DAPI (blue). Scale bar = 20 µm.
Porcine ECs form a gap junction with human ECs
Gap junctions are essential intercellular channels that facilitate the transport of ions and molecules, are formed by connexon proteins on cell membranes. Connexins establish connections with adjacent cells, enabling direct interactions and transfer of various molecules, including miRNAs26,34. We performed immunofluorescent detection of connexin 43 to ascertain whether pECs establish gap junctions with human ECs. Porcine ECs (SUVECs and differentiated pECs) were stained to distinguish them from HUVECs. Connexin 43 from SUVECs formed junctions with HUVECs (Fig. 5A). Similarly, connexin 43 from differentiated pECs formed channels with HUVECs (Fig. 5B). Additionally, we investigated connexin protein–protein interactions between humans and porcines using co-immunoprecipitation (Co-IP). Human connexin 43 (hCx43) interacted with porcine connexin 43 (pCx43) (Fig. 5C). We transfected small nucleotides tagged with Cy3 into HUVECs to validate the functionality of these gap junctions as intercellular channels for the transport of small molecules. We performed co-culture of HUVECs with porcine ECs (SUVECs and differentiated pECs). Afterward, we observed only small nucleotides transferred to porcine EC by removing HUVECs. Subsequent co-culture of HUVECs with porcine ECs (SUVECs and differentiated pECs) revealed the presence of Cy3 in SUVECs and differentiated pECs (Fig. 5D,E). These findings affirm that differentiated pECs establish gap junctions with human ECs, creating functional cell–cell channels for small-molecule transport.

GAP junction formation between porcine and human ECs. (A) Immunofluorescence image illustrating connexin 43 (green) in HUVECs and SUVECs, with SUVECs stained using PKH 26 (red). Nuclei was stained with DAPI (blue). (B) Immunofluorescence depicting connexin 43 (green) in HUVECs and differentiated pECs, with differentiated pECs stained using PKH 26 (red). Nuclei was stained with DAPI (blue). (C) Western blotting data for co-IP showing the interaction between Myc-pCx43 and HA-hCx43 in HEK293T cells transfected with Myc-pCx43 and HA-hCx43. (D) Fluorescence image displaying small nucleotide tagged Cy3 (red) in SUVECs, transported by connexin channels from HUVECs. (E) Fluorescence image showing small nucleotide tagged Cy3 (red) in differentiated pECs, transported by connexin channels from HUVECs. Scale bar = 20 µm.
Discussion
We previously developed a method for differentiating endothelial cells (ECs) from porcine epiblast stem cells (pEpiSCs) using vascular endothelial growth factor (VEGF) and refined ECs from differentiated pEpiSCs using a MACS assay with CD-31 antibody. We subsequently demonstrated that differentiated pECs function as endothelial cells and established the culture conditions30. However, it was necessary to determine whether the differentiated pECs settled in vivo and interacted with human ECs.
To overcome the limitations of cellular therapy using human stem cells, we focused on porcine cells as functional endothelial cells that can replace human stem cells. However, Immune rejection is an issue that must be resolved xenotransplantation as well as allogeneic transplantation. The immune response to xenotransplantation can be more pronounced than to allotransplantation, necessitating the removal of foreign antigens by the host immune system. Strategies such as addressing galactose-α-1,3-galactose deficiency, modifying B-2-microglobulin, and MHC class 1 molecules aim to mitigate immune responses. Despite these efforts, the success of xenotransplantation remains limited. A recent attempt to transplant a pig heart into a human has demonstrated advancements in overcoming hyperacute rejection; however, challenges persist35,36. Our study did not use immunosuppressed porcine stem cells, highlighting the need for further investigations into the differentiation of ECs from immunosuppressed pEpiSCs. Future studies should explore the functionality and vascular network of differentiated pECs in animal models. This study lays a foundation for the development of artificial blood vessels using porcine cells.
In this study, we demonstrated that differentiated pECs have viability in vivo and exhibit angiogenic functions comparable to human ECs, forming vascular structures and facilitating xenogeneic intercellular communication through gap junctions. Furthermore, we found that differentiated pECs have relatively better angiogenic ability than HUVECs. Impairment of angiogenesis may lead to poor wound healing, peripheral arterial disease, and ischemic heart disease. Aging alters various signaling pathways related to angiogenesis within endothelial cells37. Angiogenesis is stimulated by various chemical factors. Aged endothelial cells become resistance to these factors38, thereby reducing their angiogenic ability, which can contribute to the development and worsening of vascular diseases. Although various cellular therapy strategies have been attempted for angiogenesis, most methods involve extracting stem cells from patients and injecting them. However, the possibility of tumorigenesis cannot be ruled out when administering stem cells39. These therapies do not lead to good results in all patients. Although bone marrow derived mononuclear cells were transplanted in critical limb ischemia patients, it failed to elicit positive effects from endpoint stage patients40. Our results suggest that differentiated pECs, with their outstanding angiogenic ability, could serve as a potential treatment strategy for various cardiovascular diseases and wound recovery resulting from impaired angiogenesis.
We investigated the gap junction channels between human and porcine ECs by detecting connexin 43. Endothelial cells expressed connexins 37, 40, and 43. Porcine ECs also expressed connexin 37 and 43; however, connexin 40 expression was not detected at the protein level41. Gap junctions can comprise various connexin hemichannels with connexin 37; for instance, interacting with itself and with connexins 40 and connexin 4342. Our findings demonstrated the formation of gap junctions between HUVECs and differentiated pECs, suggesting that connexins in differentiated pECs may assemble gap junction channels with various connexin hemichannels in HUVECs. These gap junctions can transport small molecules including miRNAs. Our observations of small nucleotide transport from HUVECs to differentiated pECs indicate the potential for miRNA exchange between these two cell types. While our study focused on the movement of small RNAs without regulating specific gene expression, future investigations may delve into whether transfected miRNAs targeting specific genes induce phenotypic changes in human ECs. Studies on miRNA delivery through gap junctions for disease treatment are ongoing43, positioning differentiated pECs as potential carriers for restoring dysfunctional ECs. Administering cells loaded with miRNAs targeting angiogenesis-related genes could be a potential strategy to improve angiogenesis functions44,45. However, interspecific aspects require closer scrutiny to ensure the safe use of porcine EC-derived miRNAs in humans.
Although our study successfully demonstrated the interaction between differentiated pECs and human ECs, the practical introduction of these cells into the living body remains a challenge. Most cells (apart from those circulating in the blood or lymphatic vessels) are fixed and attached to specific areas within the tissues. Tissue engineering studies have introduced various scaffold materials to facilitate cell attachment, and stents were employed to expand narrow blood vessels and enhance blood flow. However, in-stent restenosis presents as a problem. Although drug eluting stents are widely used to solve these problems, restenosis rate ranges from 8 to 15%46. Attaching endothelial cells to stents has been studied47. Attaching these cells to a stent is a promising strategy for the precise positioning of differentiated pECs in vivo. However, additional experiments are necessary to confirm the adherence and survival of differentiated pECs to the stents.
Our study established that stably purified, differentiated pECs exhibited functional equivalence to human ECs, have angiogenic capability and could form vascular structures in vivo. The observed junction formation between differentiated pECs and human ECs, coupled with material transfer through gap junctions, suggests that transplanted ECs have the potential to function normally in dysfunctional blood vessels. The outstanding angiogenesis ability of differentiated pECs is expected to be a potential cell therapy strategy in vascular diseases that require vascular regeneration and blood flow remodeling. Intercellular interactions facilitated by gap junctions are expected to play a crucial role in supporting dysfunctional ECs and maintaining vascular homeostasis.
Data availability
No datasets were generated or analysed during the current study.
References
Filippini, A., Tamagnone, L. & D’Alessio, A. Endothelial cell metabolism in vascular functions. Cancershttps://doi.org/10.3390/cancers14081929 (2022).
Rajendran, P. et al. The vascular endothelium and human diseases. Int. J. Biol. Sci.9, 1057–1069. https://doi.org/10.7150/ijbs.7502 (2013).
Fowkes, F. G. et al. Peripheral artery disease: epidemiology and global perspectives. Nat. Rev. Cardiol.14, 156–170. https://doi.org/10.1038/nrcardio.2016.179 (2017).
Kopczak, A. et al. Complicated carotid artery plaques and risk of recurrent ischemic stroke or TIA. J. Am. Coll. Cardiol.79, 2189–2199. https://doi.org/10.1016/j.jacc.2022.03.376 (2022).
Zhu, Y. et al. Research progress on the relationship between atherosclerosis and inflammation. Biomolecules8, 80 (2018).
Xiao, P. et al. Impaired angiogenesis in ageing: the central role of the extracellular matrix. J. Transl. Med.21, 457. https://doi.org/10.1186/s12967-023-04315-z (2023).
Tarnawski, A. S., Pai, R., Tanigawa, T., Matysiak-Budnik, T. & Ahluwalia, A. PTEN silencing reverses aging-related impairment of angiogenesis in microvascular endothelial cells. Biochem. Biophys. Res. Commun.394, 291–296. https://doi.org/10.1016/j.bbrc.2010.02.161 (2010).
Deveza, L., Choi, J. & Yang, F. Therapeutic angiogenesis for treating cardiovascular diseases. Theranostics2, 801 (2012).
Jahani, M. et al. Regenerative medicine and angiogenesis: Challenges and opportunities. Adv. Pharm. Bull.10, 490–501. https://doi.org/10.34172/apb.2020.061 (2020).
Murohara, T. Autologous adipose tissue as a new source of progenitor cells for therapeutic angiogenesis. J. Cardiol.53, 155–163 (2009).
Atala, A. & Lanza, R. Handbook of Stem Cells. (Academic press, 2012).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science318, 1917–1920. https://doi.org/10.1126/science.1151526 (2007).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell131, 861–872. https://doi.org/10.1016/j.cell.2007.11.019 (2007).
Vallier, L., Alexander, M. & Pedersen, R. A. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci.118, 4495–4509. https://doi.org/10.1242/jcs.02553 (2005).
Ying, Q. L., Nichols, J., Chambers, I. & Smith, A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell115, 281–292. https://doi.org/10.1016/s0092-8674(03)00847-x (2003).
Daheron, L. et al. LIF/STAT3 signaling fails to maintain self-renewal of human embryonic stem cells. Stem Cells22, 770–778. https://doi.org/10.1634/stemcells.22-5-770 (2004).
Xi, J. et al. Genetically engineered pigs for xenotransplantation: Hopes and challenges. Front. Cell Dev. Biol.10, 1093534. https://doi.org/10.3389/fcell.2022.1093534 (2022).
Hansen-Estruch, C., Cooper, D. K. C. & Judd, E. Physiological aspects of pig kidney xenotransplantation and implications for management following transplant. Xenotransplantation29, e12743. https://doi.org/10.1111/xen.12743 (2022).
Sykes, M. Developing pig-to-human organ transplants. Science378, 135–136. https://doi.org/10.1126/science.abo7935 (2022).
Wu, J. et al. Interspecies chimerism with mammalian pluripotent stem cells. Cell168, 473-486 e415. https://doi.org/10.1016/j.cell.2016.12.036 (2017).
Lei, T. et al. Genetic engineering of pigs for xenotransplantation to overcome immune rejection and physiological incompatibilities: The first clinical steps. Front. Immunol.13, 1031185. https://doi.org/10.3389/fimmu.2022.1031185 (2022).
Huang, C. P., Chen, C. C. & Shyr, C. R. Xenogeneic cell therapy provides a novel potential therapeutic option for cancers by restoring tissue function, repairing cancer wound and reviving anti-tumor immune responses. Cancer Cell Int.18, 9. https://doi.org/10.1186/s12935-018-0501-7 (2018).
Qu, Y. & Dahl, G. Function of the voltage gate of gap junction channels: Selective exclusion of molecules. Proc. Natl. Acad. Sci. USA99, 697–702. https://doi.org/10.1073/pnas.022324499 (2002).
Lemcke, H., Steinhoff, G. & David, R. Gap junctional shuttling of miRNA—A novel pathway of intercellular gene regulation and its prospects in clinical application. Cell. Signal.27, 2506–2514 (2015).
Totland, M. Z., Rasmussen, N. L., Knudsen, L. M. & Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: Physiological and pathophysiological implications. Cell Mol. Life Sci.77, 573–591. https://doi.org/10.1007/s00018-019-03285-0 (2020).
Peng, Y. et al. Pattern of cell-to-cell transfer of microRNA by gap junction and its effect on the proliferation of glioma cells. Cancer Sci.110, 1947–1958. https://doi.org/10.1111/cas.14029 (2019).
Hong, X., Sin, W. C., Harris, A. L. & Naus, C. C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget6, 15566–15577. https://doi.org/10.18632/oncotarget.3904 (2015).
Neijssen, J., Pang, B. & Neefjes, J. Gap junction-mediated intercellular communication in the immune system. Prog. Biophys. Mol. Biol.94, 207–218. https://doi.org/10.1016/j.pbiomolbio.2007.03.008 (2007).
Jeon, S. B. et al. Endothelial cells differentiated from porcine epiblast stem cells. Cell. Reprogramming23, 89–98. https://doi.org/10.1089/cell.2020.0088 (2021).
Shin, J. H. et al. Functional characterization of endothelial cells differentiated from porcine epiblast stem cells. Cellshttps://doi.org/10.3390/cells11091524 (2022).
Shan, L. et al. Matrix metalloproteinases induce extracellular matrix degradation through various pathways to alleviate hepatic fibrosis. Biomed. Pharmacother.161, 114472 (2023).
Quintero-Fabian, S. et al. Role of matrix metalloproteinases in angiogenesis and cancer. Front. Oncol.9, 1370. https://doi.org/10.3389/fonc.2019.01370 (2019).
Liu, Y. et al. MMP-2 and MMP-9 contribute to the angiogenic effect produced by hypoxia/15-HETE in pulmonary endothelial cells. J. Mol. Cell Cardiol.121, 36–50. https://doi.org/10.1016/j.yjmcc.2018.06.006 (2018).
Harris, A. L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys.34, 325–472. https://doi.org/10.1017/s0033583501003705 (2001).
Wang, W., He, W., Ruan, Y. & Geng, Q. First pig-to-human heart transplantation. Innovation3, 100223. https://doi.org/10.1016/j.xinn.2022.100223 (2022).
Pierson, R. N. 3rd., Burdorf, L., Madsen, J. C., Lewis, G. D. & D’Alessandro, D. A. Pig-to-human heart transplantation: Who goes first?. Am. J. Transplant.20, 2669–2674. https://doi.org/10.1111/ajt.15916 (2020).
Ungvari, Z. et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol.15, 555–565. https://doi.org/10.1038/s41569-018-0030-z (2018).
Ungvari, Z. et al. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J. Gerontol. A Biol. Sci. Med. Sci.68, 877–891. https://doi.org/10.1093/gerona/gls242 (2013).
Mathew, E. et al. Mesenchymal stem cells promote pancreatic tumor growth by inducing alternative polarization of macrophages. Neoplasia18, 142–151. https://doi.org/10.1016/j.neo.2016.01.005 (2016).
Madaric, J. et al. Characteristics of responders to autologous bone marrow cell therapy for no-option critical limb ischemia. Stem Cell Res. Ther.7, 116. https://doi.org/10.1186/s13287-016-0379-z (2016).
Carter, T. D. et al. Porcine aortic endothelial gap junctions: Identification and permeation by caged InsP3. J. Cell Sci.109, 1765–1773 (1996).
Bai, D., Yue, B. & Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr.1860, 9–21. https://doi.org/10.1016/j.bbamem.2017.07.003 (2018).
Zou, Z., Yu, J., Huang, R. & Yu, J. Cx43-Delivered miR-181b negatively regulates Sirt1/FOXO3a signalling pathway-mediated apoptosis on intestinal injury in sepsis. Digestion104, 370–380. https://doi.org/10.1159/000529102 (2023).
Ding, M. H., Lozoya, E. G., Rico, R. N. & Chew, S. A. The role of angiogenesis-inducing micrornas in vascular tissue engineering. Tissue Eng. Part A26, 1283–1302. https://doi.org/10.1089/ten.TEA.2020.0170 (2020).
Yang, L. et al. MicroRNA-26b-5p inhibits mouse liver fibrogenesis and angiogenesis by targeting PDGF receptor-beta. Mol. Ther. Nucleic Acids16, 206–217. https://doi.org/10.1016/j.omtn.2019.02.014 (2019).
Pan, C., Han, Y. & Lu, J. Structural design of vascular stents: A review. Micromachineshttps://doi.org/10.3390/mi12070770 (2021).
Alferiev, I. S. et al. Intraprocedural endothelial cell seeding of arterial stents via biotin/avidin targeting mitigates in-stent restenosis. Sci. Rep.12, 19212. https://doi.org/10.1038/s41598-022-23820-7 (2022).
Funding
This study was funded by the National Research Foundation of Korea. NRF of Korea (NRF-2021R1C1C1006516 and NRF-2020R1I1A1A01072377). This work was supported by the Technology Innovation Program (20022828, Research and Development of micronized human acellular dermal matrix preserving collagen and growth factor for soft tissue filling), funded By the Ministry of Trade, Industry & Energy (MOTIE, Korea).
Author information
Authors and Affiliations
Contributions
J.-H.L. (GNU) and C.H. conceived and designed the study. B.-G.S. and I.-W.L. conducted the research and investigation and performed the experiments. B.-G.S analyzed data. O.K. and J.-H.L. (KNU) contributed to perform in vivo experiments. B.-G.S. wrote the first draft of the manuscript. H.-J.K. and Y.-J.L. reviewed the manuscript. J.-H.L. (GNU) and C.H. revised the final manuscript. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consents to participate
All procedures involving animals were performed according to the guidelines approved by both the Animal Care and Use Committee at Kangwon National University. (1) Title of the approved project: Evaluation of the differential ability of embryonic stem cells; (2) Name of the institutional approval committee: Institutional Animal Care and Use Committee (IACUC); (3) IACUC approval no. KW-220223–2; (4) Date of approval: March 02, 2022. Animal Care and Use Committee at Gyeongsang National University; (1) Title of the approved project: production and functional appraisal of vascular endothelial cells using porcine embryonic, induced pluripotent and adult stem cells-based 3D bioprinting organoid; (2) Name of the institutional approval committee: Institutional Animal Care and Use Committee (IACUC); (3) IACUC approval no. GNU-210524-P0050; (4) Date of approval: May 24, 2021.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seo, BG., Lee, IW., Kim, HJ. et al. Angiogenic properties and intercellular communication of differentiated porcine endothelial cells in vascular therapy. Sci Rep 14, 22844 (2024). https://doi.org/10.1038/s41598-024-73584-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73584-5
