
Abstract
Bract coloration is one of the key ornamental traits in Bougainvillea, yet research has predominantly focused on phenotypic color traits and pigment composition, with limited understanding of the molecular mechanisms underlying color formation. This gap hinders the improvement and innovation in bract coloration. To elucidate the regulatory mechanisms of bract coloration in Bougainvillea and to enhance the utilization of its germplasm resources, this study employed the Illumina Novaseq 6000 sequencing platform to conduct transcriptomic sequencing on 21 samples of bracts exhibiting seven distinct phenotypes. Comparative analysis against Nr, Pfam, EggNOG, GO, and KEGG databases annotated 90,279 unigenes. The highest annotation rates were achieved with the Nr (40.13%), GO (30.44%), and EggNOG (25.64%) databases. Among the species annotated, Beta vulgaris (20.08%) and Chenopodium quinoa (14.58%) shared the highest homology with Bougainvillea bract transcriptomes. WGCNA analysis identified 12 positively correlated tissue-specific modules, of which 2 are related to bract color formation. By comparing transcriptome data and genes within these specific modules against the KEGG database, a total of 321 unigenes associated with bract color formation in Bougainvillea were discovered. Among these, 220 unigenes are involved in anthocyanin synthesis, 43 unigenes are involved in betalain synthesis, 23 unigenes are annotated as Chlorophyll a-b binding protein genes, and 35 unigenes participate in carotenoid synthesis. Quantitative real-time PCR (qRT-PCR) validation of 16 differentially expressed genes (DEGs) including PAL2, CHS1, ANS, BZ1, 6GT, CDOPA5GT, ANR, CHS2, and DOPA, revealed significant expression differences among magenta, yellow, white, and cherry-colored bracts, suggesting their potential as candidate genes for bract color development. This study not only enriches the transcriptomic data of Bougainvillea but also identifies genes associated with bract coloration, providing a valuable theoretical basis for future gene cloning, genetic engineering, and breeding efforts in Bougainvillea.
Similar content being viewed by others
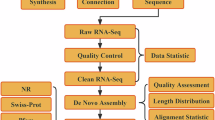


Introduction
Bougainvillea glabra Choisy, belonging to the Nyctaginaceae family and the Bougainvillea genus, is primarily a vine or shrub and one of the main woody ornamental plants1,2,3,4. It also holds significant ornamental and medicinal value5,6, and is utilized in the development of natural food colorants7, biological pest control8, environmental protection, and landscape greening9,10. The bracts of Bougainvillea, characterized by their rich colors and unique shapes, serve as the main ornamental aspect of the plant. Identifying genes related to bract coloration is of significant importance for improving the ornamental and market value of Bougainvillea bracts.
Transcriptome sequencing involves sequencing all cDNA sequences of a specific tissue and comparing these sequences with known sequences in public databases to annotate them. This process clusters differentially expressed genes with similar functions, thereby facilitating the discovery of genes and their functional prediction in non-model species11. Previous research, such as that by Wei Zhao12, has shown that the white coloration in Brassica pekinensis flowers is due to low expression of carotenoid biosynthesis genes in white petals. Li Na Sun13,14 screened key genes involved in bract development process and color regulation, as well as related enzymes involved in flavonoid metabolic pathway and betaine biosynthesis pathway by transcriptome sequencing of bracts at different periods of Bougainvillea spectabilis ‘Senjakala’. Yiqi Deng et al.15 conducted transcriptome sequencing on the leaves and bracts of B. glabra, identifying homologous genes related to plant photosynthesis in the leaves. Furthermore, transcriptome sequencing has been applied to identify flower color-related genes in other plants such as Dunnia sinensis16, Clematis florida17, and Camellia sinensis18. Extensive research indicates that anthocyanins, betalains, and carotenoids are the main pigments determining flower color in plants. The pigments of B. glabra bracts are primarily composed of flavonoids, betalains, and betaxanthins, with a potential presence of anthocyanins9,19,20. Identifying genes related to bract coloration and clarifying the biosynthetic pathways of B. glabra bract pigments are significant for regulating the color of B. glabra bracts.
Currently, research on B. glabra primarily focuses on phenotypic color traits and pigment composition, with little reported on the mechanisms of bract coloration and development. To understand the mechanisms behind bract color formation and to reveal the genetic background of B. glabra, in this study, high-throughput transcriptome sequencing was performed on B. glabra bracts. Quality control sequences were compared with annotation databases to obtain species annotations, protein and molecular function annotations for B. glabra. Combining these results with WGCNA, genes related to bract color synthesis pathways were screened from the transcriptome data and the positively correlated specific modules. Furthermore, the relative quantitative expression of genes related to betalain and anthocyanin synthesis in B. glabra was analyzed using qRT-PCR, revealing the regulatory mechanisms of betalain and anthocyanin biosynthesis from the transcriptome perspective. The findings from this study are crucial for elucidating the molecular mechanisms underlying bract color formation, enabling the regulation and utilization of bract coloration in B. glabra
Materials and methods
Experimental materials
The experimental material consisted of B. glabra bracts at full bloom, representing seven distinct phenotypic traits. These samples were collected in October 2019 from the greenhouse facility of Jicheng Garden Science & Technology Co., Ltd., located in Mile City, Yunnan Province, China (103° 04′ E, 23° 50′ N; elevation 1450 m, greenhouse temperature 31 °C), all sample specimens are stored in the Herbarium of Southwest Forestry University; The test material number is: A (GQ001); B (GQ002); C (GQ003); D (GQ004); E (GQ005); F (GQ006); G (GQ007). Each sample type was biologically replicated three times, yielding a total of 21 samples. After being washed, the samples were blotted dry with filter paper, transferred to cryovials, immediately flash-frozen in liquid nitrogen, stored on dry ice, and shipped to Meiji Biomedical Technology Company (Shanghai) for sequencing. To minimize the interference between different types of bract traits, five single-bract materials excluding types B and D were selected for gene expression analysis related to bract color formation (Fig. 1).

Materia of B. glabra with differently shaped bracts. Note: The traits of the samples were as follows: (A) single bract, magenta; (B) double bracts, magenta; (C) single bract, white with cherry blossom pink tips; (D) double bracts, white with cherry blossom pink tips; (E) single bract, white; (F) single bract, yellow; (G) butterfly-shaped, red.
Methods
RNA extraction and transcriptome sequencing
RNA was extracted from B. glabra bracts using a magnetic bead-based RNA extraction kit from Meiji Biomedical Technology Company. RNA integrity was verified by 1% agarose gel electrophoresis, and RNA concentration and purity were assessed using a NanoDrop 2000 spectrophotometer. For reverse transcription, the RNA was diluted to 50 ng/µl using a reverse transcription kit from Tiangen Biochemical Technology Company. The reaction setup included 4 µl of 5× Fast King-RT Super Mix, RNA x µl (50 ng/µl), topped up to 20 µl with RNase-Free ddH2O. The mixture was kept on ice during sample addition. The reaction protocol was set to 42 °C for 15 min (to remove genomic DNA and perform reverse transcription) followed by enzyme inactivation at 95 °C for 3 min. Sequencing libraries were constructed using the Illumina TruSeq TMRNA sample prep Kit.
Sequence acquisition and functional annotation
The raw sequencing data, containing various impurities, were subjected to stringent quality control to ensure the accuracy of subsequent analyses. Clean data were used for analysis. Transcriptome assembly was performed using the Trinity21 software suite (https://github.com/trinityrnaseq/trinityrnaseq), which integrates Inchworm, Chrysalis, and Butterfly programs to process extensive RNA-seq data. Subsequent annotations of the unigenes were performed using the BLAST22 algorithm (E-value threshold of 1e-5), aligning sequences to databases including the Non-Redundant Protein Sequence Database (Nr), Protein families (Pfam), Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups (EggNOG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG)23,24,25.
Construction of weighted gene co-expression network and screening of bract color-related genes
Using the RSEM and WGCNA packages in R version 4.2.126, genes in transcriptome samples of five different bract traits were filtered by setting the mean gene expression value to 1 and the coefficient of variation to 0.1. The filtered gene expression matrix was used as input data to identify and screen specific expression modules. Genes within the unigenes from the transcriptome assembly and the weighted gene co-expression specific modules (correlation coefficient > 0.5) were screened for bract color biosynthesis-related KO pathways based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases, thereby identifying genes involved in bract color formation. GO and KEGG enrichment results were displayed under the condition of padjust < 0.05, showing the top 20 enriched terms. The parameters for module identification were set as follows: minModuleSize = 30, minKMEtoStay = 0.3, mergeCutHeight = 0.25. Modules with a correlation coefficient > 0.65 and significance (P-value) < 0.0041 in positively correlated modules were considered specific modules.
Validation of bract coloration genes by qRT-PCR
Primer design and screening
Based on the results of gene screening, sequences of genes related to bract coloration were identified from the transcriptome assembly. Primers were designed using the Primer-BLAST tool from the National Center for Biotechnology Information (NCBI), adhering to the following criteria: target fragment length of 100–300 bp, primer length of 18–24 bp, and GC content of 40-60%. From these, 2–3 pairs of primers were selected. The primers were aligned against the transcriptome unigenes sequences using TBtools, and primers that matched the corresponding gene sequences were synthesized (Beijing Qingke Biotech Co., Ltd.—Kunming Synthesis Department), including primers for TYR, PAL, BZ1, among others, totaling 16 genes (detailed information on the quantitative fluorescent primers is provided in Supplementary Table 1). To minimize the impact of nonspecific bands on the accuracy of subsequent real-time fluorescent quantification, primers with specific bands were selected using Polymerase Chain Reaction (PCR).
qRT-PCR validation of candidate genes
The reference genes selected for normalization in B. glabra were the stably expressed F1-ATPase alpha subunit (atp1), 18 S, and 26 S genes. The sequences of these reference genes were obtained from the NCBI database’s B. glabra chloroplast genome. The primer design principles for the reference genes were the same as for the candidate genes. Specificity of the primers was first confirmed by PCR, followed by analysis of reference gene expression stability using quantitative fluorescent reactions.
Preliminary PCR reaction setup (10 µl volume): 2×Taq PCR Star Mix 5 µl, forward/reverse primers 1 µl each; cDNA template 1 µl, ddH2O 3 µl. PCR protocol: 94 °C for 5 min for initial denaturation; 35 cycles of 94 °C for 30 s, 50–60 °C for 30 s annealing, 72 °C for 30 s extension; final extension at 72 °C for 10 min. Products were analyzed using 1% agarose gel electrophoresis.
Quantitative fluorescent reaction setup (10 µl volume): SYBR Master Mix (2×) 5 µl; forward/reverse primers 0.2 µl each; Reference Dye 0.2 µl; cDNA template 1 µl, ddH2O 3.4 µl. Fluorescent quantification protocol (three-step method): initial denaturation at 95 °C for 5 min; 40 cycles of 95 °C for 15 s, 50–60 °C for 30 s annealing, 72 °C for 30 s extension; followed by a melt curve analysis cycle of 95 °C for 15 s, 50–60 °C for 60 s, 95 °C for 15 s.
The expression levels of candidate genes in the sequencing samples were validated using quantitative reverse transcription PCR (qRT-PCR), and gene expression was analyzed using the 2−ΔΔCT method as described by Jie Yang27.
Results
Transcriptome sequencing analysis of bracts
RNA-Seq and denovo a ssembly
High-throughput transcriptome sequencing yielded approximately 1256 Gb of original sequences. After data cleaning, 184.85 Gb of quality-controlled sequences were acquired. For each sample, the quality-controlled sequences surpassed 7.5 Gb, showcasing an average base error rate of 0.02%, an average Q20 base percentage of 98.36%, and an average GC content of 43.71%. These results indicate that the B. glabra sequences obtained from the Illumina Novaseq 6000 sequencing platform are of high quality and meet the requirements for subsequent bioinformatics analysis (Table 1).
A total of 220,543 unigenes were assembled from all clean data using trinity, with the longest unigene being 16,992 bp, the shortest 201 bp, an average unigene length of 815.66 bp, N50 of 1176 bp, and E90N50 of 2451 bp. Of these, 108,855 unigenes (49.36%) measured between 200–500 bp, 62,818 (28.48%) between 501–1000 bp, 31,104 (14.1%) between 1001–2000 bp, and 17,766 (8.06%) exceeded 2000 bp in length (Table 2).
Functional annotation of unigenes
Functional annotation results of the 220,543 unigenes of B. glabra transcriptome in the five major databases are shown in Fig. 2. Among them, 88,493 unigenes (40.13%) were matched in the Nr database, 24,707 (11.20%) in the KEGG database, 67,140 (30.44%) in the GO database, 56,548 (25.64%) in the EggNOG database, and 48,976 (22.21%) in the Pfam database. A total of 90,279 unigenes (40.93%) were matched in all databases, higher than the number of unigenes matched in each individual database, indicating that the same unigene could be matched in multiple databases. Further analysis revealed that 14,121 unigenes (6.40% of the total) were matched in all six databases.

Venn diagram of functional annotation of B. glabra unigenes based on five databases.
Transcriptome unigenes Nr species annotation
The NR database is a non-redundant protein sequence database. Comparing the species unigenes with the Nr database provides information on the similarity and homologous sequence functions of the species to closely related species. As shown in Fig. 3 and 220,543 unigenes of B. glabra showed significant homology in the Nr database. The highest number of homologous sequences was matched to Beta vulgaris (beet), with 17,770 sequences (20.08% of the total). This was followed by homologous sequences matched to Chenopodium quinoa (quinoa), Vitis vinifera (grape), and Spinacia oleracea (spinach), accounting for 14.58% (12905 sequences), 12.71% (11248 sequences), and 11.10% (9822 sequences), respectively. Homologous sequences matched to Nicotiana attenuata, Quercus suber (cork oak), Cajanus cajan (pigeon pea), Camellia sinensis (tea), Nicotiana tomentosiformis (fuzzy tobacco), Nicotiana sylvestris (woodland tobacco), Asparagus officinalis (asparagus), Nicotiana tabacum (tobacco), Artemisia annua (sweet wormwood), and Capsicum baccatum (pepper) accounted for 2.99% (2647 sequences), 2.53% (2239 sequences), 1.57% (1390 sequences), 1.46% (1294 sequences), 1.00% (882 sequences), 0.97% (857 sequences), 0.88% (783 sequences), 0.85% (751 sequences), 0.74% (652 sequences), and 0.73% (644 sequences), respectively. Approximately 27.81% of the homologous sequences were distributed among other species.

Nr annotation of B. glabra transcript tomeunigenes.
GO functional annotation of transcriptome unigenes
The Gene Ontology (GO) database provides classification annotations for genes and gene products at three levels: Cellular Component (CC), Molecular Function (MF), and Biological Process (BP). The GO functional annotation results are shown in Fig. 4. Among the 67,140 unigenes matched in the GO database, there were 13 subcategories in Cellular Component, 17 in Molecular Function, and 23 in Biological Process, with corresponding quantities distributed as 115,708 (52.47%), 98,168 (44.51%), and 109,658 (49.72%) respectively, indicating the most annotations in the Cellular Component category. Among the 13 subcellular localizations in Cellular Component, cell part (GO:0044464), membrane (GO:0044425), and organelle (GO:0043226) received the most annotations, accounting for 27.56% (31886), 22.00% (25454), and 16.62% (19231) of this category, respectively. In the 17 Molecular Function categories, genes related to binding (GO:0005488) and catalytic activity (GO:0003824) were most common, accounting for 46.76% (45901) and 44.02% (43210) of this category, respectively. Among the 23 subcategories in Biological Process, genes annotated as cellular process (GO:0009987) and metabolic process (GO:0008152) were more numerous, accounting for 31.35% (34378) and 29.55% (32409) of this category, respectively.

GO annotation of B. glabra transcriptome unigenes. Note: The upper horizontal axis represents the percentage of the total number of unigenes in that level of classification, while the lower horizontal axis represents the number of unigenes compared to that level of classification.
KEGG functional annotation of transcriptome unigenes
The Kyoto Encyclopedia of Genes and Genomes (KEGG) is an integrated database that amalgamates systematic information on gene functions, genomic data, and chemical properties. The comparison of B. glabra unigenes with the KEGG database, illustrated in Fig. 5, revealed that 19,562 unigenes are involved in KEGG metabolic pathways, allocated across six major categories (Level 1) and 27 subcategories (Level 2). Among these categories, genes classified under Metabolism (9,710 unigenes, 49.64%, 11 subcategories) were predominant, followed by Genetic Information Processing (4,793 unigenes, 24.50%, 4 subcategories), Cellular Processes (1,815 unigenes, 9.28%, 2 subcategories), Organismal Systems (1,232 unigenes, 6.30%, 4 subcategories), Environmental Information Processing (1,094 unigenes, 5.59%, 2 subcategories), and Human Diseases (918 unigenes, 4.70%, 4 subcategories).

KEGG annotation of B. glabra transcriptome unigenes.
Prediction of CDS in transcriptome unigenes
The transcriptome unigenes were compared with the NR protein database and the Swissprot protein database to obtain the coding sequences (CDS). For sequences that did not match with the protein databases, Trans Decoder was used to predict candidate coding regions in the transcriptome sequences. A total of 152,009 CDS were identified through comparison and prediction. As shown in Fig. 6, the most numerous CDS lengths were in the range of 201–400 bp, totaling 62,187, accounting for 40.91% of all CDS. Following this, CDS lengths of 200 bp and 401–600 bp were the next most common, with 44,841 and 17,252 instances respectively, accounting for 29.50% and 11.35% of all CDS. There were 19,586 CDS longer than 600 bp, accounting for 18.24% of the total CDS.

CDS length distribution of B. glabra transcriptome.
Weighted gene co-expression network analysis
Data preprocessing
Cluster analysis was performed on the samples to exclude outliers. As shown in Fig. 7, the repetitions of each sample clustered together, indicating no outliers, thus ensuring the accuracy of subsequent analysis.

Clustering of bract samples with different traits.
Construction of gene clustering tree and module identification
By analyzing the RNA-seq data of bracts with five different traits, genes with low variability in the expression matrix were filtered, and 40,139 genes were selected for WGCNA analysis. When the optimal power value β = 6 and R2 > 0.80, the mean connectivity tended to 0 (Fig. 8), indicating that this power value is most suitable for constructing a scale-free network. Nineteen gene co-expression modules were successfully constructed for bracts with different traits (Fig. 9), the number of genes in each module is shown in Fig. 10, with the number of genes in the modules ranging from 96 to 12,508. The three modules with the highest number of genes were turquoise (12,508), blue (7,709), and brown (4,798); the module with the fewest genes was lightyellow (96).

Selection of soft threshold. Note: (a) The horizontal axis represents the power value β, and the vertical axis represents the degree of fit between the adjacency matrix transformed by the corresponding β value and the scale-free network hypothesis R2 (the larger the R2, the more the network conforms to the scale-free distribution). (b) The horizontal axis represents the power value β, and the vertical axis represents the mean connectivity (k/degree/connectivity) of each node in the network represented by the corresponding β value transformed adjacency matrix.

Gene clustering tree and module division in Co-expression network. Note: In the clustering tree in the upper part, each branch represents a module, and each leaf on the branch represents a gene. In the module colors in the lower part, each color represents a module, and gray represents genes not assigned to any module, totaling 1390. The letters on the vertical axis in the lower part represent bract trait types: (A)—magenta; C—cherry blossom; E—white; F—yellow; G—red, same as below.

Statistical chart of the number of genes in each odule.
Through screening, 12 positively and negatively correlated tissue-specific modules were found in the five different bract traits (Fig. 11). The significantly positively correlated specific modules in different materials are: A (pink, 0.694); C (turquoise, 0.656; greenyellow and cyan, 0.694); E (brown, 0.656); F (tan, green, blue, and red, 0.694); G (purple and black, 0.694). The significantly negatively correlated specific modules are: A (brown and magenta, -0.694); E (black, greenyellow, yellow, blue, red, and pink, -0.964); F (turquoise and purple, -0.964; lightyellow, -0.656); G (salmon, -0.696). There are no significantly negatively correlated modules in trait C, and the module with the lowest correlation coefficient is lightgreen (-0.54).

Heatmap of module-sample correlations. Note: The horizontal axis represents different samples, and the vertical axis represents different modules. The numbers on the left side of the figure indicate the number of genes in the module, and the data on the right side indicate the correlation coefficient and significance P value (in parentheses) between the module and the sample.
GO enrichment analysis of specific modules
Enrichment analysis was performed on genes with a correlation coefficient > 0.5 in the significantly positively correlated specific modules of the five different bract traits (Supplementary Tables 2 and 3).
As shown in Fig. 12, GO enrichment in trait A indicated that the 733 genes in the significantly positively correlated module were mainly enriched in oxidoreductase activity (number: 49, GO:0016491), dioxygenase activity (12, GO:0051213), heme binding (9, GO:0020037), and others. KEGG enrichment revealed that the 733 genes were primarily enriched in Glycolysis / Gluconeogenesis (number: 6), alpha-Linolenic acid metabolism (5), Pyruvate metabolism (5), and Fatty acid degradation (5), among others.
In trait C, GO enrichment showed that the 2852 genes in the three significantly positively correlated modules were mainly enriched in cell wall macromolecule metabolic process (17, GO:0044036), cell wall polysaccharide metabolic process (16, GO:0010383), protein dimerization activity (23, GO:0046983), antioxidant activity (58, GO:0016209), and others. KEGG enrichment indicated that the 2852 genes were mainly enriched in Phenylpropanoid biosynthesis (22), Ribosome (31), DNA replication (9), Peroxisome (11), and others.
In trait E, GO enrichment showed that the 2593 genes in the significantly positively correlated module were mainly enriched in Golgi apparatus (51, GO:0005794), external encapsulating structure (39, GO:0030312), cell wall organization or biogenesis (51, GO:0071554), external encapsulating structure organization (50, GO:0045229), cell wall organization (45, GO:0071555), and others. KEGG enrichment revealed that the 2593 genes were primarily enriched in Amino sugar and nucleotide sugar metabolism (30), Plant hormone signal transduction (31), Phenylpropanoid biosynthesis (17), Starch and sucrose metabolism (16), and others.
In trait F, GO enrichment showed that the 1221 genes in the four significantly positively correlated modules were mainly enriched in posttranscriptional regulation of gene expression (8, GO:0010608), translation factor activity, RNA binding (11, GO:0008135), translation regulator activity, nucleic acid binding (11, GO:0090079), translation regulator activity (11, GO:0045182), and others. KEGG enrichment indicated that the 1221 genes were primarily enriched in Glycolysis / Gluconeogenesis (6), Glutathione metabolism (6), Peroxisome (5), Glycine, serine and threonine metabolism (6), and others.
In trait G, GO enrichment analysis indicated that the 1569 genes in the two significantly positively correlated modules were mainly enriched in nuclear chromosome part (8, GO:0044454), chromosomal part (19, GO:0044427), regulation of hydrolase activity (5, GO:0051336), FAD binding (8, GO:0071949), and others. KEGG enrichment revealed that the 1569 genes were primarily enriched in Amino sugar and nucleotide sugar metabolism (11), Phenylpropanoid biosynthesis (8), Ubiquinone and other terpenoid-quinone biosynthesis (6), MAPK signaling pathway - plant (5), and others.

GO and KEGG enrichment bubble plots of genes in specific modules with different traits.
Analysis of target modules
Further KEGG enrichment analysis of each significantly positively correlated module revealed that in trait C, 8 unigenes in the greenyellow and cyan specific modules were enriched in the phenylpropanoid biosynthesis process, 2 unigenes in the stilbenoid biosynthesis process, and 3 unigenes in the flavonoid biosynthesis process. In trait F, 10 unigenes in the red module were enriched in the carotenoid biosynthesis process, and 5 unigenes in the flavonoid biosynthesis process. These genes could be candidate genes regulating bract color formation (Fig. 13).

KEGG enrichment bubble plots of target modules (greenyellow, cyan, and red).
Expression analysis of bract color-related genes
Genes related to bract color formation
As shown in Table 3, a total of 321 unigenes related to the biosynthesis pathways of anthocyanins, betalains, chlorophylls, and carotenoids were detected. Among them, 220 unigenes were involved in the anthocyanin biosynthesis pathway, with 42 unigenes related to the F3’5’H (K13083) gene, 28 unigenes related to the 4CL (K01904) gene, 15 unigenes related to the CHS (K00660) gene, and 15 unigenes related to the E2.1.1.104 (K00588) gene. Additionally, key genes regulating anthocyanin biosynthesis, such as PAL, FLS, ANS, ANR, and LAR, also had corresponding unigenes.
43 unigenes were detected to be involved in the betalain biosynthesis pathway, with 10 unigenes matching the CDOPA5GT (K15776) gene. The DOPA (K15777) and TYR (K00505) genes were each annotated with 7 unigenes. Twenty-three unigenes were annotated as Chlorophyll a-b binding protein genes, and 35 unigenes were involved in the carotenoid biosynthesis pathway. These results provide a reference for the chemical composition analysis of B. glabra species and the cloning of genes regulating bract color.
Differential expression analysis of genes in different pigment biosynthesis pathways
Based on WGCNA analysis, 28 genes enriched in different pigment biosynthesis pathways were screened from Table 3. These include 16 genes involved in the anthocyanin synthesis pathway (ANR, ANS, BZ1, CHS2, PAL2, CHS1, COMT, E2.1.1.104, bglX, FLS, CYP75A, CCR, E1.1.1.195, 4CL, E3.2.1.21 and E1.11.1.7), 3 genes involved in betalain synthesis (6GT, CDOPA5GT, and DOPA), 4 genes involved in chlorophyll synthesis (LHCA4, LHCB4, LHCA3, and LHCB3), and 5 genes involved in carotenoid synthesis (AAO1, AAO3, NCED, CYP97A3, and ABA2). Using R version 4.2.028, a clustering heatmap of differential genes and bract samples with different color traits was constructed to analyze the expression patterns of pigment biosynthesis genes in different bract samples. This analysis aimed to identify candidate genes related to the formation of bracts with different colors.
As shown in Fig. 14, the expression patterns of genes in different pigment biosynthesis pathways vary across different bract traits. Five genes involved in the anthocyanin synthesis pathway (ANR, BZ1, ANS, PAL2, and CHS1), two genes involved in betalain synthesis (6GT and CDOPA5GT), and three genes involved in carotenoid synthesis (AAO1, AAO3, and NCED) exhibit higher expression levels in trait A, suggesting that multiple pigments may jointly regulate the color formation process in trait A. The anthocyanin synthesis pathway genes FLS, CYP75A, and E1.1.1.195 are highly expressed in traits C and E, while CHS2 is expressed in traits C and F, and COMT, E2.1.1.104, and bglX are highly expressed only in trait C, indicating that these genes may play a key role in the color regulation of trait C. Notably, the anthocyanin synthesis gene E1.11.1.7 shows significantly higher expression in trait E than in other traits, suggesting its indispensable role in the color formation process of trait E. The genes CCR, ABA2, and the four chlorophyll synthesis genes are highly expressed in trait F, indicating the involvement of multiple pigment genes in regulating the color formation of trait F. Similarly, the betalain synthesis gene DOPA, the carotenoid synthesis gene CYP97A3, and three anthocyanin synthesis pathway genes CYP97A3, 4CL, and E3.2.1.21 are highly expressed in trait G, with CYP97A3, 4CL, and DOPA also exhibiting some expression in traits C and E, respectively.
Among the key genes screened from the bract transcriptome for anthocyanin synthesis, the absence of the C4H and DFR genes in the anthocyanin biosynthesis pathway may result in the inability of B. glabra bracts to synthesize anthocyanins. Therefore, the annotated genes related to anthocyanin synthesis identified in this study may not fully participate in the anthocyanin biosynthesis pathway in B. glabra. These genes may only be involved in the synthesis of anthocyanin precursors but are key genes in flavonoid biosynthesis. Both anthocyanins and flavonoids are products of the flavonoid metabolic pathway, with flavonoids being synthesized upstream of anthocyanins. Thus, analyzing the expression of these genes can provide a basis for regulating the biosynthesis of flavonoids in B. glabra.
The differential expression of pigment synthesis genes in bracts with different traits indicates that the pigments contributing to the coloration of B. glabra bracts may be composed of various pigments, which together participate in and regulate the color formation process of the bracts.

Clustering heatmap of candidate genes in different pigment synthesis pathways and bracts with different traits.
Ct value analysis of candidate reference genes
The Ct values of candidate reference genes in the samples were analyzed. The Ct value ranges for the atp1, 26 S, and 18 S genes across the five samples were 16.34 to 17.95, 17.85 to 19.8, and 9.85 to 11.95, respectively, The coefficient of variation (CV) values were 0.03, 0.03, and 0.06, respectively (Table 4). Given that Ct values inversely correlate with expression levels, the 18 S gene exhibited the highest expression, followed by atp1 and 26 S. Considering the CV values, all three reference genes showed relative stability, with the 18 S gene having the highest CV value (0.06%) and therefore the least stability, with the atp1 gene demonstrating the lowest CV value (0.03%) and thus the highest stability. Therefore, the atp1 gene was chosen as the reference gene for subsequent analysis of gene expression related to bract color.
Analysis of relative expression levels of anthocyanin synthesis-related genes in different colored bracts
Quantitative Reverse Transcription PCR (qRT-PCR) validation was performed on 16 genes, including BZ1, ANR, CDOPA5GT, CHS1, CYP73A, PAL2, TYR, ANS, 6GT, DOPA, F3H1, F3H2, 5GT, LAR, 4CL, and CHS2. Notably, six of these genes—PAL2, ANR, CHS1, CHS2, ANS, and BZ1—which are related to the regulation of bract anthocyanin synthesis, showed markedly different expression levels across the samples (Fig. 15). PAL, pivotal for synthesizing phenolic precursors of anthocyanins and considered a rate-limiting enzyme in anthocyanin biosynthesis29, showed higher expression in magenta and red bracts, with lower expression in cherry blossom, white, and yellow bracts. ANR, which catalyzes anthocyanins’ conversion to flavan-3-ols and directly transfers anthocyanins to catechins (yellow)30, had the highest expression in yellow bracts, notably higher than in magenta, cherry blossom, and white bracts. CHS catalyzes the condensation of 4-coumaroyl-CoA and malonyl-CoA to form tetrahydroxychalcone31, the first key enzyme in the biosynthesis of flavonoid secondary metabolites. CHS1 gene expression was significantly higher in magenta and red bracts compared to cherry blossom, white, and yellow bracts. Conversely, CHS2 showed higher expression in cherry blossom and white bracts than in magenta, yellow, and red bracts. ANS catalyzes the oxidation of colorless anthocyanins to form colored anthocyanins, a key enzyme in the anthocyanin biosynthesis pathway; its expression level was highest in magenta bracts, significantly higher than in yellow, cherry blossom, red, and white bracts. BZ1 is involved in the conversion of pelargonidin to anthocyanin glycosides32; its expression was highest in magenta bracts, significantly higher than in yellow, cherry blossom, red, and white bracts, followed by higher expression in yellow bracts, with no significant differences in cherry blossom, red, and white bracts.

The relative expression of genes related to anthocyanin synthesis in B. glabra bracts of different colors. Note: Each bar represents the average expression level of genes across different color traits, with error bars indicating standard error of the mean (SEM). Different lowercase letters indicate significant differences (p < 0.05), while the same letters indicate no significant difference statistically, the same below.
Analysis of relative expression levels of betalain synthesis-related genes in different colored bracts
The relative expression levels of the genes 6GT, CDOPA5GT, and DOPA, which are involved in the regulation of bract betalain pigmentation, varied significantly among the samples (Fig. 16). The 6GT gene catalyzes the transfer of a glucosyl group to betalamic acid’s 6-hydroxyl33; its expression level in the five different colored bracts was in the order: magenta > red > cherry blossom > white > yellow, with significantly higher expression in magenta bracts compared to the other four colors. CDOPA5GT gene, which glycosylates cyclic DOPA to form betanidin34—thereby spontaneously generating red betalains and enriching the betalain biosynthesis pathway—displayed expression levels in the order of magenta > red > cherry blossom > white > yellow across the five colored bracts, with significantly higher expression in magenta and red bracts, indicating this gene regulated the red coloration in B. glabra bracts. DOPA, which catalyzes the cleavage of the 4,5-carbon bond of dopa to produce 4,5-seco-dopa, is one of the key enzymes in the betalain biosynthesis pathway. Its expression level is highest in magenta bracts and significantly higher than in cherry blossom, white, yellow, and red bracts.

The relative expression of genes related to betalain synthesis in B. glabra bracts of different color.
Discussion
GO and KEGG annotation
The annotation results of the bract transcriptome data indicate that out of the 220,543 unigenes obtained from de novo assembly using Trinity software, 90,279 unigenes were annotated. The 130,264 unigenes that were not matched may be due to differences in the splicing software and parameters used. Nr annotation indicated that the B. glabra bract transcriptome sequences matched the most homologous sequences of Beta vulgaris (beet) (20.08%) and Chenopodium quinoa (quinoa) (14.58%). B. glabra and beet are both model species for betalain metabolism studies; B. glabra leaf transcriptome sequences also show high homology (60.7%) with beet genes35, providing theoretical support for studying the evolutionary directions of betalain metabolism among these species. Comparison with the GO database indicated the highest annotations in the cellular component category. A large number of sequences were annotated in biological processes to cell processes (GO:0009987) and metabolic processes (GO:0008152), potentially involved in the biosynthesis of bract pigments. Zheng et al.36 pointed out that related DEGs are enriched in pigmentation processes, and this study also found sequences with pigmentation functions (GO:0043473), which can serve as candidate genes for regulating bract color formation.A total of 97 metabolic pathways are involved in KEGG annotation, among the 27 subcategories, genes annotated to Translation and Carbohydrate Metabolism were the most numerous, accounting for 45.44% and 21.84% of their respective pathways, respectively. These findings are consistent with the research by Zhao37 and Zhao38. Similarly, Jing Li39 study on Panax japonicus highlighted a significant number of unigenes annotated to the Carbohydrate Metabolism category in the KEGG database. The abundance of unigenes related to Genetic Information Processing and Metabolic Processes in the bracts suggests active involvement in bract pigment synthesis. These results can provide references for studying the biosynthetic pathways of bioactive components in B. glabra bracts, aiding in the functional analysis of related genes.
Bract pigment composition
In this study, transcriptome sequencing was performed on bracts of five different colored B. glabra, combined with WGCNA, identifying a total of 220 unigenes involved in the anthocyanin biosynthesis pathway, 43 unigenes related to the betalain biosynthesis pathway, 23 unigenes annotated as Chlorophyll a-b binding protein, and 35 unigenes involved in carotenoid biosynthesis. Bract color is an important phenotypic trait affecting the ornamental value of B. glabra. Flavonoids, carotenoids, flavonols, and chlorophylls are the main pigments in flavonoid plants40,41. Brockington et al.42 proposed that betalains and anthocyanins are mutually exclusive within the same plant species. In contradiction to this study, it is possible that the bracts accumulate betalains while also expressing enzymes related to the biosynthesis of anthocyanins, or that there is a co-presence of biosynthetic enzyme genes for both anthocyanins and betalains during their synthesis processes43,44; KEGG functional annotation identified seven unigenes annotated as DOPA genes. The presence of DOPA genes suggests that plants accumulating anthocyanins can also produce betalain pigments45, resulting in the coexistence of both pigments in B. glabra bracts. Previous research has confirmed the presence of DOPA genes in B. glabra bracts46,47. Betalains and anthocyanins are common water-soluble pigments in plants44, with betalains giving plants yellow, purple, and red colors, while anthocyanins can also produce natural blue. While betalains have been extracted from B. glabra bracts, flavonoid substances identified in B. glabra bracts have merely been hypothesized to be anthocyanins48,49. Yuqian Kang10 and Huihui Zhang20 also did not extract anthocyanins from B. glabra bracts, so it is not clear whether anthocyanins exist in B. glabra bracts. This study discovered 42 unigenes annotated as F3’5’H, which is crucial for dihydroflavonol biosynthesis in plants50. Dihydroflavonols can form colorless proanthocyanidins (one of the precursors of anthocyanins) under the action of Dihydroflavonol-4-reductase (DFR) and can be catalyzed by Anthocyanidin synthase (ANS) to form blue proanthocyanidins51,52. Through in-depth analysis of the B. glabra bract transcriptome, it was found that the anthocyanin biosynthesis pathway in B. glabra lacks the C4H and DFR genes53, which may result in the synthesis of only anthocyanin precursors in the bracts.
He et al.54 identified 28 co-expression modules in sweet potato using WGCNA and analyzed the modules significantly enriched in flavonoid and anthocyanin metabolism, successfully identifying genes related to anthocyanin synthesis. This study screened a total of 12 positively correlated tissue-specific modules in five different bract traits using WGCNA, and the genes in these modules may participate in and regulate the color formation process of different bracts. Relevant studies have shown that, except for white B. glabra bracts, which do not contain betalains, the pigments in other B. glabra bracts are composed of chlorophylls, carotenoids, flavonols, and betalains20. In this study, 23 unigenes in the yellow trait (F) red module were annotated as members of the light-harvesting complex (LHC) gene family, specifically Chlorophyll a-b binding protein. Chlorophyll a-b binding protein is a component of LHC, where light energy is received by pigment molecules such as chlorophyll and carotenoids located in the LHC55,56. The clustering heatmap indicates that these four genes are highly expressed in trait F, suggesting that chlorophyll may be involved in the color regulation process of trait F. The study found that two abscisic aldehyde oxidase genes (AAO1 and AAO3) and one carotenoid cleavage dioxygenase 3 gene (NCED), involved in carotenoid biosynthesis57,58, are highly expressed in trait A, possibly contributing to the formation of magenta color in the bracts.Additionally, in the white trait (E) brown module, 10 unigenes were annotated as the E1.11.1.7 gene and were highly expressed in trait E. This gene has been shown to be involved in the bleaching process of betacyanin and betaxanthin during betalain biosynthesis59,60,61,62. Therefore, this study suggests that this gene plays an indispensable role in regulating the formation of the white color in trait E. It has been reported that high expression of the SGR, NOL, and ABA2 genes may lead to reduced chlorophyll and increased carotenoid accumulation, resulting in red and purple B. glabra bracts63. This study only annotated one ABA2 gene, which is highly expressed in yellow bract F, possibly due to the lack of co-expression with SGR and NOL genes.
Multiple studies have shown that CYP76AD5/6/1 are key genes for the initial hydroxylation of tyrosine to L-DOPA and the conversion of L-DOPA to cDOPA in the betalain biosynthesis pathway64,65,66,67,68. This study annotated four genes encoding cytochrome P450 (CYP75B1, CYP73A, CYP75A, and CYP97A3), which may be involved in the bract color formation process.
Therefore, this study suggests that the pigments contributing to the color of B. glabra bracts are mainly composed of flavonoids and betalains, and may include chlorophylls and carotenoids, although the presence of anthocyanins remains unclear.
Candidate gene qRT-PCR validation
Among the 16 candidate genes for bract color validated through qRT-PCR expression analysis, 9 genes exhibited significant differences in relative expression across the five B. glabra bracts. Among them, PAL2, CHS1, CHS2, ANS, 6GT, CDOPA5GT, and DOPA had fluorescence quantitative analysis relative expression levels similar to, but not entirely consistent with, transcriptome analysis expression trends. Of these genes, CHS1 had the highest relative expression level (0.23 ~ 1.05), followed by PAL2 (0.05 ~ 0.12), and then BZ1 (0.01 ~ 0.04); PAL2, CHS1, ANS, BZ1, 6GT, and CDOPA5GT showed significantly high expression in magenta bracts; the expression of ANR was significantly higher in yellow bracts than in other colors; CHS2 was mainly expressed in cherry blossom and yellow bracts; and DOPA was significantly highly expressed in magenta bracts. The study also found that CDOPA5GT and 6GT genes were consistently expressed in the five B. glabra bracts, with the expression order: magenta > red > cherry blossom > white > yellow, which may be related to their involvement in the downstream betalain biosynthesis pathway. In general, Relative to genes associated with betalain biosynthesis, those linked to anthocyanin biosynthesis demonstrated higher expression levels; all 9 genes had relatively low expression levels, This might be because the expression levels of the candidate genes were lower than those of the reference genes, positioning them as potential regulators of bract color formation.
Conclusions
Transcriptome sequencing and analysis of color-related genes in B. glabra bracts indicate that genes annotated to cellular processes and metabolic pathways with pigment deposition functions can serve as candidate genes for regulating color formation in B. glabra bracts. KEGG annotations suggest that genetic information processing and metabolic processes are likely involved in bract pigment synthesis. Deletion of C4H and DFR genes in the anthocyanin biosynthesis pathway may result in the inability of delphinium bracts to synthesize anthocyanins; The specific modules significantly positively correlated with different bract traits identified through WGCNA analysis provide new clues and a theoretical basis for further exploration of the genes regulating B. glabra bract color. The pigments contributing to B. glabra bract coloration are mainly composed of flavonoids and betalains, and may include chlorophylls and carotenoids, but the presence of anthocyanins remains unclear. Quantitative reverse transcription PCR (qRT-PCR) validated the expression of 16 genes, with PAL2, CHS1, ANS, BZ1, 6GT, CDOPA5GT, ANR, CHS2, and DOPA showing differential expression in magenta, yellow, cherry blossom-colored, and yellow bracts. These nine genes are identified as potential candidates for influencing color formation in B. glabra bracts.
Data availability
All data are available in the main text or the supplementary materials.The transcriptome sequence data have been uploaded to the NCBI database, BioProject ID: PRJNA1143692; access link: http://www.ncbi.nlm.nih.gov/bioproject/1143692.
References
Chen, C. Z. Study on germplasm resources investigation and variety classification of Bougainvillea. (Master’s Thesis, South China Agricultural University, Guangzhou, (2017).
Lin, G. S. Progress in the study of Taiwan kudzu. Taiwan. Agric. Res. 1, 24–25. https://doi.org/10.16006/j.cnki.twnt.1991.01.010 (1991).
Kumar, P. P. et al. Molecular characterization and cultivar identification in Bougainvillea spp. using SSR markers. Indian J. Agric. Sci. 84, 1124–1130. https://doi.org/10.56093/ijas.v84i9.43474 (2014).
Walter, L. H. The discovery, naming and typification of Bougainvillea spectabilis (Nyctaginaceae). Willdenowia. 42, 117–126. https://doi.org/10.3372/wi.42.42114 (2012).
Ghogar, A., Jiraungkoorskul, K. & Jiraungkoorskul, W. Paper Flower, Bougainvillea spectabilis: Update properties of traditional medicinal plant. J. Nat. Remedies. 16, 82. doi10.18311/jnr/2016/5703 (2016).
Ibtisam, H. Genetic variation among Bougainvillea glabra cultivars (Nyctaginaceae) detected by rapd markers and isozymes patterns. RJABS. 5, 63–71 (2009).
Celli, G. B. & Brooks, M. S. Impact of extraction and processing conditions on betalains and comparison of properties with anthocyanins—A current review. Food Res. Int. 100, 501–509. https://doi.org/10.1016/j.foodres.2016.08.034 (2017).
Sangthong, S., Suksabye, P. & Thiravetyan, R. Air-borne xylene degradation by Bougainvillea Buttiana and the role of epiphytic bacteria in the degradation. Ecotoxicol. Environ. Safe. 126, 273–280. https://doi.org/10.1016/j.ecoenv.2015.12.017 (2016).
Kang, Y. Q. et al. Identification of color phenotype and pigment species of bracts of Bougainvillea ‘Pictorial’. North. Hortic. 21, 66–73. https://doi.org/10.11937/bfyy.20221209 (2022).
Kang, Y. Q. et al. Analysis of bract color phenotype and pigment composition of different Bougainvillea species. Plant Sci. J. 40, 714–723. https://doi.org/10.11913/PSJ/2095-837.50714 (2022).
Zhou, H., Zhang, X., Liu, T. Y. & Yu, F. X. Data analysis and gene discovery by high-throughput transcriptome sequencing. Jiangxi Sci. 30, 607–611. https://doi.org/10.3969/j.issn.1001-3679.2012.05.015 (2012).
Zhang, F. Z. Transcriptome analysis of flower color in Brassica pekinensis. (Master’s Thesis, Shenyang Agricultural University, Liaoning, 2020). https://doi.org/10.27327/d.cnki.gshnu.2020.000765.
Sun, L. N. et al. Study on the gene expression patterns related to bract color of Bougainvillea spectabilis ‘Senjakala’ based on transcriptome sequencing. Guangxi For. Sci. 52, 1–13. https://doi.org/10.19692/j.issn.1006-1126.20230101 (2023).
Sun, L. N. Molecular identification and bract transcriptome analysis of Bougainvillea spectabilis. (Ph.D. Thesis, China Academy of Forestry Sciences, Beijing, 2019).
Deng, Y. Q., He, X. J. & Yu, Y. Comparative transcriptomics of petaloid bracts and leaves of Leafy flowers. Acta Bot. Boreali-Occidentalia Sinica. 38, 1039–1047. https://doi.org/10.7606/j.issn.1000-4025.2018.06.1039 (2018).
Jiang, J. H., Huang, C., Lin, Z. M., He, B. S. & Li, X. Y. Analysis of differentially expressed genes in the calyx of Dunnia sinensis and screening of color-regulated transcription factors. J. Trop. Subtrop. Bot. 1–11 (2024). http://kns.cnki.net/kcms/detail/44.1374.Q.20231213.1538.002.html
Zhou, S. J. et al. Mining and expression analysis of Clematis flower color-related genes based on transcriptome analysis. Mol. Plant Breed. 1–13. (2024). http://kns.cnki.net/kcms/detail/46.1068.s.20240326.1427.010.html
Zhang, T. Metabolomics combined with transcriptomics analyzes the mechanism of flower color formation of Camellia sinensis pink tea tree flowers. (Master’s Thesis, Guizhou University, Gui Yang, (2023).
Zhong, X. Y., Liu, S., Sun, R. & Zhang, Y. Analysis of color parameters and pigment content in bracts of Bougainvillea spectabilis and screening of blue transgenic receptors. Acta Bot. Boreali-Occidentalia Sinica. 42, 646–655 (2022).
Zhang, H. H. et al. Analysis of pigment components in bracts of different Bougainvillea cultivars. Mol. Plant. Breed. 21, 929–940. https://doi.org/10.13271/j.mpb.021.000929 (2023).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. https://doi.org/10.1038/nbt.1883 (2011).
Karlin, S. & Altschul, S.F. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. 87 2264–2268 https://doi.org/10.1073/pnas.87.6.2264 (1990).
Minoru, K. H. & Susumu, G. K. E. G. G. Kyoto Encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51, 587–592. https://doi.org/10.1093/nar/gkac963 (2023).
J Duan, M. Transcriptome and target metabolome analysis of carotenoid regulatory factors in Capsicum annuumr fruit. J. Hortic. 1-19, 2024–2006. https://doi.org/10.16420/j.issn.0513-353x.2023-0374 (2024).
Yang, J. Analysis of differential genes between green leaf and white leaf Brassica napus based on transcriptome sequencing. (Master’s Thesis, Huazhong Agricultural University, Wuhan, 2021).
Jeon, J. E. et al. A Pathogen-Responsive Gene Cluster for highly modified fatty acids in Tomato. Cell. 180, 176–187.e19. https://doi.org/10.1016/j.cell.2019.11.037 (2020).
Zhang, X. Y., Zhang, S. L., Ye, L. W., Luo, J. & Li, S. C. Analysis of the influencing factors of different color fruit bags on plum fruit coloring and anthocyanin synthesis. J. Fruit Sci. 24, 39–44. https://doi.org/10.13925/j.cnki.gsxb.2007.05.031 (2007).
Zhu, Y., Peng, Q. Z., Li, K. G. & Xie, D. Y. Molecular cloning and functional characterization of the anthocyanidin reductase gene from Vitis bellula. Planta. 240, 381–398. https://doi.org/10.1007/s00425-014-2094-2 (2014).
Shao, L. & Xiao, S. H. Effect of chalcone synthase gene on flower color and fertility of transgenic plants. Acta Bot. Sinica. 38, 517–524. https://doi.org/10.1007/BF02951625 (1996).
Ford, C. M., Boss, P. K. & Hoj, P. B. Cloning and characterization of Vitis vinifera UDP-glucose: Flavonoid 3-O-glucosyltransferase, a homologue of the enzyme encoded by the maize Bronze-1 locus that may primarily serve to glucosylate anthocyanidins in vivo. J. Biol. Chem. 273, 9224–9233. https://doi.org/10.1074/jbc.273.15.9224 (1998).
Vogt, T. Substrate specificity and sequence analysis define a polyphyletic origin of betanidin 5- and 6-O-glucosyltransferase from Dorotheanthus Bellidiformis. Planta. 214, 492–495. https://doi.org/10.1007/s00425-001-0685-1 (2002).
Sasaki, N. et al. Isolation and characterization of cDNAs encoding an enzyme with glucosyltransferase activity for cyclo-DOPA from four o’clocks and feather cockscombs. Plant. Cell. Physiol. 46, 666–670. https://doi.org/10.1093/pcp/pci064 (2005).
Alsamadany, H. De novo leaf transcriptome assembly of Bougainvillea spectabilis for the identification of genes involves in the secondary metabolite pathways. Gene. 746, 144660. https://doi.org/10.1016/j.gene.2020.144660 (2020).
Zheng, X. T., Zhang, B., Zhang, Y. W., Zhong, H. A. & Wu C.X. Transcriptome analysis of feather follicles reveals candidate genes and pathways associated with pheomelanin pigmentation in chickens. Sci. Rep. 10, 12088. https://doi.org/10.1038/s41598-020-68931-1 (2020).
Zhao, T. Development of SSR molecular markers and construction of in vitro regeneration system of Bougainvillea spectabilis transcriptome. (Master’s Thesis, Hainan University, Haikou, (2019).
Zhao, F. Y. Study on genes related to numb-taste substances in Zanthoxylum armatum based on transcriptome sequencing. (Master’s Thesis, Sichuan Agricultural University, Chengdu, 2022).
Li, J. Correlation between transcriptome gene mining and saponin accumulation in response to stress in Panax japonicus. (Master’s Thesis, Guizhou Normal University, Gui Yang, 2023).
Kumar, S. N. A., Ritesh, S. K., Sharmila, G. & Muthukumaran, C. Extraction optimization and characterization of water soluble red purple pigment from floral bracts of Bougainvillea glabra. Arab. J. Chem. 10, S2145–S2150. https://doi.org/10.1016/j.arabjc.2013.07.047 (2017).
Mahendran, G. B. et al. Green preparation of reduced graphene oxide by Bougainvillea glabra flower extract and sensing application. Mater. Sci. Mater. Electron. 31, 14345–14356. https://doi.org/10.1007/s10854-020-03994-4 (2020).
Brockington, S. F., Walker, R. H., Glover, B. J., Soltis, P. S. & Soltis, D. E. Complex pigment evolution in the caryophyllales. New. Phytol. 190, 854–864. https://doi.org/10.1111/j.1469-8137.2011.03687.x (2011).
Huang, H., Hu, H. X., Jiang, L. H., Wang, L. S. & Ren, H. X. Research progress of plant beet pigment. Bull. Biol.. 49, 1–4 (2014).
Li, J., Li, T. G., Wang, T. X. & Wang, J. Research progress on the mutual exclusion mechanism of plant anthocyanins and beet pigments. J. Plant. Geneti. Resour. 24, 1515–1526. https://doi.org/10.13430/j.cnki.jpgr.20230505002 (2023).
Xu, S. X., Lin, C. S., Huang, Q. Y., Zeng, B. Y. & Lin, F. C. Isolation and expression of beet pigment metabolizing enzyme 4,5-DOPA-dioxygenase gene from Bougainvillea spectabilis. Chin. J. Trop. Crops. 34, 2214–2221. https://doi.org/10.3969/j.issn.1000-2561.2013.11.024 (2013).
Harris, N. N. et al. Betalain production is possible in anthocyanin-producing plant species given the presence of DOPA-dioxygenase and L-DOPA. BMC Plant. Biol. 12, 34. https://doi.org/10.1186/1471-2229-12-34 (2012).
Zhang, W. P. et al. Transcriptome analyses shed light on floral organ morphogenesis and bract color formation in Bougainvillea. BMC Plant Biol. 22, 1–13. https://doi.org/10.1186/s12870-022-03478-z (2022).
Wybraniec, S., Jerz, G., Gebers, N. & Winterhalter, P. Ion-pair high-speed countercurrent chromatography in fractionation of a high-molecular weight variation of acyl-oligosaccharide linked betacyanins from purple bracts of Bougainvillea glabra. CHROMB. 878, 538–550. https://doi.org/10.1016/j.jchromb.2009.12.029 (2010).
Liu, J. X., Hu, H. B. & Zhao, G. L. Study on extraction and stability of red pigment from Bougainvillea spectabilis. Food Sci.27, 98–102. https://doi.org/10.3321/j.issn:1002-6630.2006.08.019 (2006).
Fan, K. et al. Nitrogen regulation in flavonoid biosynthesis in plants. Plant. Physiol. J. 52, 843–850. https://doi.org/10.13592/j.cnki.ppj.2015.0534 (2016).
Hou, Z. H., Wang, S. P., Wei, S. D., Liu, Z. X. & Fang, Z. W. Research progress on biosynthesis and regulation of plant anthocyanins. Guihaia. 37, 1603–1613. https://doi.org/10.11931/guihaia.gxzw201705002 (2017).
Gao, J. F., Ke, L. P. & Sun, Y. Q. Research progress in the cultivation of blue flowers based on anthocyanin metabolism. Chin. J. Biotechnol. 36, 678–692. https://doi.org/10.13345/j.cjb.190335 (2020).
Luo, J. J., Li, H., Bai, B. B., Yu, H. Q. & You, J. Effect of light on anthocyanin biosythesis and expression of CHS and DFR genes in Rosa chinensis‘Spectra’. Mol. Plant. Breed. 11, 126–131. https://doi.org/10.3969/mpb.011.000126 (2013).
He, L. H. et al. Construction of co-expression network of Dioscorea esculenta anthocyanin synthesis-related genes and mining of core genes based on caseomes and WGCNAs. Chin. J. Bioeng. 41, 27–36. https://doi.org/10.13523/j.cb.2106048 (2021).
Yang, J. H., Zhang, X. C. & Z, Z. Molecular cloning and analysis of HbCAB2, a chlorophyll a/b-binding protein-encoding gene from Hevea brasiliensis. J. Southwest. For. Univ. (Nat. Sci.). 39, 88–94. CNKI:SUN:YNLX.0.2019-01-011 (2019).
Zhong, S. T. et al. Advances of light-harvesting chlorophyll a/b binding protein(CAB) in higher plants. J. Agric. Biotechnol. 32, 444–457. https://doi.org/10.3969/j.issn.1674-7968.2024.02.016 (2024).
Colasuonno, P., Marcotuli, I., Lozito, M. L., Simeone, R. & Blanco, A. Characterization of aldehyde oxidase (AO) genes involved in the accumulation of carotenoid pigments in wheat grain. Front. Plant Sci. 8, 863. https://doi.org/10.3389/fpls.2017.00863 (2017).
Xia, Y., Chen, W., Xiang, W., Wang, D. & Liang, G. Petal Color-Transition in lonicera japonica is mainly associated with increase of the carotenoid content and carotenoid biosynthetic gene expression. Res. Square Platf. LLC. https://doi.org/10.21203/rs.3.rs-62167/v1 (2020).
Kugler, F., Stintzing, F. C. & Carle, R. Characterisation of betalain patterns of differently coloured inflorescences from Gomphrena globosa L. and Bougainvillea sp. by HPLC-DAD-ESI-MSn. Anal. Bioanal. Chem. 387, 637–648. https://doi.org/10.1007/s00216-006-0897-0 (2007).
Wasserman, B. P., Eiberger, L. L. & Guilfoy, M. P. Effect of hydrogen peroxide and phenolic compounds on horseradish peroxidase-catalyzed decolorization of betalain pigments. Food Sci. 49, 536–538. https://doi.org/10.1111/j.1365-2621.1984.tb12461.x (2010).
Manchali, S., Murthy, K. N. C., Nagaraju, S. & Neelwarne, B. Stability of Betalain Pigments of Red Beet. Springer US. 55–74. https://doi.org/10.1007/978-1-4614-3458-0_3 (2013).
Pedre, O., Escribano, J. & M.A. & Studying the oxidation and the antiradical activity of betalain from beetroot. J. Biol. Educ. 35, 49–51. https://doi.org/10.1080/00219266.2000.9655736 (2000).
Sato, T., Shimoda, Y., Matsuda, K., Tanaka, A. & Ito, H. Mg dechelation of chlorophyll a by stay-green activates chlorophyll b degradation through expressing non-yellow coloring 1 in Arabidopsis thaliana. Plant. Physiol. 222, 94–102. https://doi.org/10.1016/j.jplph.2018.01.010 (2018).
Tanaka, Y., Sasaki, N. & Ohmiya, A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 54, 733–749. https://doi.org/10.1111/j.1365-313X.2008.03447.x (2008).
Gandıa-Herrero, F. & Garcıa-Carmona, F. Biosynthesis of betalains: yellow and violet plant pigments. Trends Plant. Sci. 18, 334–343. https://doi.org/10.1016/j.tplants.2013.01.003 (2013).
Ohno, S., Makishima, R., Doi, M. & Sunkar, R. Post-transcriptional gene silencing of CYP76AD controls betalain biosynthesis in bracts of bougainvillea. J. Exp. Bot. 72, 6949–6962. https://doi.org/10.1093/jxb/erab340 (2021).
Tossi, V. E., Martınez Tosar, L., Pitta-Alvarez, S. I. & Causin, H. F. Casting light on the pathway to betalain biosynthesis: A review. Environ. Exp. Bot. 186, 104464. https://doi.org/10.1016/j.envexpbot.2021.104464 (2021).
Huang, H. et al. Pantranscriptome combined with phenotypic quantification reveals germplasm kinship and regulation network of bract color variation in Bougainvillea. Front. Plant Sci. 13, 1018846 (2022).
Acknowledgements
We thank Zhenghai Sun and Peiyao Xin professor for editing this document, and Mr. Jianyun Li., Mr.Zihan Li, Mr. Wen Zhu for technical assistance.
Funding
This work was funded by Key Technologies Research for the Germplasm of Important Woody Flowers in Yunnan Province (No. 202302AE090018).
Author information
Authors and Affiliations
Contributions
Conceptualization, P.X. and Z.S.; validation, F.W. and G.Y.; formal analysis, F.W.; G.Y.; J.L.; Z.L. and W.Z.; investigation, F.W.; G.Y and P.X.; resources, Z.S. and P.X.; writing—original draft preparation, F.W. and G.Y.; writing—review and editing, P.X. and Z.S.; supervision, P.X. and Z.S.; project administration, P.X.; funding acquisition, P.X. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Collection permission statement for B. glabra
All test materials used in this study were collected with permission from the Bougainvillea Base of Jicheng Garden Technology Co., Ltd., located in Mile City, Yunnan Province, China. All collected samples are stored in the Herbarium of Southwest Forestry University, Yunnan Province, China.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, F., Yao, G., Li, J. et al. Mining and expression analysis of color related genes in Bougainvillea glabra bracts based on transcriptome sequencing. Sci Rep 14, 24491 (2024). https://doi.org/10.1038/s41598-024-73964-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-73964-x
