
Abstract
Percutaneous coronary intervention (PCI) combined with stent implantation is currently one of the most effective treatments for coronary artery disease (CAD). However, in-stent restenosis (ISR) significantly compromises its long-term efficacy. Mitophagy plays a crucial role in vascular homeostasis, yet its role in ISR remains unclear. This study aims to identify mitophagy-related biomarkers for ISR and explore their underlying molecular mechanisms. Through differential gene expression analysis between ISR and Control samples in the combined dataset, 169 differentially expressed genes (DEGs) were identified. Twenty-three differentially expressed mitophagy-related genes (DEMRGs) were identified by intersecting with mitophagy-related genes (MRGs) from the GeneCards, and functional enrichment analysis indicated their significant involvement in mitophagy-related biological processes. Using Weighted Gene Co-expression Network Analysis (WGCNA) and three machine learning algorithms (Logistic-LASSO, RF, and SVM-RFE), LRRK2, and ANKRD13A were identified as mitophagy-related biomarkers for ISR. The nomogram based on these two genes also exhibited promising diagnostic performance for ISR. Gene Set Enrichment Analysis (GSEA) as well as immune infiltration analyses showed that these two genes were closely associated with immune and inflammatory responses in ISR. Furthermore, potential small molecule compounds with therapeutic implications for ISR were predicted using the connectivity Map (cMAP) database. This study systematically investigated mitophagy-related biomarkers for ISR and their potential biological functions, providing new insights into early diagnosis and precision treatment strategies for ISR.
Similar content being viewed by others


Introduction
Coronary artery disease (CAD) is a leading cause of death and disability worldwide1. Percutaneous coronary intervention (PCI) with stent implantation is currently one of the most effective treatments for CAD, significantly improving patient prognosis. However, In-stent restenosis (ISR) remains a common and challenging complication post-PCI, severely impacting the long-term efficacy of the procedure2,3. Despite drug-eluting stents (DES) which combine a metal stent platform with the release of anti-proliferative drugs, significantly reducing ISR rates, the incidence of ISR in one-year post-PCI still ranges between 2–10%2,3. Given the millions of stents implanted globally each year, ISR poses a serious public health and economic burden.
Currently, coronary angiography remains the gold standard for diagnosing ISR. However, this method is invasive, costly, and cannot detect ISR in its early stages4. Treatment options for ISR include repeat PCI, often with drug-coated balloons (DCB) or newer-generation DES, and in severe cases, coronary artery bypass grafting (CABG). However, these treatments are associated with risks of recurrence and complications4,5. Therefore, there is an urgent need to develop new diagnostic biomarkers and therapeutic targets to enable accurate early-stage diagnosis of ISR and provide new strategies for its prevention and treatment.
The development of ISR is a complex process involving vascular injury, inflammatory responses, and subsequent neointimal hyperplasia (NIH)6. During this progression, the importance of mitochondria in maintaining vascular homeostasis and disease is increasingly recognized7. Mitochondria not only play a critical role in energy metabolism but also regulate cell apoptosis, oxidative stress, and cellular metabolism7. Mitophagy, a form of selective autophagy, specifically identifies and degrades damaged or dysfunctional mitochondria, crucial for maintaining quality control and quantity balance within cells, emerging as a novel and potential therapeutic target8. Mitophagy is involved in regulating various pathophysiological processes that contribute to the progression of ISR. PINK1/Parkin-mediated mitophagy has been shown to mitigate endothelial cell damage induced by a variety of stimuli9,10,11. Research indicates that mitophagy plays a pivotal role in regulating the proliferation and migration of vascular smooth muscle cells (VSMCs)12,13,14, critical factors in the onset of ISR. Furthermore, mitophagy is closely linked to ISR-related inflammatory responses and cell apoptosis15,16. Mitophagy influences these processes by modulating inflammasome activation and cytochrome c release, among other pathways17. However, systematic studies on mitophagy in ISR are currently lacking. Identifying potential biomarkers related to mitophagy in ISR could enhance understanding and exploration of their roles in ISR pathogenesis, providing new insights into early diagnosis and treatment for ISR patients.
This study utilized two ISR-related datasets from the Gene Expression Omnibus (GEO) database, combined with mitophagy related genes (MRGs) obtained from the Genecard database, to identify mitophagy related biomarkers in ISR through differential gene expression analysis, Weighted Gene Co-expression Network Analysis (WGCNA), and machine learning methods (Logistic Regression with Least Absolute Shrinkage and Selection Operator (LASSO), Random Forest (RF), and support vector machine-recursive feature elimination (SVM-RFE)). Subsequently, a nomogram was established and evaluated using receiver operating characteristic (ROC) curves, calibration curves, and decision curve analysis (DCA). Furthermore, using single sample Gene Set Enrichment Analysis (ssGSEA) algorithm, we compared differences in immune infiltration between ISR patients and controls, and explored the correlation with mitophagy-related biomarkers. Additionally, using the connectivity MAP (cMAP) database, we identified potential small molecule compounds that may impact ISR based on differentially expressed genes (DEGs). Overall, the findings of this study may provide significant insights for the diagnosis, mechanistic understanding, and treatment of ISR.
Materials and methods
Data pretreatment
Figure 1 illustrates the workflow of this study. Two ISR-related datasets were downloaded from the GEO database: GSE46560, and GSE48060. All the two datasets were derived from Homo sapiens and sourced from whole blood samples. The platform for dataset GSE46560 was GPL15207 Affymetrix Human Gene Expression Array, containing 5 ISR samples and 6 controls. The platform for dataset GSE48060 was GPL570 Affymetrix Human Genome U133 Plus 2.0 Array, containing 5 ISR samples and 26 controls. The GSE46560, and GSE48060 datasets were merged and batch-normalized using the R packages “sva”18 and “limma”19, resulting in a combined dataset comprising 10 ISR samples and 32 controls. Additionally, based on the handling of outlier samples in the WGCNA analysis described below, one outlier sample (GSM1167076) was removed from the combined dataset, resulting in a final combined dataset consisting of 10 ISR samples and 31 control samples. Detailed information about the datasets is provided in Supplementary Table S1.

The flowchart of the study. DEGs, differentially expressed genes; MRGs, mitophagy-related genes; cMAP, connectivity Map; DEMRGs, differentially expressed mitophagy-related genes; GO, Gene Ontology; WGCNA, weighted correlation network analysis; LASSO, least absolute shrinkage and selection operator; SVM, Support Vector Machine; GSEA, Gene Set Enrichment Analysis; ssGSEA, single sample Gene Set Enrichment Analysis.
We screened MRGs in the GeneCards database20 using the keyword “mitophagy” and retained only the protein-coding genes with a relevance score of ≥ 2. A total of 1,639 MRGs were identified.
Differential gene expression analysis
Subsequently, differential gene expression analysis between the ISR group and the control group in the combined dataset was conducted using the R package limma. The criteria for selecting DEGs were |log2 fold change (logFC)| > 0.5 and p-value < 0.05. The results of the DEGs were visualized using a volcano plot generated by the R package ggplot2. Differentially expressed mitophagy-related genes (DEMRGs) were obtained by intersecting DEGs with MRGs, and the results were displayed using Venn diagrams and heatmaps.
Gene ontology (GO) enrichment analysis
To explore the biological functions of DEMRGs, GO enrichment analysis was performed using the R package clusterProfiler21. This analysis aimed to reveal the significant enrichment of these genes in biological processes (BP), cellular components (CC), and molecular functions (MF). The selection criteria for terms were p-value < 0.05, with p-value correction performed using the Benjamini-Hochberg (BH) method.
WGCNA
To identify important modules genes in ISR, WGCNA was conducted using the R package WGCNA22. Initially, samples were clustered to remove outliers, and one outlier sample (GSM1167076) was removed. The correlation coefficient between any two genes was calculated. Then a scale-free co-expression network was built. A hierarchical clustering tree was constructed based on the correlation coefficients between genes. Different branches of the clustering tree represented different gene modules, with different colors denoting distinct modules. Module significance was subsequently calculated. The top 8000 genes with the highest median absolute deviation (MAD) of all genes in the combined dataset were included in the WGCNA. The minimum module gene number was set to 40, the optimal soft power was set to 9, and the module merging cut height was set to 0.25. Modules with p-value < 0.05 were selected, and all genes within these modules were identified as highly related to ISR. Common DEMRGs were obtained by intersecting these genes with DEMRGs.
Hub gene identification
The Common DEMRGs were selected as candidate genes, and hub genes were identified using three machine learning algorithms: Logistic Regression with LASSO (Logistic-LASSO)23, SVM-RFE24, and RF25, based on the normalized expression levels of these genes. The objective of the machine learning feature selection was to identify key genes that distinguish between ISR and control groups. A portion of the combined dataset was selected as the training set, which included the expression levels of DEMRGs and the corresponding ISR/control group information. The remaining data were used as the test set to validate and evaluate the performance of the diagnostic prediction model we developed. The division of the training and test sets was performed randomly, ensuring that both sets were representative and balanced, in order to maintain the accuracy and robustness of the model. Logistic-LASSO was performed to reduce model overfitting by adding a penalty term. This was implemented using the glmnet26 package with parameters set as set.seed (1) and family = “binomial”. The L1 penalty for LASSO was selected based on the cross-validation results that minimized the classification error. SVM-RFE is an efficient feature selection technique. The e1071 package27 was used to determine the number of genes with the highest accuracy and lowest error rate. RF is an algorithm that integrates multiple decision trees through ensemble learning, commonly used for classification problems. It was implemented using the randomForest package. The criterion for feature importance was IncNodePurity > 0.5, and this value was selected to retain features that contributed the most to classification accuracy. Cross-validation (10-fold or 5-fold) was performed to optimize the hyperparameters of each machine learning model. The genes identified by these three algorithms were overlapped and visualized using Venn diagrams to select the hub genes. The chromosomal locations of the hub genes were mapped using the RCircos package28.
Construction and evaluation of nomogram
First, the diagnostic efficiency of the hub genes was assessed by plotting ROC curves and calculating the area under the curve (AUC) using the pROC package. An AUC > 0.7 was considered indicative of high diagnostic performance. Based on the expression levels of the hub genes in the combined dataset, a nomogram was constructed using the rms package in R. The nomogram model evaluates the relationships between variables in the predictive model. Subsequently, the diagnostic efficacy, predictive ability, and clinical net benefit at different risk thresholds of the nomogram were assessed using ROC curves, calibration curves, and DCA plot, respectively, created with the pROC, rms, and ggDCA packages.
Gene set enrichment analysis (GSEA)
To further investigate the potential roles of the hub genes, GSEA was performed using the clusterProfiler package. Samples were divided into high-expression and low-expression groups based on the median expression levels of the hub genes, and GSEA was conducted on the differentially expressed genes. The reference gene set used was " h.all.v2023.2.Hs.symbols.gmt”. The parameters were set to a minimum of 10 and a maximum of 500 genes, with p-value correction performed using the BH method. Pathways were considered significantly enriched if the adjusted p-value (P.adj) < 0.05 and the false discovery rate (FDR, q.value) < 0.05.
Immune infiltration analysis
ssGSEA was used to analyze differences in immune cell infiltration between the ISR and control groups. The results were visualized using boxplots created with the ggplot2 package. Subsequently, the correlations between different immune cells and the hub genes were calculated using the Spearman algorithm, and these relationships were visualized using correlation heatmaps and lollipop charts.
Identification of small molecular therapeutic compounds
The connectivity MAP (cMap, https://clue.io/) database29 was utilized to screen and identify small molecules associated with ISR. To identify candidate small molecule compounds, the DEGs were introduced into the cMAP database for gene set enrichment analysis. The top six compounds with highest negative scores were selected as potential therapeutic agents for ISR.
Statistical analysis
All statistical analyses were conducted using R software (version 4.3.3). Comparisons between two groups of continuous variables following a normal distribution were performed using the student t-test, while comparisons of non-normally distributed variables were conducted using the Wilcoxon rank-sum test. Unless otherwise specified, Spearman correlation analysis was used to calculate correlation coefficients between different variables. All statistical p-values were two-sided, with p-value < 0.05 considered statistically significant.
Results
Data pretreatment
We downloaded two datasets (GSE46560, and GSE48060) from the GEO database and merged them. Batch effects were removed using the combat function from the sva package, followed by normalization with the normalizeBetweenArrays function from the limma package. The results showed significant batch effects between different datasets in the combined dataset before normalization, leading to dispersed distribution in principal component analysis (PCA) diagram (Fig. 2A-B). After normalization, batch effects between different datasets were significantly reduced, and the expression distribution of samples became more consistent (Fig. 2C-D). This normalization enabled the combined dataset to be used for subsequent integrated analyses.

Data pretreatment. (A) Boxplot illustrating the distribution of gene expression levels in combined dataset before batch effect correction and normalization. (B) PCA plot before batch effect correction and normalization. (C) Boxplot showing the normalized gene expression levels after batch effect correction of combined dataset. (D) PCA plot after batch effect correction and normalization. PCA, Principal component analysis.
Identification of DEMRGs
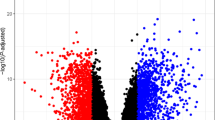
Through differential gene expression analysis between ISR and Control samples in the combined dataset, 169 DEGs were identified, with 31 upregulated and 138 downregulated in ISR (Fig. 3A). By intersecting these 169 DEGs with the 1639 MRGs, 23 DEMRGs (with 9 genes upregulated and 14 genes downregulated) were obtained (Fig. 3B): GPS1, PPM1G, LRRK2, XIAP, ANKRD13A, SND1, CKB, HSPA6, HSDL2, PPP3CA, LAMP2, POLD1, PRKAR1A, KIAA0232, OSBPL11, AMFR, DNAJC3, PKN2, PARK7, TOMM40, SLC12A6, STAM2, and RPL36. The detailed parameters of these genes were presented in Supplementary Table S2. These genes may play significant roles in the development and progression of ISR. The expression profiles of these genes across different samples are shown in the heatmap (Fig. 3C).

Identification of DEMRGs. (A) Volcano plot of DEGs. Red and blue dots represent significantly upregulated and downregulated genes, respectively. (B) Venn diagram showing the overlap between DEGs and MRGs. (C) Heatmap of DEMRGs with hierarchical clustering. DEGs, differentially expressed genes; MRGs, mitophagy-related genes; DEMRGs, differentially expressed mitophagy-related genes.
GO enrichment analysis
To analyze the biological functions of these 23 DEMRGs, GO enrichment analysis was performed. The results showed significant enrichment in several categories. In BP, the DEMRGs were notably enriched in phosphorylation, negative regulation of gene expression and negative regulation of endoplasmic reticulum stress-induced intrinsic apoptotic signaling pathway. In CC, significant enrichments were found in membrane, cytosol, cytoplasm, autolysosome and perinuclear region of cytoplasm. In MF, the DEMRGs were significantly enriched in ubiquitin protein ligase binding, kinase activity, ubiquitin-specific protease binding and enzyme binding. Detailed pathway information is provided in Supplementary Table S3. The results were presented in a bar plot (Fig. 4A). As expected, many of these terms are directly or indirectly involved in the regulation of mitophagy. Additionally, the relationships between the genes and their significantly enriched BP and MF terms were illustrated using chord diagrams (Fig. 4B-C).

GO Enrichment Analysis. (A) The bar chart shows the significant enrichment of DEMRGs in GO terms. (B) The chord diagram illustrates the relationships between DEMRGs and significantly enriched BP terms. (C) The chord diagram illustrates the relationships between DEMRGs and significantly enriched MF terms. GO, Gene Ontology; DEMRGs, differentially expressed mitophagy-related genes; MF, Molecular Function; BP, Biological Process.
WGCNA
To identify gene modules highly associated with ISR, WGCNA was performed on the combined dataset. By setting the scale-free R^2 to 0.85, the optimal soft power was determined to be 9, ensuring the network’s scale-free topology and high gene connectivity (Fig. 5A). A total of 11 modules were identified (Fig. 5B). Module-trait correlation analysis revealed that the brown module was significantly positively correlated with ISR (Fig. 5C), suggesting that genes within this module may play critical roles in ISR pathogenesis. By intersecting the genes within the brown module with the DEMRGs, 13 Common DEMRGs were identified (Fig. 5D). These genes—ANKRD13A, DNAJC3, HSDL2, HSPA6, KIAA0232, LAMP2, LRRK2, OSBPL11, PKN2, PPP3CA, PRKAR1A, SLC12A6 and STAM2 —were all significantly downregulated in the ISR group compared to the control group (all p-value < 0.05, Fig. 5E).

WGCNA. (A) The curves for the scale-free topology model fit index (left) and mean connectivity (right) change with different soft threshold values. (B) The gene clustering dendrogram shows different gene modules in distinct colors. (C) The heatmap shows the correlation and significance levels between gene modules and the ISR phenotype. (D) Venn diagram of DEGs and brown module genes. (E) Boxplot of expression levels for Common DEMRGs genes in the ISR and control groups. WGCNA, weighted correlation network analysis; ISR, in-stent restenosis. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
Hub genes identification
To further identify the feature genes in ISR, three machine learning algorithms were employed. Based on the normalized gene expression levels of these 13 common DEMRGs in the combined dataset, a Logistic-LASSO model was constructed (Fig. 6A-B), which identified 4 non-zero coefficient genes: LRRK2, ANKRD13A, HSPA6 and PPP3CA. The Random Forest algorithm was applied next, using IncNodePurity > 0.5 as the selection criterion, and 8 Common DEMRGs were identified as feature genes: ANKRD13A, PRKAR1A, OSBPL11, DNAJC3, LRRK2, LAMP2, SLC12A6 and HSDL2 (Fig. 6C-D). Subsequently, the SVM-RFE algorithm was employed to minimize classification error and maximize accuracy, resulting in the inclusion of 3 common DEMRGs: ANKRD13A, PKN2 and LRRK2 (Fig. 6E-F). Traditional classification metrics for each model such as accuracy, precision, recall, and F1 score were included in the Supplementary Table S4. The intersection of the genes identified by these three algorithms, as illustrated by the Venn diagram, ultimately highlighted LRRK2 and ANKRD13A as the feature hub genes for ISR (Fig. 6G). Additionally, the chromosomal locations of these two hub genes were mapped (Fig. 6H).

Hub Genes Identification. (A) The Logistic-LASSO model shows the change in regression coefficients across different λ values. (B) Cross-validation determines the optimal λ value for the Logistic-LASSO model. (C) The error curve of the random forest model across different numbers of trees. (D) The importance scores of genes in the random forest model (IncNodePurity). (E) The classification error of the SVM-RFE method with different numbers of features. (F) The classification accuracy of the SVM-RFE method with different numbers of features. (G) The Venn diagram shows the overlap of feature genes selected by Logistic-LASSO, Random Forest, and SVM-RFE methods. (H) Chromosomal localization plots for Hub MRGs. Logistic-LASSO: Logistic Regression with Least Absolute Shrinkage and Selection Operator; SVM, Support Vector Machine; MRGs, mitophagy-related genes.
Construction and evaluation of nomogram
First, the biomarker potential of the hub genes LRRK2 and ANKRD13A was assessed by plotting ROC curves. The results showed that LRRK2 had an AUC of 0.787 (Fig. 7A), and ANKRD13 had an AUC of 0.755 (Fig. 7B), indicating good diagnostic performance for these genes in ISR. Subsequently, a nomogram model was constructed based on the expression of LRRK2 and ANKRD13A (Fig. 7C). The model assigns a score to each gene, calculates the total score, and predicts the risk of ISR. The ROC curve of the nomogram model demonstrated an AUC of 0.787, suggesting excellent diagnostic efficiency (Fig. 7D). The calibration curve indicated a high concordance between the predicted probabilities and actual outcomes (Fig. 7E). The DCA plot showed a high clinical net benefit across different risk thresholds (Fig. 7F). These findings suggest that the model has strong predictive power and that the two diagnostic biomarkers play crucial roles in ISR development.

Construction and Evaluation of Nomogram. (A-B) ROC curves for LRRK2, and ANKRD13A. (C) The nomogram model, based on LRRK2, and ANKRD13A genes, predicts the risk score for ISR. (D) ROC curve for the nomogram model. (E) The calibration curve for the nomogram model shows the consistency between predicted probabilities and actual outcomes. (F) The decision curve analysis of the nomogram model evaluates the clinical net benefit at different risk thresholds. ROC, Receiver Operating Characteristic; ISR, in-stent restenosis.
Single-gene GSEA of hub genes
To investigate the potential mechanisms of the two biomarkers in ISR, GSEA was conducted. LRRK2 was predominantly associated with Heme Metabolism, Interferon Alpha Response, Interferon Gamma Response, Myc Targets V1 and TNF-α Signaling Via NF-κB (Fig. 8A). ANKRD13A was primarily linked to Androgen Response, E2F Targets, Heme Metabolism, Myc Targets V1 and Protein Secretion (Fig. 8B). A comprehensive list of pathways related to these genes is provided in Supplementary Table S5. Notably, all of these pathways are directly or indirectly involved in immune and inflammatory processes.

GSEA for hub genes. GSEA analysis for LRRK2 (A) and ANKRD13A (B) in ISR. GSEA, gene set enrichment analysis; ISR, in-stent restenosis.
Analysis of immune cell infiltration and its correlation with hub genes
To further explore the landscape of immune cell infiltration in ISR and its correlation with Hub genes, we conducted a comprehensive investigation using the ssGSEA algorithm to assess the abundance of immune cell types across different groups. The results revealed significant differences in the abundance of CD56dim natural killer cells, Neutrophils and Plasmacytoid dendritic cell between ISR and control groups, suggesting their potential roles in the pathogenesis of ISR (Fig. 9A). A correlation heatmap unveiled complex interactions among immune cell types (Fig. 9B). Strong correlations were observed among specific cell types such as activated CD4 T cell and central memory CD4 T cell. Analysis of the direct correlations between the expression of the two hub genes in ISR samples and various immune cells highlighted intriguing correlations. The lollipop charts prominently displayed the relationships between immune cell types and Hub genes (Fig. 9C-D).

Immune Cell Infiltration Analysis. (A) Boxplot of relative abundance of immune cell types in ISR and control groups. ns, p-value ≥ 0.05; * p-value < 0.05; ** p-value < 0.01. (B) Heatmap of correlations between immune cell types. (C) Lollipop charts for the correlation between various immune cell types and LRRK2. (D) Lollipop charts for the correlation between various immune cell types and ANKRD13A. ISR, in-stent restenosis.
Identification of small molecular therapeutic compounds
To predict small molecule compounds capable of reversing gene expression changes in ISR, DEGs from ISR and control groups were input into the cMAP database. Varenicline, calyculin, cobalt(II)-chloride, genipin, bimatoprost and phorbol-12-myristate-13-acetate (PMA) emerged as the top six compounds with the highest negative scores. Table 1 provides details on these compounds including their name, characterization, and the potential mechanisms of ISR modulation.
Discussion
Coronary ISR is a common complication following coronary artery intervention, characterized by complex mechanisms involving various cellular and molecular processes that are not fully understood35. Mitophagy, a selective autophagic process responsible for mitochondrial degradation, plays a critical role in mitochondrial quality control and cellular homeostasis maintenance8. Recent studies have linked mitochondrial dysfunction to several cardiovascular diseases, including myocardial infarction and heart failure, etc36. However, the expression profile and potential biological functions of MRGs in ISR remain unclear. In this study, we identified two diagnostic biomarkers for ISR: LRRK2, and ANKRD13A. By combining MRGs with DEGs and using WGCNA and machine learning algorithms, we successfully identified these biomarkers. Based on these two hub genes, we constructed a nomogram model with strong diagnostic performance and analyzed their potential biological functions and correlations with immune cell infiltration. Additionally, we identified six small molecule compounds that may influence ISR progression.
Previous research on the mechanisms of ISR has primarily focused on traditional pathways such as vascular smooth muscle cell proliferation and inflammation. Our study reveals the critical role of mitophagy in ISR. Mitophagy is a specialized form of autophagy that involves the processes of mitochondrial damage recognition, ubiquitination and sequestration, autophagosome maturation, and degradation37,38,39. Mitophagy is regulated by various pathways, such as the PINK1-Parkin pathway, which identifies damaged mitochondria, and mitophagy receptors like FUNDC1, BNIP3, NIX, and BCL2L13, which facilitate the recognition and binding of the autophagy-promoting complex to damaged mitochondria40. Our GO enrichment analysis showed that DEMRGs were significantly enriched in biological processes including ubiquitin protein ligase binding, ubiquitin-specific protease binding, enzyme binding and phosphorylation. These processes are involved in regulating different stages of mitophagy40,41,42, suggesting that DEMRGs might influence ISR development through multiple pathways. This finding provides a direction for further investigation into the biological mechanisms of mitophagy in ISR.
Classical machine learning methods such as Logistic-LASSO, SVM-RFE, and RF are widely applied and perform well in biomedical data analysis43,44. By integrating these three machine learning algorithms with WGCNA, we identified LRRK2, and ANKRD13A as hub MRGs that may serve as potential diagnostic biomarkers for ISR. These two genes exhibited significant expression differences between the ISR and control groups, with ROC curve demonstrating good diagnostic performance. Furthermore, the nomogram based on these two genes showed high predictive accuracy, further confirming their potential as diagnostic markers for ISR. Significant progress has been made in the study of circulating mRNA as diagnostic biomarkers, offering advantages such as non-invasiveness, easy sample collection, and high reproducibility. These have led to preliminary clinical applications in various tumor and non-tumor diseases45,46,47,48. With advances in technology, the cost of mRNA analysis has significantly decreased and is becoming more affordable, with further reductions expected in the future49. LRRK2 and ANKRD13A, as diagnostic biomarkers for ISR, are poised for broader clinical application.
The expressions of LRRK2, and ANKRD13A were significantly downregulated in ISR, suggesting their potential inhibitory roles in its pathogenesis. LRRK2 (Leucine-Rich Repeat Kinase 2) is a serine/threonine kinase involved in cellular signal transduction, cell cycle regulation, and apoptosis, with gene mutations closely linked to neurodegenerative diseases like Parkinson’s disease50. The role of LRRK2 in cellular signaling and autophagy has been extensively studied. LRRK2 regulates mitophagy by phosphorylating substrate proteins, influencing autophagosome formation and mitochondrial degradation51. Liu et al. suggested that LRRK2 deficiency prevents cardiac remodeling under pressure overload by enhancing autophagy levels regulated by Bcl-2/beclin1 and Rab752, and protects mouse hearts from myocardial infarction damage via the P53/HMGB1 pathway53. This contrasts with our hypothesis of LRRK2’s protective role, indicating a need for experimental validation of LRRK2’s specific role and mechanisms in ISR.
ANKRD13A (ankyrin repeat domain 13a), is a ubiquitin-binding protein, specifically recognizing Lys-63-linked polyubiquitin chain in protein54. It is well known that ubiquitination is a key regulatory step in mitophagy. Lys63-linked polyubiquitin chains play a critical role in PINK1/Parkin-mediated mitophagy55. However, no direct studies have yet linked ANKRD13A to mitophagy. Additionally, research directly examining the role of ANKRD13A in cardiovascular diseases is extremely limited. Other members of the ANKRD family, such as ANKRD1 (also known as cardiac ankyrin repeat protein), have been extensively studied and shown to play significant roles in cardiovascular diseases56. This study reveals a correlation between ANKRD13A and ISR, suggesting a potential role for ANKRD13A in the pathological process of ISR.
Mitophagy plays a crucial role in the inflammatory response57. Damaged mitochondria release mitochondrial DNA (mtDNA) and reactive oxygen species (ROS), both of which can trigger inflammation57. By clearing damaged mitochondria, mitophagy serves to mitigate excessive inflammation and maintain cellular homeostasis58. In this study, single-gene GSEA analysis revealed that the two hub MRGs (LRRK2, and ANKRD13A) are associated with multiple inflammatory pathways, further underscoring the interplay between mitophagy and inflammation. For example, LRRK2’s role in TNF-α signaling through NF-κB suggests it may influence chronic inflammatory responses, a key contributor to ISR progression59,60. Similarly, ANKRD13A’s association with E2F targets implicates it in vascular smooth muscle cell proliferation and migration, processes essential for neointimal hyperplasia in ISR61,62. These findings suggest that inflammatory responses play critical roles in ISR development, consistent with previous reports63. Moreover, immune cell infiltration analysis revealed significant differences in the abundance of CD56dim natural killer cells, Neutrophils and Plasmacytoid dendritic cells between ISR and control groups. A strong correlation was observed between hub genes and various immune cells, indicating that these hub genes may regulate ISR through a complex interaction between mitophagy, immune response, and inflammation. These findings collectively highlight the critical role of inflammatory and immune pathways in ISR pathogenesis, providing potential avenues for early diagnosis and therapeutic intervention.
Additionally, through cMAP database, six small molecule compounds were predicted to potentially reverse the gene expression changes in ISR: Varenicline, calyculin, cobalt(II)-chloride, genipin, bimatoprost and phorbol-12-myristate-13-acetate (PMA). The regulatory effects of these small molecules on ISR require further experimental validation. Varenicline, primarily used as a smoking cessation aid, could potentially mitigate cardiovascular risks associated with smoking, offering clinical benefits in ISR patients who smoke30. Bimatoprost, a prostaglandin analog, currently treats glaucoma and might influence blood flow velocity33. Cobalt(II)-chloride and genipin have been shown to affect vascular remodeling in different disease contexts31,32, suggesting they might also regulate ISR through similar mechanisms. Calyculin and phorbol-12-myristate-13-acetate, both protease modulators, have demonstrated clear regulatory effects on VSMCs proliferation and migration, which are crucial in ISR development34,64. These small molecules, especially those with known effects on vascular biology, present promising therapeutic avenues for ISR. Future research should focus on experimentally validating their regulatory roles and mechanisms in ISR, potentially leading to their incorporation into clinical treatment protocols. By integrating these compounds into experimental designs, researchers can explore their potential to not only mitigate ISR progression but also improve patient outcomes post-PCI.
We acknowledge several limitations in our study. First, our analysis relied on publicly available gene expression data and lacked validation with additional clinical samples, which limits the identified biomarkers and potential mechanisms to a preliminary exploratory stage. Second, although differential RNA expression was identified, this does not necessarily reflect changes at the protein level, which is crucial for functional impact. Protein-level validation, such as proteomics or Western blot analysis, would be essential to confirm the biological significance of the identified hub genes. Third, the diagnostic biomarkers and potential therapeutic targets identified here still require extensive experimental research to validate their functions and roles. Lastly, the relatively small sample size in this study may introduce some bias, limiting the generalizability of our findings.
Conclusions
This study identified two mitophagy-related biomarkers (LRRK2, and ANKRD13A) for ISR using integrated bioinformatics analyses. Based on these hub genes, a nomogram demonstrated excellent diagnostic efficacy was constructed. Their potential biological functions and associations with immune cell infiltration were also provided. Additionally, six small molecule compounds with potential effects on ISR progression were identified. To our knowledge, this study is the first to systematically reveal the expression profile and potential biological functions of MRGs in ISR. These findings offer new perspectives for the early diagnosis and precise treatment of ISR, and lay a foundation for further exploration of the pathophysiological processes of ISR and the development of novel therapeutic strategies. Future research should focus on validating the diagnostic potential of LRRK2 and ANKRD13A through clinical samples, investigating their functional roles in ISR development, and exploring the efficacy of the identified small-molecule compounds in mitigating ISR progression. These studies will be crucial in translating bioinformatics findings into clinical applications, potentially leading to the development of more effective treatments for ISR.
Data availability
The datasets used for analysis in this study (GSE46560, and GSE48060) were derived from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The codes presented in the study are publicly available. This data can be found here: https://github.com/yeyue0077/bioinformatics.
References
Roth, G. A. et al. Global Burden of Cardiovascular diseases and Risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 76, 2982–3021. https://doi.org/10.1016/j.jacc.2020.11.010 (2020).
Giacoppo, D., Mazzone, P. M. & Capodanno, D. Current management of In-Stent restenosis. J. Clin. Med. 13 https://doi.org/10.3390/jcm13082377 (2024).
Giustino, G. et al. Coronary In-Stent restenosis: JACC state-of-the-art review. J. Am. Coll. Cardiol. 80, 348–372. https://doi.org/10.1016/j.jacc.2022.05.017 (2022).
Bajeu, I. T., Niculescu, A. G., Scafa-Udriste, A. & Andronescu, E. Intrastent Restenosis: a Comprehensive Review. Int. J. Mol. Sci. 25. https://doi.org/10.3390/ijms25031715 (2024).
Alfonso, F., Coughlan, J. J., Giacoppo, D., Kastrati, A. & Byrne, R. A. Management of in-stent restenosis. EuroIntervention. 18, e103–e123. https://doi.org/10.4244/EIJ-D-21-01034 (2022).
Costa, M. A. & Simon, D. I. Molecular basis of restenosis and drug-eluting stents. Circulation. 111, 2257–2273. https://doi.org/10.1161/01.CIR.0000163587.36485.A7 (2005).
Atici, A. E., Crother, T. R. & Noval Rivas, M. Mitochondrial quality control in health and cardiovascular diseases. Front. Cell. Dev. Biol. 11, 1290046. https://doi.org/10.3389/fcell.2023.1290046 (2023).
Forte, M. et al. Mitophagy modulation for the treatment of cardiovascular diseases. Eur. J. Clin. Invest. e14199. https://doi.org/10.1111/eci.14199 (2024).
Fan, Y. et al. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles. J. Nanobiotechnol. 20, 149. https://doi.org/10.1186/s12951-022-01338-4 (2022).
Wu, W. et al. PINK1-Parkin-mediated Mitophagy protects mitochondrial Integrity and prevents metabolic stress-Induced Endothelial Injury. PLoS One. 10, e0132499. https://doi.org/10.1371/journal.pone.0132499 (2015).
Xi, J. et al. Scutellarin ameliorates high glucose-induced vascular endothelial cells injury by activating PINK1/Parkin-mediated mitophagy. J. Ethnopharmacol. 271, 113855. https://doi.org/10.1016/j.jep.2021.113855 (2021).
Chen, Z. et al. MCU-dependent mitochondrial calcium uptake-induced mitophagy contributes to apelin-13-stimulated VSMCs proliferation. Vascul Pharmacol. 144, 106979. https://doi.org/10.1016/j.vph.2022.106979 (2022).
He, L. et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKalpha and exacerbates atherosclerotic lesions. J. Cell. Physiol. 234, 8668–8682. https://doi.org/10.1002/jcp.27527 (2019).
Huynh, D. T. N. & Heo, K. S. Role of mitochondrial dynamics and mitophagy of vascular smooth muscle cell proliferation and migration in progression of atherosclerosis. Arch. Pharm. Res. 44, 1051–1061. https://doi.org/10.1007/s12272-021-01360-4 (2021).
Gwon, J. G. & Lee, S. M. Role of PTEN-Induced protein kinase 1 as a mitochondrial dysfunction Regulator in Cardiovascular Disease Pathogenesis. Vasc Specialist Int. 40, 9. https://doi.org/10.5758/vsi.230116 (2024).
Marek-Iannucci, S. et al. Autophagy-mitophagy induction attenuates cardiovascular inflammation in a murine model of Kawasaki disease vasculitis. JCI Insight. 6 https://doi.org/10.1172/jci.insight.151981 (2021).
Diao, R. Y. & Gustafsson, A. B. Mitochondrial quality surveillance: mitophagy in cardiovascular health and disease. Am. J. Physiol. Cell. Physiol. 322, C218–C230. https://doi.org/10.1152/ajpcell.00360.2021 (2022).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Stelzer, G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinf. 54:1 (30), 31–313033. https://doi.org/10.1002/cpbi.5 (2016).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform.9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Cai, W. & van der Laan, M. Nonparametric bootstrap inference for the targeted highly adaptive least absolute shrinkage and selection operator (LASSO) estimator. Int. J. Biostat. https://doi.org/10.1515/ijb-2017-0070 (2020).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19, 432. https://doi.org/10.1186/s12859-018-2451-4 (2018).
Liu, Y. & Zhao, H. Variable importance-weighted Random forests. Quant. Biol. 5, 338–351 (2017).
Engebretsen, S. & Bohlin, J. Statistical predictions with glmnet. Clin. Epigenetics. 11, 123. https://doi.org/10.1186/s13148-019-0730-1 (2019).
Yoon, S. & Kim, S. AdaBoost-based multiple SVM-RFE for classification of mammograms in DDSM. BMC Med. Inf. Decis. Mak. 9 (1), S1. https://doi.org/10.1186/1472-6947-9-S1-S1 (2009).
Zhang, H., Meltzer, P. & Davis, S. RCircos: an R package for Circos 2D track plots. BMC Bioinform. 14, 244. https://doi.org/10.1186/1471-2105-14-244 (2013).
Subramanian, A. et al. A Next Generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell. 171, 1437–1452e1417. https://doi.org/10.1016/j.cell.2017.10.049 (2017).
Havard, A. et al. Comparison of Cardiovascular Safety for Smoking Cessation Pharmacotherapies in a Population-based cohort in Australia. JAMA Netw. Open. 4, e2136372. https://doi.org/10.1001/jamanetworkopen.2021.36372 (2021).
Buttyan, R. et al. Acute intravesical infusion of a cobalt solution stimulates a hypoxia response, growth and angiogenesis in the rat bladder. J. Urol. 169, 2402–2406. https://doi.org/10.1097/01.ju.0000058406.16931.93 (2003).
Furdella, K. J. et al. In-vivo assessment of a tissue engineered vascular graft computationally optimized for target vessel compliance. Acta Biomater. 123, 298–311. https://doi.org/10.1016/j.actbio.2020.12.058 (2021).
Inan, U. U. et al. The comparative cardiovascular, pulmonary, ocular blood flow, and ocular hypotensive effects of topical travoprost, bimatoprost, brimonidine, and betaxolol. J. Ocul Pharmacol. Ther. 20, 293–310. https://doi.org/10.1089/1080768041725344 (2004).
Zhou, H. et al. Down-regulation of protein kinase C-epsilon by prolonged incubation with PMA inhibits the proliferation of vascular smooth muscle cells. Cell. Physiol. Biochem. 40, 379–390. https://doi.org/10.1159/000452553 (2016).
Aoki, J. & Tanabe, K. Mechanisms of drug-eluting stent restenosis. Cardiovasc. Interv Ther. 36, 23–29. https://doi.org/10.1007/s12928-020-00734-7 (2021).
Ajoolabady, A., Chiong, M., Lavandero, S., Klionsky, D. J. & Ren, J. Mitophagy in cardiovascular diseases: molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med. 28, 836–849. https://doi.org/10.1016/j.molmed.2022.06.007 (2022).
Guardia-Laguarta, C. et al. PINK1 content in Mitochondria is regulated by ER-Associated Degradation. J. Neurosci. 39, 7074–7085. https://doi.org/10.1523/JNEUROSCI.1691-18.2019 (2019).
Kruppa, A. J. & Buss, F. Actin cages isolate damaged mitochondria during mitophagy. Autophagy. 14, 1644–1645. https://doi.org/10.1080/15548627.2018.1486152 (2018).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705. https://doi.org/10.15252/embj.2020104705 (2021).
D’Arcy, M. S. Mitophagy in health and disease. Molecular mechanisms, regulatory pathways, and therapeutic implications. Apoptosis. https://doi.org/10.1007/s10495-024-01977-y (2024).
Lu, G. et al. Suppression of autophagy during mitosis via CUL4-RING ubiquitin ligases-mediated WIPI2 polyubiquitination and proteasomal degradation. Autophagy. 15, 1917–1934. https://doi.org/10.1080/15548627.2019.1596484 (2019).
Wang, L. et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell. Res. 28, 787–802. https://doi.org/10.1038/s41422-018-0056-0 (2018).
Guo, H., Xiao, K., Zheng, Y. & Zong, J. Integrating bioinformatics and multiple machine learning to identify mitophagy-related targets for the diagnosis and treatment of diabetic foot ulcers: evidence from transcriptome analysis and drug docking. Front. Mol. Biosci. 11, 1420136. https://doi.org/10.3389/fmolb.2024.1420136 (2024).
Wei, C. et al. Identification and verification of diagnostic biomarkers in recurrent pregnancy loss via machine learning algorithm and WGCNA. Front. Immunol. 14, 1241816. https://doi.org/10.3389/fimmu.2023.1241816 (2023).
Chen, J. et al. Prediction of preeclampsia using maternal circulating mRNAs in early pregnancy. Arch. Gynecol. Obstet. 310, 327–335. https://doi.org/10.1007/s00404-024-07486-2 (2024).
Danila, D. C. et al. Clinical validity of detecting circulating Tumor cells by AdnaTest Assay compared with direct detection of Tumor mRNA in stabilized whole blood, as a Biomarker Predicting overall survival for metastatic castration-resistant prostate Cancer patients. Cancer J. 22, 315–320. https://doi.org/10.1097/PPO.0000000000000220 (2016).
Paik, S. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl. J. Med. 351, 2817–2826. https://doi.org/10.1056/NEJMoa041588 (2004).
Sole, C., Arnaiz, E., Manterola, L., Otaegui, D. & Lawrie, C. H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin Cancer Biol. 58, 100–108. https://doi.org/10.1016/j.semcancer.2019.01.003 (2019).
Dakal, T. C. et al. Emerging methods and techniques for cancer biomarker discovery. Pathol. Res. Pract. 262, 155567. https://doi.org/10.1016/j.prp.2024.155567 (2024).
Iannotta, L. & Greggio, E. LRRK2 signaling in neurodegeneration: two decades of progress. Essays Biochem. 65, 859–872. https://doi.org/10.1042/EBC20210013 (2021).
Singh, F. & Ganley, I. G. Parkinson’s disease and mitophagy: an emerging role for LRRK2. Biochem. Soc. Trans. 49, 551–562. https://doi.org/10.1042/BST20190236 (2021).
Liu, Y. et al. Leucine-rich repeat kinase-2 deficiency protected against cardiac remodelling in mice via regulating autophagy formation and degradation. J. Adv. Res. 37, 107–117. https://doi.org/10.1016/j.jare.2021.07.004 (2022).
Liu, Y. et al. LRRK2 deficiency protects the heart against myocardial infarction injury in mice via the P53/HMGB1 pathway. Free Radic Biol. Med. 191, 119–127. https://doi.org/10.1016/j.freeradbiomed.2022.08.035 (2022).
Zhou, W., Xu, S., Deng, T., Zhou, R. & Wang, C. LncRNA USP30-AS1 promotes the survival of acute myeloid leukemia cells by cis-regulating USP30 and ANKRD13A. Hum. Cell. 35, 360–378. https://doi.org/10.1007/s13577-021-00636-7 (2022).
Gao, B. et al. Parkin overexpression alleviates cardiac aging through facilitating K63-polyubiquitination of TBK1 to facilitate mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 165997. https://doi.org/10.1016/j.bbadis.2020.165997 (2021).
Murphy, N. P., Lubbers, E. R. & Mohler, P. J. Advancing our understanding of AnkRD1 in cardiac development and disease. Cardiovasc. Res. 116, 1402–1404. https://doi.org/10.1093/cvr/cvaa063 (2020).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23, 159–173. https://doi.org/10.1038/s41577-022-00760-x (2023).
Ameenudeen, A. A. M. et al. Emerging role of Mitophagy in Inflammatory diseases: Cellular and Molecular episodes. Curr. Pharm. Des. 26, 485–491. https://doi.org/10.2174/1381612826666200107144810 (2020).
Han, K. A. et al. Leucine-rich repeat kinase 2 (LRRK2) stimulates IL-1beta-Mediated Inflammatory Signaling through Phosphorylation of RCAN1. Front. Cell. Neurosci. 11, 125. https://doi.org/10.3389/fncel.2017.00125 (2017).
Lopez de Maturana, R. et al. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J. Neuroinflammation. 13, 295. https://doi.org/10.1186/s12974-016-0761-x (2016).
Ahn, J. D. et al. Novel E2F decoy oligodeoxynucleotides inhibit in vitro vascular smooth muscle cell proliferation and in vivo neointimal hyperplasia. Gene Ther. 9, 1682–1692. https://doi.org/10.1038/sj.gt.3301849 (2002).
Giangrande, P. H. et al. Distinct roles of E2F proteins in vascular smooth muscle cell proliferation and intimal hyperplasia. Proc. Natl. Acad. Sci. U S A. 104, 12988–12993. https://doi.org/10.1073/pnas.0704754104 (2007).
Drachman, D. E. & Simon, D. I. Inflammation as a mechanism and therapeutic target for in-stent restenosis. Curr. Atheroscler Rep. 7, 44–49. https://doi.org/10.1007/s11883-005-0074-5 (2005).
Skaletz-Rorowski, A. et al. Lovastatin blocks basic fibroblast growth factor-induced mitogen-activated protein kinase signaling in coronary smooth muscle cells via phosphatase inhibition. Eur. J. Cell Biol. 80, 207–212 https://doi.org/10.1078/0171-9335-00155 (2001).
Acknowledgements
Not applicable.
Funding
This research was supported by the Joint Special Fund for Application and Basic Research of Kunming Medical University(202301AY070001-102).
Author information
Authors and Affiliations
Contributions
MS and YY contributed to conception and design of the study. MS and MC collected the datasets and conducted the analyses. YC contributed to the visualization and validation of the study. MS, MC and YY wrote and revised the manuscript. MS and MC contributed equally to the article and share first authorship. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shen, M., Chen, M., Chen, Y. et al. Mitophagy related diagnostic biomarkers for coronary in-stent restenosis identified using machine learning and bioinformatics. Sci Rep 14, 24137 (2024). https://doi.org/10.1038/s41598-024-74862-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-74862-y
