
Abstract
We investigated whether miR143#12, a synthesized chemically modified miR-143-3p derivative, exerts therapeutic effects on acute myocardial infarction (AMI). Sprague–Dawley rats and Japanese white rabbits underwent 30 min of coronary occlusion followed by 2 weeks of reperfusion. The rat AMI model was intravenously administered with control miRNA (9 μg/kg), 3 μg/kg or 9 μg/kg of miR143#12 1 h after reperfusion, while the rabbit AMI model was intravenously administered with control miRNA (9 μg/kg) or 9 μg/kg of miR143#12. In the rat and rabbit AMI models, 9 μg/kg of miR143#12 significantly reduced infarct sizes and significantly improved cardiac function including LVEF and LVFS at 2 weeks. The tissue miR143 levels in infarct areas significantly decreased after AMI in both models. Electron microscopic study and immunohistochemistry suggested that miR143#12 suppressed autophagic cell death caused by AMI and induced neoangiogenesis in the infarct border. In cultured rat H9c2 cells, miR143#12 significantly inhibited H2O2-induced autophagic cell death by decreasing ROS levels and increased viable cell numbers more than the control by silencing COX-1, -2, and ATG7. Replacement treatment with miR143#12 in the infarct areas, where the expression levels of miR143 were significantly decreased, has a beneficial effect on AMI by silencing COX-1 and -2.
Similar content being viewed by others

Introduction
Acute myocardial infarction (AMI) is one of the most common causes of morbidity and mortality worldwide1. First-line therapy for AMI involves performing percutaneous coronary intervention (PCI) as soon as possible to reperfuse the occluded coronary artery and salvage viable cardiomyocytes. However, failed or delayed reperfusion results in the loss of many cardiomyocytes, which leads to extensive myocardial necrosis and the deterioration of left ventricular (LV) function, followed by LV dilation, and, ultimately, heart failure. A previous study reported that AMI patients with a lower LV ejection fraction (LVEF) (≤ 45%) had a poorer prognosis than those with higher LVEF (> 45%)2. Furthermore, the recovery of LVEF has been associated with a better long-term prognosis3 and LV dilation after AMI with a worse long-term prognosis4. The roles of miRNAs in this process are not well understood and therapeutic options are still underrepresented.
MicroRNAs (miRNAs) are highly conserved RNAs of approximately 21–24 nucleotides that are transcribed from DNA as non-coding genes, negatively regulate the expression of proteins by inhibiting translation or cleaving plural target messenger RNA (mRNA), and play a role in biological processes, such as proliferation, differentiation, and cell death5. Since several miRNAs have been linked to AMI6, miRNAs have potential as therapeutic strategies against AMI. miRNA143-3p (miR143) and miRNA145-5p (miR145) are from the same primary miRNA and regulate the differentiation and proliferation of vascular smooth muscle cells7. miR143 expresses rich in amount in normal tissues, especially in blood vessels7. In addition, miRs143 and 145 knock out mouse demonstrated the phenotypes of abnormal blood vessel healing and abnormal neointima formation8. Either miRNA has been suggested to inhibit the dysregulation of multiple gene expression profiles, not only in cardiomyocytes, but also in endothelial cells affected by AMI, and, thus, repair damaged tissues8. It is expected that the replacement treatment with miR143#12 would be effective because of dysregulation of miR143 in AMI. So far, we have reported post-infarct effects of pancaspase inhibitor by using a rat AMI model9, and also reported that the intravenous administration of miR145 after AMI reduced myocardial infarct sizes and improved LV function in the rabbit model of AMI10. We recently found that increases in plasma miR143 levels during the acute phase were positively associated with LV functional recovery in the chronic phase in the patients with AMI11. Under these conditions, we developed a chemically modified miR143 called miR143#1212, the lipoplex form of which was significantly stable in mouse blood in an in vivo experiment. The purpose of the present study was to establish whether miR143#12 exerts therapeutic effects on damaged cardiac tissue, reduces myocardial infarct sizes, and improves LV function in rat and rabbit AMI models and to elucidate the underlying mechanisms.
Results
The RNA sequence of chemically modified miR143#12 used in this study and its stability in mouse blood are shown in Fig. S1. The naked and lipoplexed miR143#12 were considerably stable in culture medium and mice blood than those of wild type miR143 from Ambion, respectively (dose: 1.4 mg/kg).
In vivo study
Effects of miR143#12 on infarct size and cardiac function in rat and rabbit AMI models
We prepared rat and rabbit AMI models by ligation of coronary artery for 30 min followed by reperfusion. miRNA lipoplexes were intravenously administered an hour after the reperfusion. All rats survived for 2 weeks in 9 μg/kg of control miRNA (n = 7), 3 μg/kg (n = 7) and 9 μg/kg of miR143#12 (n = 7) groups. In rabbit AMI model, one rabbit died after coronary reperfusion in the control miRNA and 9 μg/kg of miR143#12 groups, respectively. The other rabbits survived for 2 weeks in the control (n = 8) and miR143#12 groups (n = 8). No significant differences were observed in mortality rates between the 2 groups.
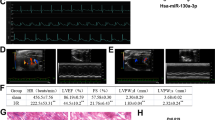
To determine the infarct sizes, we performed Masson-Trichrome-staining of tissue samples from each infarct heart in both models. Typical images of Masson-Trichrome-stained LV cross-sections from rats and rabbits are shown in Fig. 1A,B, respectively. Myocardial infarct sizes as a percentage of the LV in rats were significantly smaller in the 9 μg/kg miR143#12 group (4.40 ± 0.96%) and 3 μg/kg miR143#12 group (5.26 ± 0.94%) than in the control miRNA group (11.17 ± 1.59%) (Fig. 1A). Infarct sizes as a percentage of the LV in rabbits were significantly smaller in the 9 μg/kg miR143#12 group (8.4 ± 1.7%) than in the control group (24.7 ± 1.4%) (Fig. 1B).

Effects of miR143#12 on infarct sizes and cardiac function in rat and rabbit AMI models. (A) Representative cross-sectional images of the heart stained with Masson-Trichrome in the control miRNA (9 µg/kg), 3 µg/kg and 9 µg/kg of miR143#12 groups in the rat AMI model. Figures of myocardial infarct sizes as a percentage of LV in the control miRNA (9 µg/kg), 3 µg/kg and 9 µg/kg of miR143#12 groups in the rat AMI model (n = 7 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01; 1-way ANOVA with Dunnett’s post hoc test. (B) Representative cross-sectional images of the heart stained with Masson-Trichrome in the control miRNA (9 µg/kg) and 9 µg/kg of miR143#12 groups in the rabbit AMI model. Figures of myocardial infarct sizes as a percentage of LV in the control miRNA and miR143#12 groups in the rabbit AMI model (n = 7 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01; 1-way ANOVA with Dunnett’s post hoc test. (C) Cardiac function before surgery or before administration with miRNA and at 2 weeks after treatment in rat AMI model. The LV ejection fraction (EF) in the control miRNA, 3 mg/kg and 9 mg/kg of miR143#12 groups were 90.9 ± 0.8, 90.1 ± 0.5, and 90.0 ± 0.4%, respectively, before surgery, and 53.3 ± 0.9, 52.5 ± 0.8, and 52.5 ± 0.5%, respectively, before administration. There was no significant difference in EF among the 3 groups before surgery or administration. LV fractional shortening (FS) in the control, 3 mg/kg and 9 mg/kg of miR143#12 groups were 55.1 ± 1.3, 53.8 ± 0.7, and 53.5 ± 0.6%, respectively, before surgery, and 22.4 ± 0.5, 22.0 ± 0.4, and 22.0 ± 0.3% before administration. There was no significant difference in FS among the 3 groups before surgery or administration. The assignment was validated by similar LVEF and LVFS before administration 1 h after AMI among the 3 groups. These results suggest that infarct sizes before the administration of miR143#12 were similar among the 3 groups. In the cases of treatment, the parameters, LVEF and LVFS (%) in the control miRNA (9 µg/kg), 3 µg/kg and 9 µg/kg of miR143#12 groups (n = 7 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01; 1-way ANOVA with Dunnett’s post hoc test. (D) Cardiac function at 2 weeks after AMI in rabbit model. The parameters, LVEF LVFS, +dP/dt, −dP/dt, LVDd, and LVDs in the control miRNA (9 µg/kg) and 9 µg/kg of miR143#12 groups (n = 7 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; 2-tailed, unpaired Student’s t test. In the rat and rabbit AMI models, infarct sizes were analyzed three times from one short-axis cross-sectional image at the papillary muscle level and the average value was adopted.
As to cardiac function in rat AMI model, LVEF was significantly higher in the 9 μg/kg miR143#12 group (54.3 ± 2.9%) than in the control group (43.4 ± 1.2%) and 3 μg/kg miR143#12 group (45.6 ± 1.9%), but there was no significant difference between the control and 3 μg/kg miR143#12 group (Fig. 1C). LVFS was also significantly higher in the 9 μg/kg miR143#12 group (23.1 ± 1.6%) than in the control group (17.3 ± 0.6%) and 3 μg/kg miR143#12 group (18.4 ± 1.0%), but there was no significant difference between the control and 3 μg/kg miR143#12 group (Fig. 1C). On the other hand, no significant differences were observed in HR among the three groups 2 weeks after AMI. Furthermore, LVEF and LVFS did not significantly differ among the 3 groups before surgery or administration (Fig. 1C).
To further verify to the effects of 9 μg/kg miR143#12, we performed the same experiment in rabbit AMI model. As shown in Fig. 1D, 2 weeks after AMI, LVEF was significantly higher in the 9 μg/kg miR143#12 group (68.8 ± 1.5%) than in the 9 μg/kg control miRNA group (52.2 ± 2.4%). LVFS was also significantly higher in the miR143#12 group (34.5 ± 1.2%) than in the control group (24.2 ± 1.4%). +Dp/dt was significantly higher in the miR143#12 group (6229.0 ± 190.5%) than in the control group (5029 ± 304.7%). Furthermore, −dp/dt was significantly higher in the miR143#12 group (-6190 ± 607%) than in the control group (-4053 ± 487.2%). LVDd was significantly smaller in the miR143#12 group (9.0 ± 0.5 mm) than in the control group (12.8 ± 0.9 mm). LVDs was also significantly smaller in the miR143#12 group (5.9 ± 0.3 mm) than in the control group (9.7 ± 0.8 mm). Thus, miR143#12 had beneficial effects on AMI from the viewpoint of infarct size and cardiac function in both models.
Electron microscopic findings in infract and its border areas
To investigate the cell death in the infarct areas in the rat and rabbit AMI models, we performed electron microscopic examination. The result revealed the characteristic findings of autophagy, such as autophagosomes, autolysosomes, and mitophagy, in many cardiomyocytes in infarct areas 2 weeks after AMI (Fig. 2A). These results indicated that autophagic cell death was a central cause of cell death induced by AMI. Especially, mitophagy was a characteristic finding in the infarct areas. While the quantitative assessment by EM is difficult, the number of autophagic cells with mitophagy seemed to be lower in the miR143 treated cases than control group (Fig. 2A).

Electron microscopic findings and immunostaining of parameters affected by miR143#12 in cardiac tissues from AMI rats. (A) Electron microscopic findings of infarct areas 2 weeks after AMI. (a,b) Many autophagosomes and autolysosomes which were indicated by yellow arrows were observed in the infarct area in the rat (a) and rabbit (b) AMI models. Mitophagy is indicated by a red arrow. The representative figures were shown. Electron microscopic findings. Control miRNA: Not only autophagosomes and autolysosomes but also mitophagy are seen in the infarct area of the control group in rat and rabbit AMI. miR143#12: Small vacuoles are seen in the cells of the infarct area and apoptosis is seen in fibroblast in rat and rabbit AMI. A few normal cardiac cells appeared in the infarct areas in miR-143#12 treated AMI rat and rabbit. (B) Immunostaining of autophagy-related LC3B (9 µg/kg of control miRNA vs 9 µg/kg of miR143#12). LC3B-positive cardiomyocytes in the infarct area of both groups. A typical LC3B-positive cardiomyocyte with striation was showed in the magnified film in the figure (red arrowheads and arrows). (C) Immunostaining of angiogenesis-related CD31 and α-Smooth muscle actin (α-SMA) (9 µg/kg of control miRNA vs 9 µg/kg of miR143#12). From 4 animals in each group, 4 sections from each animal were stained with CD31 or α-smooth muscle actin. The representative images were shown. The number of positive stained vessels in each case was counted. Results are shown as the mean ± SE. ***p < 0.001; 2-tailed, unpaired Student’s t test.
Immunohistochemistry of biological parameters related to autophagy and angiogenesis, and their quantitative analyses of positive vessel in the rat AMI tissue
Based on the electron microscopic findings, we investigated the protein expression in terms of autophagic cell death (LC3B) by performing immunostaining of the tissues from infarct border areas 2 weeks after AMI (Fig. 2B). The expression between the 9 μg/kg of control miRNA and the 9 μg/kg of miR143#12 groups were compared. The results were as follows: LC3B-positive cardiomyocytes were observed in the infarct border areas (Fig. 2B). There were more strong-stained cells in the control cases than miR143#12 treated cases. In the magnified film, the granular dense-stained cells with striation were frequently observed in controls. Previously, we reported that the topical injection with miR143#12 on the tumor inoculated in the back of mouse induced neoangiogenesis (Fig. S2)13. Therefore, we performed immunostaining of angiogenesis-related CD31 and α-smooth muscle actin (α-SMA) (Fig. 2C). As a result, the number of CD31-positive microvessels was significantly higher in the miR143#12 group (133.9 ± 8.6/mm2) than in the control group (42.5 ± 4.9/mm2). α-SMA-positive vessels were observed in the infarct border areas. The number of vessels was significantly higher in the miR143#12 group (40.8 ± 3.4/mm2) than in the control group (12.8 ± 2.2/mm2). These results suggested that miR143#12 increased the number of not only capillary but also small vessel.
Plasma and cardiac tissue miR143 expression levels evaluated by qRT-PCR and in situ hybridization in the rat AMI
Sixty minutes after the administration of 9 µg of miR143#12, the mean plasma miR143 level was 282 pM in the miR143#12 group and 0.38 pM in the control group (4 cases in each group) (Fig. 3A). This finding indicated that miR143#12 molecule was detectable by qRT-PCR and confirmed the increase in the levels of plasma miR143 after the treatment. However, no significant differences in the plasma miR143 levels between the treatment with control miRNA and miR143#12 were observed 24 h after the administration of miR143#12 (data not shown). Importantly, 24 h after AMI, the tissue levels of miR143 were extremely lower in infarct areas than in remote non-infarct areas in both groups (6 cases in control group, 7 cases in miR143#12 group) (Fig. 3B), which was evaluated by qRT-PCR. The difference in the expression levels of miR143 in non-infarct areas between both groups was not significant (Fig. 3B). These results indicated that the administrated miR143#12 molecules did not affect the levels of plasma and tissue miR143 by qRT-PCR at 24 h after AMI. On the other hand, in situ hybridization (ISH) detected miR143 molecules rich in cardiomyocytes and vessels of the non-infarct areas of both groups, however, some signals were found in the cardiomyocytes and vessels in the infarct areas of the miR143 treated case, but not at all in the control (Fig. 3C). These findings showed that miR143#12 could have effective action in AMI even at 24 h after the treatment.

Plasma and cardiac tissue miR143 levels after the administration of miR143#12 in the rat AMI model. (A) Plasma miR143 expression levels 1 h after the administration of 9 µg/kg of miR143#12 or control miRNA (9 µg/kg) in the rat AMI model (N = 3 in each). (B) Cardiac tissue miR143 levels 24 h after the administration of 9 µg/kg miRNA (cases 1–6), 9 µg/kg miR143#12 (cases 11–17) in the rat AMI model. Tissue miR143 levels in infarct and non-infarct areas from each case in both the control miRNA and miR143#12 groups were evaluated by qRT-PCR (at least n = 3 in each). The miR143 levels of infarct and non-infarct areas from each case in both groups were totally evaluated (right figure). Of note, the differences in the expression levels of non-infarct areas of both groups were not significant. (C) Detection of cardiac tissue miR143 by in situ hybridization (ISH) 24 h after the administration of 9 µg/kg of control miRNA and miR143#12 (#12) in the rat AMI model. Blue signals were detected in the cardiac tissues of the rat AMI model. Representative cases in the control miRNA and miR143#12 groups were shown. The number of signals in cardiomyocytes and vessels was estimated in the infarct areas of the miR143#12 and control miRNA groups. Red arrows indicate miR143 molecules (n = 8 in each). The miR143-negative vessels in the infarct areas of controls are also shown in dot rings. Results are shown as the mean ± SE. **p < 0.01, ***p < 0.001, ****p < 0.0001; 2-tailed, unpaired Student’s t test.
In vitro study
Effects of miR143#12 on H2O2-treated H9c2 rat cardiomyocytes
Based on the results of the in vivo study, we examined the target genes of miR143#12 by performing H2O2 exposure experiments on rat H9c2 cells, because we considered oxidative stress by ischemia to be a pivotal event inducing autophagic cell death in both animal models of AMI (Fig. 2). In cultured H9c2 cells, the number of viable H9c2 cells was gradually decreased up to 72 h after exposure to H2O2 (38 μM) for 30 min compared to without exposure (Fig. 4A). Then, the surviving cells transfected with control miRNA or miR143#12 started to grow. However, the number of viable H2O2-exposed H9c2 cells transfected with 3 or 5 nM miR143#12 was approximately 1.5-fold higher than that of H9c2 cells transfected with control miRNA (5 nM) at 120 h after H2O2 exposure (Fig. 4B). The ability of miR143#12 to enhance the growth of H2O2-exposed cells was markedly greater than that of miR143-3p commercially obtained from Ambion (Fig. 4B). On the other hand, anti-miR143-3p partly blocked growth induced by miR143#12 (Fig. 4C). In the same experiment, the expression level of miR143 24 h after exposure to H2O2 significantly decreased (Fig. 4D). These results indicated that the replacement treatment with miR143#12 1 h after exposure to H2O2 more strongly blocked the decrease in the viable cell number at 72 h compared to control miRNA. Morphologically, a few vacuoles were observed in H2O2-exposed cells after 48 h (Fig. 4E). Biochemically, the Western blot analysis indicated that the LC3B transition from I to II, an autophagy marker, markedly increased in control cells 18 h after exposure to H2O2 (Fig. 4F); however, this transition was strongly inhibited in miR143#12-transfected cells 24 h after exposure. In addition, the autophagy inhibitor, 3-methyladenine (3-MA), strongly blocked the progression of autophagic cell death (Fig. 4G). The viable cell number for the dose of 400 μM 3-MA was approximately twofold higher than that for the control (DMSO). Based on these results, we investigated whether miR143#12 reduced ROS levels after exposure to H2O2. Twenty-four hours after transfection, H2O2-induced increases in ROS levels were significantly lower in cells transfected with miR143#12 than in control cells (Fig. 4H). This result corresponded to those on LC3B immunostaining of in vivo samples (Fig. 2B).

Effects of miR143#12 on cell growth, LC3B transition, autophagic changes, and ROS elicited by H2O2 in H9c2 cells. (A) Growth suppression of H9c2 cells in time course up to 120 h after exposure to H2O2 at a concentration of 38 μM for 30 min (n = 4 in each). The viable cell number at the exposure to H2O2 is indicated as 100%. Result is shown as the mean ± SE. ***p < 0.001; 2-tailed, unpaired Student’s t test. (B) Effects of the ectopic expression of miR143#12 or miR143 from Ambion (Amb-miR143) on the viability at 120 h after exposure to H2O2. Transfection was performed 1 h after exposure to H2O2 (n = 4 in each). Result is shown as the mean ± SE. *p < 0.05, ***p < 0.001; 1-way ANOVA with Dunnett’s post hoc test. (C) The co-transfection of anti-miR143 with miR143#12 partly canceled regrowth elicited by the transfection of miR143#12. The evaluation was performed 120 h after exposure to H2O2 (n = 4 in each). Result is shown as the mean ± SE. **p < 0.01, ***p < 0.001; 1-way ANOVA with Dunnett’s post hoc test. (D) miR143 expression levels in H2O2-exposed cells evaluated by qRT-PCR significantly decreased 24 h after exposure (38 μM H2O2) (n = 3 in each). Result is shown as the mean ± SE. ***p < 0.001; 2-tailed, unpaired Student’s t test. (E) Microscopic appearance of cells 48 h after exposure to H2O2. Red arrows indicate cells with vacuoles in the cytoplasm. (F) The transition of LC3B II in cells was shown by a Western blot analysis 18 and 24 h after exposure to H2O2. Band intensities were evaluated by densitometric values as 100 in non-treated cells (NT). The transition was significantly suppressed by the transfection of miR143#12 (#12) compared to that of control miRNA transfected cells (C). The band intensity of each protein was indicated as a value that was assessed using densitometry (ImageJ: version 2.0.0-rc-69/1.52p, NIH). Densitometric values below the bands are the ratios of protein/β-actin calculated with the non-treated sample as 100. (G) Cell growth inhibition after exposure to H2O2 was strongly blocked by the autophagy inhibitor, 3-MA (n = 4). C: DMSO solution. Result is shown as the mean ± SE. ***p < 0.001; 1-way ANOVA with Dunnett’s post hoc test. (H) H2O2-exposed cells were stained with a CellROX probe 24 h after transfection with miR143#12 (#12). miR-143#12 and control miRNA were transfected 1 h after exposure to H2O2. The green signals of CellROX were evaluated in the Merge film and by the intensity of signals. The nucleus was stained with Hoechst 33342 (n = 5 in each). Result is shown as the mean ± SE. **p < 0.01; 1-way ANOVA with Tukey’s post hoc test. NT: No treatment, C: Control miRNA (5 nM).
Regarding the target genes of miR143#12, we searched for antioxidant-related genes in the miRNA database (https://www.mirbase.org). We found that the expression levels of cyclooxygenase-1 (COX-1) and -2 were elevated from 18 up to 24 h after the exposure to H2O2, but were down-regulated 24 h after the transfection with miR143#12, which was estimated by a Western blot analysis (Fig. 5A). The mRNA levels of COX-1 and -2 simultaneously decreased in the same samples (Fig. 5B). Furthermore, ATG7 (E1 enzyme), which regulates the initiation of autophagy, was down-regulated at the mRNA and protein levels (Fig. 5A,B). These results suggested that the down-regulation of oxidative stress and silencing of ATG7 enabled miR143#12-transfected cells to evade autophagic cell death, leading to regrowth. siRNAs for COX-1 and -2 may also have induced regrowth significantly more in cells with reduced viability after their exposure to H2O2 than in control cells (Fig. 5C). In addition, the luciferase reporter assay targeting COX-1 and -2 by miR143#12 was performed, and their activities were found to be decreased by the ectopic expression of miR143#12 in H9c2 cells, but were partly canceled in cases with a mutated binding site at 3’UTR in COX-1 and COX-2 mRNAs (Fig. 5D).

Expression profiles of miR143#12-silenced genes after transfection in H2O2-exposed cells. (A) Time-dependent protein expression of COX-1, COX-2, and ATG7 examined by a Western blot analysis. The band intensity of each protein in the same blot was indicated as a value assessed using densitometry (ImageJ: version 2.0.0-rc-69/1.52p, NIH). Densitometric values below the bands are the ratios of protein/β-actin calculated with the non-treated sample as 100. NT: No treatment, C: Control miRNA (5 nM). (B) The expression levels of COX-1 and -2 mRNA after transfection with miR143#12 (#12), estimated by qRT-PCR (n = 3 in each). Results are shown as the mean ± SE. **p < 0.01, ***p < 0.001; 1-way ANOVA with Dunnett’s post hoc test. (C) The silencing of COX-1 and COX-2 by siRs-COX-1 or COX-2 after exposure to H2O2 increased the viable cell rate more than that by control miRNA (n = 4 in each). Result is shown as the mean ± SE. **p < 0.01, ***p < 0.001; 1-way ANOVA with Tukey’s post hoc test. (D) miR143#12 (#12) bound to COX-1 and COX-2 mRNAs, as shown by the luciferase assay. The 3′UTR region of human COX-1 or COX-2 mRNA and the predicted binding site for miR143#12 (the sequence 4515–4521 or 1585–1591 for the seed sequence, respectively) were inserted into a pMIR-REPORT Luciferase miRNA Expression Reporter Vector (wild). The mutant-type pMIR vector was inserted with the mutated seed sequence (from CTGC to AGTG) for miR143#12. Luciferase activities were measured after co-transfection with control RNA (C) (10 nM) or miR143#12 (10 nM) and the wild-type or mutant pMIR vector (n = 4 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001; 2-tailed, unpaired Student’s t test.
We then investigated whether COX-1, COX-2, and ATG7 were the target genes of miR143#12 in cardiac tissue samples 24 h after AMI in the rat model. Difficulties were associated with examining the target genes of miR143#12 because of marked decreases in gene expression levels in infarct tissues, which differed from those in the in vitro experiment. As shown in Fig. 6A, the Western blot analysis indicated that COX-1, COX-2, and ATG7, which were up-regulated in the in vitro study, were up-regulated in infarct areas in the control and miR143#12 groups. β-actin was also up-regulated. Importantly, this result indicated the validity of examining target genes in in vitro experiments on H2O2 exposure. We attempted to identify which protein was suitable as an internal control, but were unsuccessful because of differences in the effects of miR143#12 on infarct areas between both groups; therefore, we compared expression profiles from the viewpoint of protein levels. The expression of GAPDH and HPRT, which are essential metabolic enzymes and potential biomarkers for viable cells, was markedly down-regulated in both infarct areas (Fig. 6A). These expression levels were significantly higher in infarct areas in the miR143#12 group than in the control group from the viewpoint of the ratio of expression levels in the infarct area to the non-infarct area, which indicates the efficacy of miR143#12 in infarct areas even 24 h after AMI (Fig. 6B), as shown by the expression level of GAPDH in H9c2 cells (Fig. 6A). Based on these results, we defined the ratio of the expression level in infarct areas to that in non-infarct areas in the control as 1 in each case (Fig. 6B). To assess degree of decreases in the expression levels of COX-1, COX-2, and ATG7 following the treatment with miR143#12, we compared the ratios of COX-1, COX-2, and ATG7 expression levels to up-regulated metabolic enzymes. The relative ratios of COX-1, COX-2, and ATG7 expression levels to those of GAPDH or HPRT were lower in the miR143#12 group than in the control group (Fig. 6A,B). These results indicated the potential of COX-1, COX-2, and ATG7 as target genes of miR143#12 in rat AMI models. On the other hand, COX-1 and COX-2 mRNA levels in infarct areas in both groups were markedly increased (Fig. 6C), which was consistent with the results obtained on protein levels, as shown in Fig. 6A. The ratio of the expression level of COX-2 mRNA in infarct areas to non-infarct areas appeared to be lower in the miR143#12 group than in the control group due to RNA interference (Fig. 6C, right column). These results showed that miR143#12 exerted therapeutic effects on rat AMI by silencing COX-1, COX-2, and ATG7 in the rat AMI model, similar to the results of the in vitro experiment.

Examination of miR143#12 target genes in the rat AMI model. (A) Western blot analysis of COX-1, COX-2, and ATG7, which were identified as target genes of miR143#12 (#12) in the in vitro experiment and others in tissue samples from AMI rats. Tissue sampling was performed 24 h after AMI. The protein samples were extracted from the typical infarct and non-infarct tissues in each case of control miRNA (9 mg/kg) or miR143#12 (9 mg/kg) treated group. Western blot analysis of GAPDH in H2O2-exposed H9C2 cells was shown as a reference. (B) Band intensities with the same amount of protein loaded were evaluated by a densitometric analysis and the expression ratio of the infarct to non-infarct area in each protein was evaluated by the intensity value in each case. The ratios in miR143#12 cases were compared to those in control cases, which were defined as 1 (n = 4–7 in each). (C) The ratios of the mRNA expression levels of COX-1 or COX-2 to GAPDH evaluated by qRT-PCR in the same samples as in (A) were also shown. The ratios in miR143#12 cases were compared to those in control cases, which were defined as 1 (n = 4–7 in each). Results are shown as the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001; 2-tailed, unpaired Student’s t test.
To compare the transfection efficiency of cells exposed to H2O2 with those not exposed to H2O2, we transfected H2O2-exposed cells with FITC-labeled oligo double-stranded RNAs. As shown in Fig. 7A,B, H2O2-exposed cells showed stronger FITC signals than those in cells not exposed to H2O2 18 h after transfection. This result indicated that the transfection efficiency of H2O2-exposed cells to double-stranded RNAs was higher than that of non-exposed cells, which suggested the selectivity of introducing miRNA/liposomes (lipoplexes) into cells in the infarct area in the in vivo experiment.
Interestingly, tube formation was significantly greater by cultured HUVEC transfected with 5 nM of miR143#12 (15.8 ± 0.2/mm2) than by those transfected with 5 nM control miRNA (9.2 ± 0.2/mm2) (Fig. 7C), which was consistent with the result of in vivo experiment in Fig. 2C.

Transfection efficiency of double-stranded oligo RNAs in cells with or without H2O2 exposure, and miR143#12 induced increases in the number of tubes after its transfection into HUVEC. (A) The transfection of FITC-labeled BLOCK-IT was performed 1 h after exposure to H2O2 and observations were conducted 18 h after exposure. (B) FITC-labeled dots were detected in a cell. (C) Tube formation assay for HUVEC after the treatment with miR143#12 or control miRNA. (n = 4 in each). Results are shown as the mean ± SE. ***p < 0.001; 2-tailed, unpaired Student’s t test. ***p < 0.001. The representative figures were shown. NT: No treatment, C: Control miRNA (5 nM).
Discussion
Chemically modified miR143#12 significantly prevented expansion of infarct sizes and improved LV function 2 weeks after AMI in rat AMI model, which was accomplished by silencing COX-1, -2, and ATG7. Interestingly, miR143#12 induced neoangiogenesis, which was shown by the increased number of CD31-positive capillaries in the infarct border areas in miR143#12 AMI rats and promoted tube formation in cultured HUVEC cells. These effects of miR143#12 were expected from the study of miRs143 and 145 knock out mouse8.
From such a point of view, the expression levels of miR143#12 were markedly decreased in infarct areas in AMI rats (Fig. 3B,C), which situation is similar to that of miR143 knock-out mouse. Therefore, the replacement of miR143#12 may theoretically protect the damaged heart because increased expression levels of COX-1 and COX-2 in infarct areas caused by the down-regulation of miR143 elevated ROS levels. MiR143#12 blocked this vicious cycle toward increase in ROS level in both in vitro and in vivo experiments.
The expression of GAPDH and HPRT metabolic enzymes was up-regulated in the miR143#12 group based on the expression ratio of infarct to non-infarct areas. This result indicated the effectiveness of miR143#12 against AMI 24 h after its administration. Nevertheless, COX-1 and -2 and ATG7 expression levels, which increased in infarct areas, were lower in the miR143#12 group than in the control miRNA group based on the ratios of their expression levels in infarct areas to non-infarct areas (Fig. 6A,B).
In the rat AMI model, LVEF and LVFS, parameters of cardiac function, significantly improved in the 9 μg/kg of miR143#12 group, but not in the 3 μg/kg of miR143#12 group. In the 9 μg/kg of miR143#12 group, LVEF and LVFS (indicators of LV systolic function) significantly improved in rats and rabbits, while +dP/dt (an indicator of LV systolic function) and −dP/dt (an indicator of diastolic function) improved in rabbits, suggesting that the post-infarct intravenous injection of 9 μg/kg of miR143#12 improved cardiac function (Fig. 1). Therefore, 9 μg/kg of miR143#12 may be an optimal dose. In the rabbit AMI model, LVDd and LVDs were significantly smaller in the miR143#12 group than in the control miRNA group at 2 weeks, suggesting that miR143#12 may attenuate LV remodeling in terms of echocardiology. One possible explanation for the discrepancy in the effects of miR143#12 on LVDd and LVDs between rats and rabbits may be differences in the initial infarct size between rats and rabbits. Since infarct sizes were larger in rabbits than in rats, LV remodeling was accelerated while it was not accelerated in AMI rats, because infarct sizes are closely associated with LV dilatation14.
In the rat AMI model, the number of both CD31- and α-smooth muscle-positive vessels in the infarct border areas was significantly higher in the miR143#12 group than that in the control miRNA group, suggesting that miR143#12 promoted the regeneration of microvessels and larger vessels with vascular smooth muscle. Furthermore, in mouse subcutaneous tumor model topical administration of miR143#12 on the tumor induced many vessels immediately beneath the injection (Fig. S2). In our preliminary study, we showed that miR143#12 promoted sprouting angiogenesis in a blood vessel-on-a chip 3D model. This is consistent with previous findings demonstrating that miR143 promoted angiogenesis in mouse bone marrow endothelial cells15. Similarly, exosomes, which contained miR143 secreted by H9c2 cells cultured under ischemic conditions, were found to stimulate the formation of capillary-like structures when added to endothelial cells16. In cultured HUVEC in the present study, miR143#12 accelerated tube formation, suggesting that it induces neoangiogenesis. The infarct border area may have been supplied with greater blood flow through angiogenesis in the miR143#12 group than in the control miRNA group. This angiogenic effect of miR143#12 may have, at least in part, contributed to the prevention of infarct expansion and improvements in LV function. MiRs143 and 145 cooperatively suppressed the expression levels of transcription factors including Klf4, myocardin, and Elk-1 to promote differentiation and repress proliferation of vascular smooth muscle7. Mice lacking both miR143 and miR145 which are processed from the same primary miRNA did not show overt abnormality in smooth muscle cell differentiation, however, neointima formation in response to vascular injury was considerably impeded8. Furthermore, the tissue reconstitution after intestine epithelial injury by dextran sulfate sodium was profoundly disturbed in miR143/145 deficit mice17. Thus, miR143 could contribute to tissue regeneration by neoangiogenesis.
AMI caused by occlusion of coronary artery and reperfusion produces ROS18. The in vitro study indicated that miR143#12 targeted COX-1 and -2, leading to decreases in ROS levels in transfected cells. These results are supported by previous findings showing that the up-regulation of miR143-3p attenuated oxidative stress in cardiomyocytes19. ROS production due to exposure to H2O2 increased the expression of COX-1 and -2, which resulted in further increases in ROS levels. This COX-ROS axis establishes a positive feedback loop of ROS production20. Excessive ROS causes oxidative stress and induces cell death, such as apoptosis and autophagy, in cardiovascular diseases20,21. A comparison of the recovery rate of viable cells exposed to H2O2 between the transfection of miR143#12 and siRs-COX-1 or COX-2 revealed that COX-1 and COX-2 are major target genes that prevent autophagic cell death by reducing ROS levels (Fig. 5C,D). The COX-2 inhibitor, parecoxib, was previously reported to exert protective effects against AMI by regulating antioxidant and anti-inflammatory mechanisms in rat models22. There were a few reports that miR143 silenced COX-2 in cancer cells23,24.
The electron microscopic analysis revealed that ultrastructural autophagic cell death in cardiomyocytes was frequently observed 2 weeks after AMI in both models. Immunostaining demonstrated that miR143#12 seemed to inhibit autophagic cell death in cardiomyocytes induced by AMI. Furthermore, the in vitro study showed that cultured H9c2 cells exposed to H2O2, which causes autophagic cell death, decreased the number of viable cardiomyocytes and the LC3B transition from I to II was prevented by transfection with miR143#12. Therefore, the inhibitory effects of miR143#12 on autophagic cell death by reducing ROS level may have contributed to reductions in infarct sizes. Our recent research on a malnutritional state in H9c2 cells indicated longer survival after the ectopic expression of miR143#12.
In the present study, miR143#12 lipoplexes were selectively taken into cells in the affected areas (Fig. 7A,B). Furthermore, ISH results (Fig. 3C) suggested the following: (1) the expression of miR143 was markedly lower in the infarct area than in the non-infarct area in all groups, (2) more cells survived in the infarct area in the miR143#12 group, and (3) endothelial cells were maintained and may proliferate in the infarct area in the miR143#12 group.
In conclusion, replacement treatment with miR143#12 reduced myocardial infarct sizes and improved cardiac function by inhibiting the processes that contribute to autophagic cell death, targeting the oxidative stress-related genes COX-1, -2 and ATG7, and partly by promoting neovascularization. miR143#12 has potential as a novel strategy for the treatment of AMI. In the near future, understanding the mechanism of neoangiogenesis by miR143#12 will be needed and it is currently progress.
Materials and methods
The authors declare that all data that support the results of this study are available within the article and its Online Data Supplement. The study protocol was approved by the Ethical Committee of Gifu University School of Medicine, Gifu, Japan (permission number: 2019-257, 2020-146). Experiments were performed according to the ARRIVE Guidelines (https://www.nc3rs.org.uk/arrive-guidelines).
In vivo study
Surgical preparation of the rat AMI model
All male rats received human care in accordance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH publication 85-23, revised 1996). We prepared a rat AMI model by coronary ligation for 30 min followed by reperfusion as previously reported9. Male Sprague-Dawley rats weighing between 300 and 370 g (Jackson Laboratory, Japan Inc.) 10–12 weeks of age were anesthetized by the inhalation of 2% isoflurane (Mylan Inc., Tokyo, Japan) and then ventilated with a volume-cycled small-animal respirator (SN-480-7, Shimano Manufacturing Co., Ltd., Tokyo, Japan). The heart was exposed through left thoracotomy, and the left coronary artery was occluded with a Prolene 6-0 suture and reperfused after 30 min of ligation. Myocardial ischemia was confirmed by regional cyanosis and electrocardiographic changes in the ST elevation. We confirmed reperfusion by myocardial blush over the risk area and a decrease in the ST elevation after releasing the ligation. Positive end-expiratory pressure was applied to fully inflate the lungs, and the chest was closed in layers. Rats were added anesthetics when necessary to reduce the pain based on the ethics. Rats had free access to standard laboratory food and tap water. After 2 weeks, rats were euthanized by exsanguination under anesthesia by the inhalation of 2% isoflurane at the end of the study, and the heart was excised. In the rat AMI model, some rats were euthanized 24 h after AMI for a Western blot analysis and in situ hybridization (ISH). In the treatment with miRNA, 3 µg/kg or 9 µg/kg of miR143#12 or vehicle control miRNA (9 µg/kg) lipoplexes with cationic liposomes (lipoplexes: 1 mL of solution each) was intravenously administered via the tail vein 60 min after reperfusion and then, rats were followed up for 2 weeks. Twenty-one rats were randomly assigned to 3 µg/kg or 9 µg/kg of miR143#12 group (n = 7), and 9 µg/kg of control miRNA group (n = 7).
Surgical preparation of the rabbit AMI model
Male Japanese white rabbits (Japan LSC Inc., Shizuoka) (weighing ~ 2 kg) were used in the present study. To avoid hormonal effects, only male rabbits were used. AMI rabbits were randomly assigned by sealed envelopes to the vehicle or miR143#12 group. AMI was induced by ligating the coronary artery for 30 min followed by 14 days of reperfusion as previously reported10,25,26. Briefly, rabbits were anesthetized with an intravenous injection of ketamine (10 mg/kg) and xylazine (3 mg/kg), and when required, additional doses were given during the experiment. Rabbits were then intubated and ventilated with room air supplemented with a low flow of oxygen by a mechanical ventilator at a respiratory rate of 20 to 30/min and tidal volume of 25 to 35 mL (model SN-480-5, Shimano, Tokyo, Japan). Surgery was performed under sterile conditions. The carotid artery and jugular vein were cannulated and used to monitor peripheral arterial pressure and to administer anesthetics, respectively. Thoracotomy was performed at the left fourth intercostal space, and the heart was exposed after excising the pericardium. A 4-0 silk suture on a small, curved needle was passed through the myocardium beneath the middle segment of the large coronary arterial branch, which was in the middle segment of the anterolateral surface of the LV. Both ends of the silk suture were then passed through a small vinyl tube, and the coronary artery was occluded for 30 min by pulling the snare. The snare was fixed by clamping the tube with a mosquito hemostat. We confirmed myocardial ischemia by a ST segment elevation on ECG and regional myocardial cyanosis. We confirmed reperfusion by myocardial blush over the risk area and a decrease in the ST elevation after releasing the snare. The chest was then closed and rabbits were allowed to recover. Rabbits were added anesthetics when necessary to reduce the pain based on the ethics. Standard laboratory food and tap water were accessible to rabbits. Two weeks after surgery, rabbits were euthanized by an i.v. overdose of pentobarbital (150 mg/kg) at the end of the study. In the treatment with miRNA lipoplexes (1 mL solution each), 9 µg/kg of miR143#12 (miR143#12 group) (n = 8) or 9 µg/kg of control miRNA (control group) (n = 8) was intravenously administered via ear vein 60 min after reperfusion and then, rabbits were followed up for 2 weeks.
Measurement of infarct sizes
The hearts of rats and rabbits were excised, and the LV was sectioned into five (rat) and seven (rabbit) transverse slices parallel to the atrioventricular ring. The slice at the papillary muscle level was used to measure infarct sizes. Each slice was fixed in 10% buffered formalin for 4 h, embedded in paraffin, and cut into 4-μm-thick sections with a microtome. Transverse LV slices at the papillary muscle level were stained with Masson-Trichrome (SRL, Inc., Tokyo Japan). LV wall areas, infarct areas, and non-infarct areas were calculated using image analysis software (Win ROOF, version 7.4, Mitani Corporation, Tokyo, Japan) connected to a light microscope (BZ-8000, KEYENCE, Osaka). Infarct sizes were calculated as the infarct area/LV wall area × 100 (%). The infarct size was obtained within 7 days after completion of Masson-Trichrome staining.
Cardiac function
In the rat AMI model, echocardiography was performed to measure cardiac function on day 14 after AMI. The LV ejection fraction (LVEF), LV fractional shortening (LVFS), LV end-diastolic dimension (LVDd), LV end-systolic dimension (LVDs), and heart rate (HR) were measured by transthoracic echocardiography (SSA-660A, Xario, Canon Medical Systems Corporation, Tokyo, Japan) with a 7.5-MHz sector scan probe.
In the rabbit AMI model, on day 14 after AMI, arterial blood pressure and HR were measured via a catheter introduced into the carotid artery. Echocardiography (SSD2000, Aloka Co., Ltd., Tokyo, Japan) was performed and LVEF, LVFS, LVDd, and LVDs were measured. A micromanometer-tipped catheter (SPR 407, Millar Instruments, Edwards Lifesciences LLC, CA, USA) was inserted into the carotid artery and LV, and we recorded blood pressure, HR, and ±dP/dt. The reproducibility of LVEF measurements by echocardiography was confirmed by assessing the same measurement three times (the average coefficient of variation was 0.00728 in rats, n = 5, and 0.02908 in rabbits, n = 5).
Sample collection for biochemical analysis
Tissue samples in AMI animals were taken from non-infarct (remote) and infarct border (ischemic) areas where were relatively close to the non-infarct areas.
Electron microscopic observations
Hearts (n = 3 in each group) were excised 2 weeks after AMI. Cardiac tissue was quickly cut into 1-mm-thick cubes, immersion-fixed with 2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4) at 4 °C overnight, and then postfixed in 1% buffered osmium tetroxide. Specimens were then dehydrated through a graded ethanol series and embedded in epoxy resin. Ultrathin sections (thickness of 90 nm) double-stained with uranyl acetate and lead citrate were examined under an electron microscope (H-800, Hitachi, Tokyo, Japan). The electron microscopic analysis was directed at the infarct border area.
Immunohistochemistry
After the deparaffinization of rat cardiac tissue embedded in paraffin, 4-μm-thick sections were stained. The primary antibodies used were an anti-CD31 rabbit monoclonal antibody (Abcam ab182981: 0.2 µg/mL), anti-α-smooth muscle actin rabbit antibody (Abcam ab5694: 1:2,000) and anti-LC3B mouse monoclonal antibody (Cell Signaling Technology [CST], 0.2 µg/mL). The secondary antibodies used were goat anti-chicken IgY H&L (Alexa Fluor 488, Abcam) and polyclonal goat anti-rabbit immunoglobulins/biotinylated (Dako E0432, 1:600). CD31-positive vessels (C: 14 images, miR143#12: 14 images) and α-SMA-positive vessels (C:15 images, miR143#12: 17 images) were observed in the infarct border area. From 4 animals in each group, 4 sections from each animal were stained with CD31 or α-smooth muscle actin.
Measurement of plasma and cardiac tissue miR143 levels after AMI
Total circulating RNA was extracted from plasma samples using the NucleoSpin® miRNA Plasma kit (MACHEREY-NAGEL, Düren, Germany). A 300-µL volume of the sample was centrifuged at 11,000×g for 3 min to remove residual cell debris. After protein precipitation and adjustments to binding conditions with isopropanol, RNAs in the solution were bound to a miRNA collection column and eluted into 10 μL of RNase-free water. The same amount of the plasma sample (300 μL) was used in each extraction to normalize the yield of RNA. Purified miRNA was immediately used for the subsequent RT process to prevent degradation. The expression of miR143-3p (Assay ID: 002249) was assessed by performing TaqMan MicroRNA Assays (Applied Biosystems) with a THUNDERBIRD probe qPCR Mix (TOYOBO, Osaka, Japan) and the Thermal Cycler Dice Real-Time System II (TaKaRa, Otsu, Japan). Relative expression was normalized to miR16-5p (Assay ID: 000391, Applied Biosystems) by the 2−ΔCt method. Each measurement was performed in triplicate. RNA integrity was assessed by measuring circulating miR16, which is one of the most abundantly and stably expressed miRNAs in plasma, and its levels may reflect the degree of degradation and quality of miRNAs27,28.
In cardiac tissue, the collected tissue samples were quickly sonicated in the lysis buffer containing RNase inhibitor (MACHEREY-NAGEL) before RNA extraction. Total RNA was extracted from the samples using NucleoSpin miRNA (MACHEREY-NAGEL) according to the standard protocol. Relative expression was normalized to the RNU6B endogenous control (AB Assay ID 001093, Applied Biosystems) by the 2−ΔCt method. Each measurement was performed in triplicate.
ISH for miRNA 24 h after AMI
ISH for miRNA was performed using a hsa-miR143-3p miRCURY LNA miRNA Detection probe (YD00610001, QIAGEN, Hilden, Germany) and miRCURY LNA miRNA ISH Buffer and Controls (339459, QIAGEN, Hilden, Germany). All solutions were prepared under RNase-free conditions. Snap-frozen samples with OCT Compound (Sakura Finetek Japan, Tokyo, Japan) were sectioned at a thickness of 10 µm and fixed for 20 min in 4% neutral-buffered paraformaldehyde. Slides were incubated at 51 °C for 1 h on a Dako hybridizer (Dako, Agilent Technologies, CA, USA) with 50 µL of the probe at 40 nM. After 3 washes at 51 °C for 10 min with 0.2× saline-sodium citrate (SSC) buffer (UltraPure™ SSC 20×, 15557-044, Thermo Fisher Scientific, MA, USA), sections were blocked for 10 min with 2.5% normal horse serum (S-2012-50, Vector Laboratories, CA, USA) and incubated at room temperature for 60 min with 200 µL of anti-DIG-AP antibody solution (11093274910, Roche, Basel, Switzerland, 1:400). Sections were incubated at 30 °C for 2 h with NBT/BCIP solution (SIGMAFAST™ BCIP®/NBT, B5655-5TAB, Sigma-Aldrich, MA, USA) supplemented with levamisole (0.2 mM, 31742-250MG, Sigma-Aldrich, MA, USA). Slides were counterstained for 1 min using Nuclear Fast Red (H-3403, Vector Laboratories, CA, USA). Sections were dehydrated by passaging through a graded series of alcohol, cleared in lemosol, and mounted with MOUNT-QUICK (Daido Sangyo, Tokyo, Japan).
Cell culture
H9c2 cells (rat cardiomyoblast cells) and human umbilical vein endothelial cells (HUVEC) were obtained from the Cell Bank of ATCC and the Thermo Fisher, Scientific (A1460901), respectively, and grown at a density of 6–9 × 104 cells/cm2 as a monolayer. H9c2 cells were cultured in Dulbecco’s modified Eagle medium (GIBCO, Cat. No. 11960-077, USA) supplemented with 10% v/v fetal bovine serum (Nichirei Biosciences Inc., Cat. No.174012) under an atmosphere of 5% CO2 saturated with water vapor at 37 °C. Medium was replaced with fresh medium twice a week. HUVEC from a single donor were cultured in Medium 200 (M-200-500, GIBCO, USA) supplemented with large vessel endothelial supplement (LVES; A14608-01, GIBCO). The transfection efficiencies of double-stranded RNA in both cell lines were more than 80%.
Cell viability assay and H2O2 exposure
The number of viable cells was assessed by performing the trypan blue dye exclusion test (GIBCO™, Thermo Fisher). The number of viable cells was counted at 120 h after exposure to H2O2 at a concentration of 37–38 μM.
Cell transfection with double-strand RNAs
H9c2 rat cardiomyoblasts for transfection with miR143#12, a synthetic chemically modified miR143 derivative, and others were seeded in the wells of six-well plates at a concentration of 6–7 × 104 cells/well on the day before transfection. The mature type of control miR (control), hsa-miR143-3p (wild type: mirVana miRNA mimic; Ambion, Foster city, CA), miR143-3p inhibitor (anti-miR143: Ambion), miR143#12 (F/Ome type), or siR-COX-1 or siR-COX-2 (Invitrogen, Carlsbad, CA) was used for the transfection of cells, which was achieved using cationic liposomes with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA), according to the manufacturer’s lipofection protocol.
The miR143 wild-type sequence was S: 3′-GGUCUCUACGUCGUGACGUGGAGU-5′, AS: 5′-UGAGAUGAAGCACUGUAGCUCAGG-3′.
The miR143#12 sequence was S: 3′-GGUCUCUACGUCGUGACGUGGAGU-5′, AS: 5′-U^G^AGAUGAAGCACUGUAGCUCA^dT^dT-3′.
The siR-COX-1 sequence targeted 5′-AUCCCAUGUAGGAUGCUCCUCCUUC-3′.
The siR-COX-2 sequence targeted 5′-UUUCCUUUCACACCCAUGGGAGUUG-3′.
The non-specific control miRNA (HSS, Hokkaido, Japan) sequence was S: 5′-GUAGGAGUAGUGAAAGGCC-3′, AS: 5′-GGCCUUUCACUACUCCUAC dtdt-3′, which was used as a control for non-specific effects. Transfection was performed 1 h after exposure to H2O2. The concentration of control RNA used was the highest dose of miRNA or siRNA transfected in each experiment. BLOCK-iT™ Fluorescent Oligo (Invitrogen) was used to assess transfection efficiency in cells exposed to H2O2.
Immunoblot
To prepare cell lysates, H9c2 cells treated for 18 and 24 h were lysed in ice-cold RIPA buffer. Tissue samples were sonicated in 1% SDS buffer (Wako). These lysates were then centrifuged at 13,000×g for 20 min. The protein concentrations of the supernatants were measured using the DC Protein Assay Kit (Bio-Rad, CA, USA). Protein samples (1 µg/lane) were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins in the gel were transferred to 0.45-µm polyvinylidene fluoride membranes (Immobilon-P Membrane, EMD Millipore, MA, USA). These membranes were blocked with PVDF Blocking Reagent for Can Get Signal (TOYOBO, Osaka, Japan) for 1 h, incubated at 4 °C overnight with the primary antibodies at a 1:2000 dilution, and washed 3 times with Tris-buffered saline containing 0.1% Tween 20 (TBST). They were then incubated with a HRP-conjugated sheep anti-mouse or donkey anti-rabbit IgG antibody (CST Inc., Danvers, MA, USA) at room temperature and washed 3 times with TBST. Immunoblots were visualized using the Luminata Forte Western HRP substrate (EMD Millipore)29. The band intensity of each protein was indicated as a value assessed using Densitometry (ImageJ: version 2.0.0-rc-69/1.52p, NIH). Densitometric values are the ratios of protein/β-actin calculated with the non-treated sample as 100. The primary antibodies used were as follows: anti-COX2 (#12282), anti-GAPDH (#2118), and LC3B (#3868; CST); anti-α-Tubulin (#PM054; MEDICAL & BIOLOGICAL LABORATORIES Co., Ltd., Tokyo, JP); anti-α-actin (SC-32251) and HPRT (SC-376938; Santa Cruz Biotechnology, Santa Cruz, CA, USA); and anti-COX1 (ab109025) and anti-ATG7 antibodies (ab133528; Abcam, Cambridge, UK). The amount of protein loaded as the control was assessed with an anti-β-actin mouse monoclonal antibody (clone AC-74, #A5316, Sigma-Aldrich). Anti-rabbit IgG (#7074) and anti-rabbit IgG, HRP-linked (#7076; CST) antibodies were used as secondary antibodies.
RNA extraction and a miRNA reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA from cell pellets after centrifugation was extracted using the NucleoSpin® miRNA kit (MACHEREY-NAGEL, Düren, Germany). The concentration and purity of RNA were assessed by UV spectrophotometry. Twenty-five nanograms of total RNA was reverse transcribed to cDNA using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Thermo Fisher Scientific). The expression of miR143-3p (Assay ID: 002249) was then assessed by performing TaqMan MicroRNA Assays (Applied Biosystems) with the THUNDERBIRD probe qPCR Mix (TOYOBO, Osaka, Japan) and Thermal Cycler Dice Real-Time System II (TaKaRa, Otsu, Japan). Relative expression was normalized to the RNU6B endogenous control (AB Assay ID 001093, Applied Biosystems) by the 2-ΔCt method. Each measurement was performed in triplicate.
Luciferase reporter assay
Using the TargetScan 7.2 database (http://www.targetscan.org/), we identified algorithm-based binding sites for miR143-3p in the 3′ UTR of Ptgs1 (Cox1) or Ptgs2 (Cox2) mRNA at position 4515–4521 or 1585–1591, respectively. The sequence region containing the putative miR143-3p-binding sequence was inserted into the pMIR-REPORT™ Luciferase MIRNA Expression Reporter Vector (Applied Biosystems) according to the manufacturer’s protocol. Other pMIR constructs encompassing the mutated seed sequence of miR143-3p (wild type, CAUCUCA; mutant, CAAGACA) were created using the PrimeSTAR® Mutagenesis Basal Kit (TaKaRa). Vector mutations were confirmed by a sequence analysis. The pRL-TK Renilla Luciferase Reporter vector (Promega) was used as an internal control vector. H9c2 cells were seeded the day before transfection at a concentration of 0.5 × 104 per well in 96-well plates. Cells were co-transfected with the reporter vector (0.01 μg each/well) and 10 nM miR143#12 or non-specific siRNA (Dharmacon, Tokyo, Japan). Luciferase activities were measured 36 h after co-transfection using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol. Luciferase activities were reported as the firefly luciferase/renilla luciferase ratio.
Detection of oxidative stress in cells
CellROX® Green Reagent (Thermo Fisher, Japan) is a novel fluorogenic probe for measuring oxidative stress in live cells. The cell-permeant dye is weakly fluorescent while in a reduced state and exhibits bright green photostable fluorescence upon oxidation by reactive oxygen species (ROS) and subsequent binding to DNA. Cells exposed to H2O2 were transfected with MIR143#12, treated with CellROX green reagent (5 μM) at 37 °C for 30 min, and then fixed with 3.7% formaldehyde for 15 min. Signals were observed by fluorescent microscopy.
Transfection efficiency of double-stranded RNA in H2O2-exposed cells
BLOCK-iT™ Fluorescent Oligo was transfected into H2O2-exposed cells with Lipofectamine RNAiMAX at a concentration of 10 nM. After transfection, the incorporated orange signal of BLOCK-iT™ Fluorescent Oligo was observed 18 h after transfection by fluorescent microscopy.
Tube formation assay on HUVEC
Tube formation assays were performed using the Angiogenesis Starter Kit (A1460901, Thermo Fisher Scientific) according to the manufacturer’s protocol. HUVEC were cultured in Medium 200 (M-200-500, GIBCO, USA) supplemented with LVES (A14608-01, GIBCO). LVES contained fetal bovine serum, hydrocortisone, human epidermal growth factor, basic fibroblast growth factor, heparin, and ascorbic acid. HUVEC were seeded at a concentration of 1.5 × 105 cells/dish in 6-cm dishes the day before the transfection of control miRNA or miR143#12, and control miRNA or miR143#12 was transfected at a concentration of 5 nM the next day. Cells were re-seeded 48 h after transfection into 24 wells coated by the Geltrex LDEV-free reduced growth factor basement membrane matrix (A14132-02, GIBCO) and assayed.
Normality testing and statistical analysis
Each result was obtained from more than 3 independent experiments which data was performed normality testing and the representative results among the experiments were shown. Values are given as the mean ± SE. The significance of differences between the 2 groups was assessed by a paired or unpaired parametric Student’s t-test. The significance of differences among more than 3 groups was evaluated by a one-way ANOVA followed by Dunnett’s or Tukey’s post hoc test. A difference of p < 0.05 was considered to be significant. Differences of p < 0.01, p < 0.001, and p < 0.0001 were considered to be highly significant. All statistical analyses were performed using GraphPad Prism7.
Data availability
Data that support the present results are available from the corresponding author upon reasonable request. Some data may not be made available due to privacy or ethical restrictions.
References
Pfeffer, M. A. & Braunwald, E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81, 1161–1172. https://doi.org/10.1161/01.cir.81.4.1161 (1990).
Kasama, S. et al. Prognostic value of cardiac sympathetic nerve activity evaluated by [123I]m-iodobenzylguanidine imaging in patients with ST-segment elevation myocardial infarction. Heart 97, 20–26. https://doi.org/10.1136/hrt.2010.204149 (2011).
Chew, D. S. et al. Change in left ventricular ejection fraction following first myocardial infarction and outcome. JACC Clin. Electrophysiol. 4, 672–682. https://doi.org/10.1016/j.jacep.2017.12.015 (2018).
Bolognese, L. et al. Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications. Circulation 106, 2351–2357. https://doi.org/10.1161/01.cir.0000036014.90197.fa (2002).
Ambros, V. The functions of animal microRNAs. Nature 431, 350–355. https://doi.org/10.1038/nature02871 (2004).
Sun, T. et al. The role of microRNAs in myocardial infarction: from molecular mechanism to clinical application. Int. J. Mol. Sci. 18. https://doi.org/10.3390/ijms18040745 (2017).
Cordes, K. R. et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460, 705–710. https://doi.org/10.1038/nature08195 (2009).
Xin, M. et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 23, 2166–2178. https://doi.org/10.1101/gad.1842409 (2009).
Hayakawa, K. et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation 108, 104–109. https://doi.org/10.1161/01.CIR.0000074225.62168.68 (2003).
Higashi, K. et al. MicroRNA-145 repairs infarcted myocardium by accelerating cardiomyocyte autophagy. Am. J. Physiol. Heart Circ. Physiol. 309, H1813–H1826. https://doi.org/10.1152/ajpheart.00709.2014 (2015).
Satake, A. et al. Plasma microRNA-143 levels increase in the acute phase and may contribute to improvement of the cardiac function in the chronic phase in patients with acute myocardial infarction. Circulation (2022).
Akao, Y. et al. Impairment of K-Ras signaling networks and increased efficacy of epidermal growth factor receptor inhibitors by a novel synthetic miR-143. Cancer Sci. 109, 1455–1467. https://doi.org/10.1111/cas.13559 (2018).
Sugito, N., Heishima, K. & Akao, Y. Chemically modified MIR143-3p exhibited anti-cancer effects by impairing the KRAS network in colorectal cancer cells. Mol. Ther. Nucleic Acids 30, 49–61. https://doi.org/10.1016/j.omtn.2022.09.001 (2022).
Chareonthaitawee, P., Christian, T. F., Hirose, K., Gibbons, R. J. & Rumberger, J. A. Relation of initial infarct size to extent of left ventricular remodeling in the year after acute myocardial infarction. J. Am. Coll. Cardiol. 25, 567–573. https://doi.org/10.1016/0735-1097(94)00431-O (1995).
Wang, R. et al. miR-143 promotes angiogenesis and osteoblast differentiation by targeting HDAC7. Cell Death Dis. 11, 179. https://doi.org/10.1038/s41419-020-2377-4 (2020).
Ribeiro-Rodrigues, T. M. et al. Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc. Res. 113, 1338–1350. https://doi.org/10.1093/cvr/cvx118 (2017).
Chivukula, R. R. et al. An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration. Cell 157, 1104–1116. https://doi.org/10.1016/j.cell.2014.03.055 (2014).
Granger, D. N. & Kvietys, P. R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 6, 524–551. https://doi.org/10.1016/j.redox.2015.08.020 (2015).
Song, Y., Cai, W. & Wang, J. Upregulation of miR-143-3p attenuates oxidative stress-mediated cell ferroptosis in cardiomyocytes with atrial fibrillation by degrading glutamic-oxaloacetic transaminase 1. Biocell 45, 733–744. https://doi.org/10.32604/biocell.2021.013236 (2021).
Kumar, D. & Jugdutt, B. I. Apoptosis and oxidants in the heart. J. Lab. Clin. Med. 142, 288–297. https://doi.org/10.1016/S0022-2143(03)00148-3 (2003).
Morales, C. R., Pedrozo, Z., Lavandero, S. & Hill, J. A. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxid. Redox Signal. 20, 507–518. https://doi.org/10.1089/ars.2013.5359 (2014).
Wu, F. et al. Effect of parecoxib sodium on myocardial ischemia-reperfusion injury rats. Med. Sci. Monit. 27, e928205. https://doi.org/10.12659/MSM.928205 (2021).
Pham, H. et al. miR-143 decreases COX-2 mRNA stability and expression in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 439, 6–11. https://doi.org/10.1016/j.bbrc.2013.08.042 (2013).
Wu, X. L. et al. MicroRNA-143 suppresses gastric cancer cell growth and induces apoptosis by targeting COX-2. World J. Gastroenterol. 19, 7758–7765. https://doi.org/10.3748/wjg.v19.i43.7758 (2013).
Minatoguchi, S. et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation 109, 2572–2580. https://doi.org/10.1161/01.CIR.0000129770.93985.3E (2004).
Yamada, Y. et al. S1P–S1PR2 axis mediates homing of muse cells into damaged heart for long-lasting tissue repair and functional recovery after acute myocardial infarction. Circ. Res. 122, 1069–1083. https://doi.org/10.1161/CIRCRESAHA.117.311648 (2018).
Zhelankin, A. V. et al. Elevated plasma levels of circulating extracellular miR-320a-3p in patients with paroxysmal atrial fibrillation. Int. J. Mol. Sci. 21. https://doi.org/10.3390/ijms21103485 (2020).
Yamano, T. et al. Assessment of circulating microRNA specific for patients with familial adenomatous polyposis. PLoS One 16, e0250072. https://doi.org/10.1371/journal.pone.0250072 (2021).
Sugito, N., Heishima, K., Ito, Y. & Akao, Y. Synthetic MIR143-3p suppresses cell growth in rhabdomyosarcoma cells by interrupting ras pathways including PAX3-FOXO1. Cancers (Basel) 12. https://doi.org/10.3390/cancers12113312 (2020).
Acknowledgements
The present study was funded by Grants-in-Aid from the Japan Agency for Medical Research and Development under Grant Number JP21km0908001. We thank Mrs. Noriko Endo for her technical assistance. The authors declare that all data that support the results of this study are available within the article and its Online Data Supplement. The study protocol was approved by the Ethical Committee of Gifu University School of Medicine, Gifu, Japan (permission number: 2019-257, 2020-146).).
Author information
Authors and Affiliations
Contributions
Y.A., Shinya M, and H.O. performed data curation. Shingo M, K.H., Y.I., R.N. and N.S. performed the formal analyses. Shinya M and Y.A. raised the funds, supervised the project, and wrote the draft. Shingo M and NS were responsible for the majority of the analyses. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Minatoguchi, S., Sugito, N., Heishima, K. et al. Restoration effect of chemically modified microRNA-143-3p on acute myocardial infarction in animal models. Sci Rep 15, 1107 (2025). https://doi.org/10.1038/s41598-024-76429-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-76429-3
Keywords
This article is cited by
-
Advances in MicroRNA-Based Therapies for Cardiac Repair: Emerging Strategies for Treating Myocardial Infarction
Molecular and Cellular Biochemistry (2026)
