
Abstract
Spinal cord injury (SCI) is a profound affliction of the central nervous system that often remains inadequately addressed. Prior research has indicated that endoplasmic reticulum stress (ERS), associated with apoptotic signaling, plays a part in subsequent injuries post-SCI. However, the exact mechanisms are still unclear. ERS-related genes and SCI-associated datasets were sourced from the Genecard and GEO databases. We identified 68 ERSDEGs and pinpointed 6 marker genes vital for SCI diagnosis (CYBB, PRDX6, PTGS1, GCH1, TLR2 and PIK3CG) which were all upregulated in SCI based on bioinformatics and qRT-PCR. The nomogram exhibited that these genes could effectively predict the occurrence of SCI. Functional analysis revealed the potential roles of these genes was closely related to neuron cells and immune response. Immune infiltration research underscored the substantial roles of macrophage and CD56 dim NK cells in SCI. The ceRNA network analysis further revealed the complex interplay among marker genes, lncRNAs and miRNAs in SCI. We screened six marker genes with great diagnostic value, and found that these genes may affect the occurrence of SCI by affecting the immune response and recovery of neurons.
Similar content being viewed by others

Introduction
Spinal cord injury (SCI), results in profound impairment, incurring both financial and emotional burdens1. Annually, over 12,000 individuals suffer from spinal cord injuries in the U.S., leading to a rough count of 270,000 such incidents across North America2. Despite global efforts, present surgical and pharmacological treatments yield inadequate results, often leading to suboptimal patient recovery3. Hence, a comprehensive understanding of SCI’s pathological progression becomes indispensable for devising effective therapeutic strategies.
The underlying physiological processes of SCI can be segmented into two stages: an initial injury, mainly due to physical trauma to the spine, followed by a subsequent phase characterized by a series of cellular and biological reactions. The latter encompasses a myriad of events including hemorrhage, ischemia, oxidative stress, inflammation, neuronal death, demyelination, and scar formation. Recent single-cell transcriptome analyses highlight that the most significant changes post-SCI occur between days 3 and 14, marked by microglia activation and cellular transformations4. The initial injury triggers microglial activity in the vicinity, compromising the stability of the barrier between the blood and spinal cord. This attracts circulating immune cells like neutrophils, lymphocytes, and macrophages into the spine area. As these cells emit agents that are either inflammatory or modulatory in nature, they have a complex impact on immunity, typically hindering the recovery from SCI5,6. Nevertheless, the precise role of immunity, spanning from damage amplification to regenerative potential, remains a contentious topic.
The endoplasmic reticulum (ER), serving as a cellular hub, is actively involved in both protein production and calcium storage, and it participates in diverse cell signaling pathways, including those of lipid biogenesis and calcium metabolism7,8. Acute intrinsic responses within the spinal cord post-SCI involve ER stress activation9. Notably, the demise of neuronal cells post-SCI, pivotal to the ensued neurological deficits, is predominantly attributed to secondary biochemical perturbations rather than the immediate mechanical impact10. These biochemical aberrations, including protein misfolding, instigate the ER stress response. Prolonged and intensified ER stress can perturb ER functions, culminating in cell death11. However, the detailed mechanisms by which endoplasmic reticulum stress accentuates apoptosis during secondary injuries remain elusive.
To bridge this knowledge chasm, we deployed an array of bioinformatic techniques, ranging from differential expression analysis to molecular subtyping, aiming to discern robust biomarkers pertinent to SCI diagnosis. Concurrently, we discerned dual immune regulatory modalities in SCI and constructed a competing endogenous RNA (ceRNA) network. Key gene-immune cell interactions were explored, and validation was done using datasets from single cell RNA (scRNA)-seq for gene expression, with qPCR analysis for core genes. Our findings accentuate the significance of genes associated with ER stress in SCI evolution, proffering enhanced insights into the molecular immunological frameworks orchestrating SCI pathology.
Materials and methods
Data acquisition and preprocessing
The four mRNA datasets of SCI patients (GSE5296, GSE47681, GSE132242) were obtained from GEO database, including 23 SCI and 19 Sham samples. Besides, these SCI samples were all collected on day 3 post-SCI.
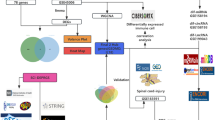
In instances where multiple probes matched a singular gene, we took the average expression, excluding the minimal values and outliers. To ensure uniformity across the datasets, we employed batch normalization with the assistance of “sva” package12. The integrity of the downloaded and processed data was corroborated through boxplots and principal component analysis (PCA) using “ggplot2” and “FactoMineR”. Separately, we retrieved 1193 ER-associated genes via the Genecard platform, with the condition that a relevance score exceeding 7. The study workflow is succinctly depicted in Fig. 1.

The normalization of datasets and identification of DEGs. (A) The boxplot of three datasets before normalization. (B) The boxplot of three datasets after normalization. (C) PCA plot of three datasets before normalization. (D) PCA plot of three datasets after normalization. (E) The volcano diagram showed the differentially expressed genes between SCI group and control group.
Differentially expressed genes (DEGs) identification
We initiated our analysis by extracting the expression matrix of genes related to ERS from the merged dataset, yielding 409 genes in common. Through the R package “limma”13, differentially expressed analysis was analyzed. Genes were deemed differentially expressed if they met the criteria of |logFC| > 1, and a significance level of p < 0.05. Visualization of these findings was facilitated using the “ggplot2” package.
Functional enrichment and pathway analysis
For a deeper understanding of the implications of the identified genes, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses with “ClusterProfiler” in R14. We set the significance thresholds at p < 0.05 and FDR < 0.25.
Biomarker identification and risk prediction modeling
To refine data dimensionality, we applied the LASSO method with the “glmnet” tool in R15. Concurrently, we employed the SVM-RFE model using the “SVM” package. The performance of model was evaluated based on mean misclassification rates across 10-fold cross-validations16. The overlapping genes recognized by both algorithms were considered optimal for SCI diagnosis. Besides, ROC curve was used to evaluate the diagnostic capabilities of our selected markers using “pROC” package.
Pathway analysis using single-gene gene set enrichment analysis (GSEA)
For clarification of pathways linked to the marker genes, we examined their relationships with the remaining genes using the GSEA algorithm. Subsequently, genes were ranked based on their correlation strengths, from highest to lowest, and this ranked list was utilized for the enrichment analysis. The KEGG pathway database was used as the reference to determine the enrichment levels within the gene set.
Assessment of immune cell infiltration in SCI
To evaluate the degree of immune cell infiltration in both SCI and control samples, the single sample GSEA (ssGSEA) method was employed. Then, an evaluation of the association among the identified genes and the infiltrated immune cells was performed based on Pearson correlation analysis using reshape2 package.
Elucidation of SCI-associated molecular subtypes
The consensus cluster plus analysis was used to determine the subtype of SCI using ConsensusClusterPlus package. Parameters for this analysis were set as follows: sample resampling ratio at 80%, number of resampling at 1,000, and a maximum of 9 clusters. By analyzing the cumulative distribution function (CDF), consensus matrix, and variations in the region beneath the CDF curve, the best cluster count was ascertained. Visualization of the molecular typing results was achieved using the pheatmap package.
Construction of the ceRNA and TF-mRNA network
In our research, a ceRNA framework was built encompassing mRNAs, miRNAs, and lncRNAs. MiRWalk(http://mirwalk.umm.uni-heidelberg.de/) was employed to forecast the targeted miRNAs for the chosen 6 mRNAs17. Subsequent identification of lncRNAs, acting as upstream regulators of miRNAs, was conducted with StarBase v2.0 18. Given that both databases include experimentally confirmed miRNA-target interactions, we ensured a robust network construct. The ceRNA framework was depicted with the assistance of Cytoscape (version 3.9.0).
Data for forming the TF-mRNA network was acquired through the TRRUST database (https://www.grnpedia.org/trrust/)19. Using the paired TFs and mRNAs, we juxtaposed the findings from RNA differential analysis, which yielded a unique TF-mRNA network. This network was subsequently visualized using the “ggplot2” and “ggalluvial” packages20 in R.
Single-cell dataset exploration and validation
The scRNA-seq count data was retrieved from GSE189070 for our exploration, comprising eight instances: seven from SCI at varying time points and one from an uninjured sample. We specifically chose GSM5694214 (from three days post-SCI) and GSM5694211 (uninjured) for our analysis. The Seurat R package21 was utilized for Quality Control (QC). Specific criteria led to the exclusion of cells: (a) fewer than 500 RNA counts or (b) mitochondrial gene expression ratios exceeding 5%. The Seurat package’s NormalizeData function was employed for data normalization. Subsequently, cell clusters were pinpointed using the FindClusters tool in Seurat (configured to a 0.5 resolution) and visualized through a 2D tSNE plot22. Using the SingleR package, we compared cells across clusters to a previously annotated reference dataset23. Cluster annotations were finalized after analyzing cell markers and assessing comparison outcomes.
Establishment of SCI mice model
Six male C57BL/6 mice were procured at the age of 6–7 weeks and accommodated in an environment that alternated between 12-hour day and night cycles, maintaining a temperature range of 20–23 degrees Celsius and a humidity level of 50%. All operational procedures adhered rigorously to the standards established by the National Institutes of Health. The mice were assigned randomly to two distinct groups: three underwent spinal cord injury surgery with a deliberate injury at the T10 segment, while the remaining three underwent a sham operation involving a laminectomy without spinal cord injury. The surgical procedure was meticulously executed as follows: the mice were anesthetized with 1% Isoflurane, sterilized using povidone-iodine, and a median dorsal incision was skillfully performed to expose the T10 segment of the spinal cord. Subsequently, the T10 laminectomy was executed to unveil the spinal cord. A spinal cord injury percussion device was then employed to deliver a blow at a height of 1 cm, utilizing a percussion rod weighing 10 g. Following the impact, both lower limbs of the mice exhibited involuntary twitching, and their tails demonstrated reflexive movements. Upon regaining consciousness from anesthesia, the mice experienced complete lower limb paralysis. The presence of dark red blood at the impact site served as an unequivocal indicator of the successful preparation of the injury model. In contrast, the sham operation group experienced immediate closure of the incision after laminectomy. The mice were humanely euthanized three days post-procedure to facilitate tissue sampling.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was meticulously isolated from mice spinal cord tissues utilizing TRIzol reagent (Solarbio, 15596-026, China). Subsequently, the RNA underwent reverse transcription, employing the HiFiScript Reverse Transcription Kit (Cwbio, CW2020M, China). To gauge gene expression levels, we conducted qRT-PCR using the Takara TB Green® Fast qPCR Mix kit (Takara, RR430A, China) in conjunction with a state-of-the-art LightCycler 7500 System. Each sample was subjected to three sets of replicate experiments to ensure data robustness. To quantify the mRNA expression levels accurately, the 2−ΔΔCt method was employed. The internal reference of mRNA was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences for the pivotal genes under investigation can be found in Table 1 for reference.
Statistical analysis
All bioinformatic and statistical evaluations were carried out via R software (version 4.2.2). Statistical contrasts were made using the t-test for two groups. Using a two-tailed perspective, we interpreted all p-values, marking a value of p < 0.05 as the cutoff for statistical significance.
Results
Identification and characterization of DE-ERSRGs
It was obvious that the gene expression distribution of samples from different dataset has some deviation (Fig. 1A). However, the gene expression level is basically in the same range after the batch removal effect treatment (Fig. 1B). Besides, PCA results show that after batch effect is removed, samples from different data sets overlap together, indicating that batch effect is effectively removed (Fig. 1C–D).
From our results, 68 genes were differentially expressed (63 upregulated genes and 5 downregulated genes) in SCI group compared with sham group (Fig. 1E).
Functional enrichment analysis of DE-ERSRGs
During the development of SCI, the DE-ERSRGs may play essential roles. As shown in Fig. 2A, the BP of GO results exhibited that DE-ERSRGs were mainly involved in the response to toxic substance, response to oxidative stress and cellular response to chemical stress. Besides, most DE-ERSRGs were existed in collagen-containing extracellular matrix, endoplasmic reticulum lumen and phagocytic vesicle (Fig. 2B). In the term of MF, these genes were involved in peroxidase activity, heme binding and Toll-like receptor binding (Fig. 2C). The KEGG results showed that DE-ERSRGs were closely related to the lipid and atherosclerosis, TNF signaling pathway and HIF-1 signaling pathway (Fig. 2D).

The GO and KEGG functional enrichment of DE-ERSRGs. (A) The GO enrichment of DE-ERSRGs in term of BP. (B) The GO enrichment of DE-ERSRGs in term of CC. (C) The GO enrichment of DE-ERSRGs in term of MF. (D) The KEGG enrichment of DE-ERSRGs.
Identification and validation of diagnostic markers for SCI
Then, we tried to determine the key genes with diagnosis value for SCI through machine learning. Through tenfold cross-validation in LASSO logistic regression, 9 key features were screened out based on the optimal lambda (Fig. 3A–B). Besides, the SVM-RFE algorithm further refined this list, targeting the optimal gene feature combination. Ultimately, 10 genes were pinpointed as the most distinguishing features with the lowest RMSE and the highest accuracy (Fig. 3C–D). An intersection of genes derived from both algorithms led to the identification of 6 key marker genes, namely CYBB, PRDX6, PTGS1, GCH1, TLR2, and PIK3CG, for subsequent analyses (Fig. 3E).

Identification of DE-ERSRGs as SCI diagnostic markers. (A, B) LASSO approach identification. (C, D) SVM-RFE algorithm filtering. (E) Venn diagram showing algorithm intersections. (F) Logistic regression model underwent ROC analysis. (G) ROC evaluation of the 6 markers.
To further discern the diagnostic utility of each gene, we illustrated individual ROC curves using the set of markers. Figure 3F–G shows that the diagnosis model and all 6 genes rendered AUC values exceeding 0.9.
Then, as shown in Fig. 4A–F, it can be seen that six hub genes were all significantly upregulated in SCI samples compared with normal samples. Moreover, leveraging the logistic regression findings, we designed a predictive nomogram to estimate the likelihood of SCI occurrence. It was obvious that PTGS1 had the highest point while PRDX6 had the lowest point (Fig. 5A). The corresponding calibration curve attested to the nomogram’s proficiency in identifying SCI with remarkable precision (Fig. 5B). Besides, the DCA curves showed that the nomogram has great net benefits in diagnosis of SCI (Fig. 5C).

The expression of six marker genes in SCI and normal group. (A) The expression of GCH1 in SCI and normal group. (B) The expression of CYBB in SCI and normal group. (C) The expression of TLR2 in SCI and normal group. (D) The expression of PTGS1 in SCI and normal group. (E) The expression of PRDX6 in SCI and normal group. (F) The expression of PIK3CG in SCI and normal group. *** (p < 0.001).

The construction and validation of nomogram based on six marker genes. (A) The construction of nomogram for SCI diagnosis. (B) The calibration curve for nomogram. (C) Decision curve analysis (DCA) was performed for the Nomogram.
Involvement of key genes in SCI-associated pathways
In an endeavor to elucidate the implications of the identified marker genes in differentiating SCI from sham samples, we conducted a GSEA-KEGG pathway analysis for individual genes. A multifaceted analysis revealed substantial associations of four highly expressed marker genes, namely GCH1, PIK3CG, PRDX6 and PTGS1, with pathways like focal adhesion and ECM receptor interaction (Fig. 6A–D). Furthermore, the downregulated expression of TLR2 were enriched in Nod like receptor signal pathway and Toll like receptor pathway (Fig. 6E). In addition, the downregulated expression of CYBB were enriched in Notch signaling pathway (Fig. 6F).

Single-gene GSEA for six marker genes using KEGG reference dataset. (A) The GSEA pathway enrichment analysis for GCH1. (B) The GSEA pathway enrichment analysis for PIK3CG. (C) The GSEA pathway enrichment analysis for PRDX6. (D) The GSEA pathway enrichment analysis for PTGS1. (E) The GSEA pathway enrichment analysis for TLR2. (F) The GSEA pathway enrichment analysis for CYBB.
Molecular subtyping and immune correlations in SCI
In our pursuit to determine the immune subtypes inherent in SCI, we employed the cumulative distribution function (CDF) to meticulously categorize SCI patients, leveraging the expression profiles of the 68 DE-ERSRGs. Optimal subtyping was achieved with k = 2, denoting the most suitable count of patterns (Fig. 7A–C). Subsequently, the discernibility of these two clusters was substantiated by principal component analysis (PCA) (Fig. 7D). Besides, boxplots and heatmaps illustrated that the expression of CYBB, PIK3CG, GCH1 and TLR2 were significantly downregulated in cluster 2 compared with cluster 1, while the difference of PRDX6 and PTGS1 were not significant between cluster 1 and cluster 2 (Fig. 7E–F).

Molecular subtypes and expression of six marker genes in SCI. (A) Cumulative distribution function (CDF) curve illustrating consensus clustering for SCI-related molecules. (B) Area variation under the CDF curve illustrating two stable molecular types. (C) Cluster heatmap for SCI molecular subtypes. (D) SCI-related molecular subtypes evaluated by Principal component analysis (PCA). (E) Expression difference in 6 markers between Cluster1 and Cluster2. (F) Heatmap of 6 markers across two subtypes.
Immune landscape in SCI
The interplay between SCI and the immune microenvironment is increasingly recognized. In light of this, the degree of immune infiltration within SCI samples was assessed by us utilizing the ssGSEA approach. Figure 8A depicts a noticeable reduction in the proportion of CD56dim natural killer (NK) cells and activated B cells among SCI samples compared to control groups. In contrast, the fractions of gamma delta T cells, MDSC, regulatory T cells, natural killer T cells, plasmacytoid dendritic cells, activated dendritic cells, natural killer cells, macrophages, mast cells, immature B cells, and type 1 T helper cells were noticeably elevated in SCI samples.

The calculation of immune landscape between different groups. (A) The infiltration levels of multiple immune cells in SCI and sham were assessed via the ssGSEA (B) The infiltration levels of multiple immune cells in cluster1 and cluster2 were assessed via the ssGSEA. ns (p > 0.05), * (p < 0.05), ** (p < 0.01), *** (p < 0.001).
Then, we also explored the disparities in immune landscape between two clusters. Notably, Cluster 1 exhibited a heightened abundance of MDSC, Macrophage, Regulatory T cells (Treg), and Immature B cells when contrasted with Cluster 2. In contrast, Cluster 2 was enriched in Activated CD8 T cells, CD56 bright NK cells, and CD56 dim NK cells (Fig. 8B).
The correlation between hub genes and Immune cells
Next, we further analyzed the correlation between hub genes and infiltration levels of immune cells to explore the potential roles in the development of SCI. From Fig. 9A, it can be seen that PRDX6 was only positively related to the activated B cells. PTGS1 was positively and negatively related to the activated B cells and activated CD8T cells, respectively (Fig. 9B). Besides, PIK3CG was positively and closely related to the immature B cell, monocyte, T follicular helper cell and macrophage (Fig. 9C). In addition, CYBB, TLR2 and GCH1 were all negatively related to CD56 dim NK cells (Fig. 9D-F).

The correlation between immune cells and six marker genes. (A) The correlation between the infiltration of immune cells and PRDX6 expression. (B) The correlation between the infiltration of immune cells and PTGS1 expression. (C) The correlation between the infiltration of immune cells and PIK3CG expression. (D) The correlation between the infiltration of immune cells and CYBB expression. (E) The correlation between the infiltration of immune cells and TLR2 expression. (F) The correlation between the infiltration of immune cells and GCH1 expression.
Constructing ceRNA networks and transcription factor predictions
To further understand the post-transcriptional regulations in SCI, we established ceRNA networks, pinpointing the intersections between the target miRNAs of lncRNAs and those of the hub genes. Among the identified miRNAs, several showcased a high frequency of connections with genes (Supplement Fig. 1A). Specifically, miRNAs such as hsa-miR-873-5p, hsa-miR-875-3p, hsa-miR-1-3p, hsa-miR-561-3p, and hsa-miR-590-3p exhibited connections with four genes, while others including hsa-miR-4305, hsa-miR-3126-3p, hsa-miR-335-3p, hsa-miR-130b-5p, and hsa-miR-33a-3p were linked to three. In total, 11 miRNAs manifested an extensive interaction network with at least two genes. Of these, hsa-miR-873-5p, hsa-miR-1-3p, and hsa-miR-875-3p displayed the highest reliability by having upstream regulatory lncRNAs, with all three targeting the gene PTGS1. Consequently, through these interactions, we identified eight lncRNAs – CDR1-AS, LINC01043, RP5-894D12.5, RP11-102K13.5, RP13-895J2.3, RP11-96L7.2, RP4-539M6.22, and RP11-333E1.2 – that target these three key miRNAs.
Building upon the findings, we harvested a comprehensive 830 pairs involving transcription factors and mRNA by referencing the TRRUST database’s variably expressed mRNAs. The intricate associations between the 318 transcription factors and corresponding mRNA sequences were effectively visualized using a Sankey diagram. Prominently, MYC, HSF1, JMJD1C, and SUMO1 emerged as the top four transcription factors exhibiting the strongest correlations (Supplement Fig. 1B).
Single-cell dataset validation
In this phase of the study, we used a scRNA-seq dataset for a more granular look into the cellular environment of SCI and uninjured samples.
From the entire dataset, 22,768 cells passed the quality control (QC) benchmarks. These cells were divided between SCI samples (12,757 cells) and uninjured samples (the remaining 10,011 cells). Through unsupervised clustering, the dataset was further categorized into 19 distinct clusters, as depicted in Fig. 10A. When exploring these clusters in depth, seven predominant cell types emerged from both the SCI and uninjured samples. These are: Gametocytes, Neurons, Dendritic Cells (DC), Bone Marrow Cells (BM), Erythroblasts, Embryonic stem cells, and Hematopoietic Stem Cells (HSC CD34+). This distribution is showcased in Fig. 10B.

The analysis of single-cell sequencing dataset. (A–B) T-Distributed Stochastic Neighbor Embedding (T-SNE) plots showcasing 19 cell clusters and 7 main cell types in pSCI3d and uninjured tissues. (C-H) Violin diagrams displaying expression of 6 marker genes across varied cell types.
The study then proceeded to scrutinize the expression patterns of the six previously identified marker genes (CYBB, PRDX6, PTGS1, GCH1, TLR2, and PIK3CG) across these cell types, in both SCI and uninjured conditions. Figure 10C-H visually depict how these identified genes are expressed among predominant cellular categories, facilitating a straightforward contrast between SCI and uninjured samples. Notably, Neurons in the SCI samples expressed the majority of these marker genes. Specifically, CYBB, PTGS1, GCH1, TLR2, and PIK3CG were recognized as distinct markers for Neurons. Additionally, CYBB, GCH1, TLR2, and PTGS1 were markedly enriched in Erythroblasts. Furthermore, PTGS1 and TLR2 were pronouncedly expressed in Gametocytes.
Validation of marker genes in vivo
Finally, we evaluated the expression of six marker genes in mouse spinal cord tissues through qRT-PCR. It was observed that the SCI group exhibited markedly elevated levels of these five key biomarkers, namely CYBB, PRDX6, PTGS1, TLR2, and PIK3CG, in comparison to the sham group, while GCH1 was downregulated in SCI group (Fig. 11A–F).

The validation of six marker genes in SCI and sham mouse samples calculating by qRT-PCR. (A) The expression of TLR2 in control and SCI groups. (B) The expression of GCH1 in control and SCI groups. (C) The expression of CYBB in control and SCI groups. (D) The expression of PTGS1 in control and SCI groups. (E) The expression of PRDX6 in control and SCI groups. (F) The expression of PIK3CG in control and SCI groups. * (p < 0.05), ** (p < 0.01), **** (p < 0.0001).
Discussion
SCI represents a grave medical condition, inflicting profound health implications on patients and imposing a significant economic strain1. Despite the rapid technological advancements, a plethora of basic and clinical research studies on SCI have been undertaken, yet the validation of effective molecularly targeted therapies remains elusive. The activation of ERS has historically been acknowledged as a component of the immediate intrinsic response after a traumatic injury to the spinal cord9. However, the intricacies of its mechanism continue to be elusive. In light of this, our study aimed to discern potential ERSRGs associated with SCI using bioinformatics analysis and further explore the influence of immune cell infiltration in SCI.
In our study, three SCI datasets (GSE5296, GSE47681, GSE132242) were chosen to mitigate the potential error rate and to bolster the reliability of our findings. We identified 68 DE-ERSRGs when comparing SCI with healthy controls: 63 of these were up-regulated, while 5 exhibited down-regulation. Both GO and KEGG analyses further substantiated a deep connection of these DEGs with an array of pathways. These pathways encompassed TNF, HIF-1, p53, NOD-like receptor and IL-17 signaling pathways. The TNF signaling pathway, specifically, plays a crucial role in modulating immune responses and facilitating T-cell activation, which can result in cellular demise24. Notably, studies have emphasized the advantageous impact of inhibiting the TNF-α signaling pathway on enhancing post-SCI functional recovery25. Additionally, Chen et al. discovered that activating the HIF-1 signaling pathway could enhance angiogenesis, neurogenesis, and the hypoxic ischemic environment to facilitate the recovery of neural functionality by regulating HIF-1α, VEGF proteins, and M2 macrophage exosomes26,27. Moreover, the role of p53, NOD-like, and IL17 in the pathogenesis of SCI is intricately linked to the inflammatory response28,29,30. Collectively, these findings underscore the pivotal significance of inflammation, immune response, oxidative stress, and neurogenesis in the initiation and progression of SCI.
In our analysis, six critical DE-ERSRGs, including CYBB, PRDX6, PTGS1, GCH1, TLR2 and PIK3CG, were identified through machine learning, demonstrating remarkable predictive accuracy for SCI. CYBB, a constituent of cytochrome B, has been implicated in inducing oxidative stress following SCI31. TLR2, a member of the Toll-like receptor (TLR) family, is essential for pathogen detection and the initiation of innate immune responses32. Its significant role is further highlighted in the pathogenesis of various autoimmune diseases, with studies indicating that TLR2 deletion may potentially hinder recovery processes post-injury33,34. GCH1 encodes for GTP cyclohydrolase I, a key enzyme in tetrahydrobiopterin synthesis, crucial for nitric oxide synthase activity35. The downregulation of GCH1 resulted in reduced microglial activation and pain36 and was associated with neuroprotection in SCI37. PIK3CG (PI3Kγ/p110γ) serves as a central regulator in inflammation and oxidation38,39. Further studies demonstrated that PIK3CG could enhance the recovery of SCI patients by promoting neuronal differentiation40. PRDX6, or peroxiredoxin-6, is an antioxidant protein. It has been detected in traumatic central nervous system injuries and linked to post-injury inflammation and apoptotic reactions41,42,43. PTGS1 encodes for cyclooxygenase-1 (COX-1), which plays a substantial role in neuronal health, with studies showing its protective capabilities post-injury44,45. In conclusion, our findings underscore the pivotal roles of these six marker genes in the evolution and progression of SCI.
The significant impact of immune cell infiltration on the progression and development of SCI is highlighted in recent research46. For example, Noble et al. demonstrated that functional recovery following SCI could be enhanced by restoring immune homeostasis47. SCI post-injury leads to reduced activity of NK cells, indicating their potential as therapeutic targets for SCI48. Furthermore, our study compared immune cells in SCI with sham samples. Notably, certain immunocytes such as macrophages, regulatory T cells, and NK cells exhibited increased infiltration in SCI samples, while the presence of CD8 T cells and CD56dim NK cells was reduced. CD8 T cells are among the most prevalent and crucial effector cells in the immune system49. Wu et al. discovered that the levels of CD8 T cells increased at 3-, 7-, and 14-day post-SCI treatment in a mouse model50. Similarly, we also observed an increase in CD8 T cell levels in the SCI group. However, PTGS1 was upregulated in the SCI group and negatively correlated with CD8 T cells, suggesting that CD8 T cells were not primarily affected by PTGS1. Additionally, PTGS1 and PRDX6 were both positively correlated with activated B cells, which showed no significant difference between the SCI and sham groups. According to GSEA results, we found that PTGS1 and PRDX6 were enriched in ECM receptor interaction and focal adhesion, which may be involved in the migration and proliferation of neural cells51,52. These findings suggest that PTGS1 and PRDX6 may play a role in the development of SCI by influencing neural cells rather than the immune response.
Macrophages typically differentiate into two subtypes: M1 macrophage and M2 macrophage. M1 macrophages are primarily associated with inflammatory responses53,54. In the mouse model of SCI, the microenvironment is predominantly influenced by M1 macrophages55. Elevated levels of M1 macrophages lead to increased production of pro-inflammatory mediators, exacerbating SCI, impeding recovery post-injury, and inducing neuronal damage27,56. Additionally, several studies have identified that PIK3CG can enhance the levels of M1 macrophages57. In our study, we observed that PIK3CG was upregulated in the SCI group and closely associated with macrophages. This suggests that PIK3CG may exacerbate SCI by promoting the polarization and infiltration of M1 macrophages.
Furthermore, CD56dim NK cells are the predominant NK cell subsets and exhibit potent cytotoxicity58. Campagnolo et al. demonstrated that NK cell cytotoxicity was significantly reduced in the SCI group compared to the control group59. Similarly, we observed a decrease in the levels of CD56dim NK cells in the SCI group in our study. Additionally, we found that the upregulated CYBB, GCH1, and TLR2 exhibited strong negative correlations with CD56dim NK cells. This suggests that CYBB, GCH1, and TLR2 may be involved in the development of SCI by inhibiting CD56dim NK cells, thereby promoting inflammation.
Nonetheless, our study has limitations. It predominantly relies on bioinformatics, necessitating further experimental and clinical validation. Data were sourced from public repositories with a limited sample size, and our clinical diagnostic model awaits external validation. In the initial phase of our study, we relied predominantly on animal data for analysis instead of patient specimens. Moving forward, the next stage of our research will involve the use of patient blood samples and other relevant specimens. In addition, our in vitro experiments included only three mice per group. Further studies should increase the number of experimental subjects to enhance the reliability of the results. This progression is anticipated to significantly enhance the clinical applicability and the informative value of our results.
Conclusion
In summary, our research discerned six pivotal marker genes tied to SCI’s onset and progression using advanced machine learning techniques. By developing a diagnostic model, we can precisely diagnose SCI based on these genes. Additionally, significant alterations in the immune landscape following SCI were uncovered in our findings, emphasizing the nuanced interplay of immune cells during post-injury reactions. Moreover, our understanding of the intricacies of ceRNA interactions and the ability to discern varying immune response patterns in SCI paves the way for personalized therapeutic strategies. Collectively, our findings offer enhanced insights into SCI’s molecular foundations and unveil promising avenues for therapeutic intervention.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Singh, A., Tetreault, L., Kalsi-Ryan, S., Nouri, A. & Fehlings, M. G. Global prevalence and incidence of traumatic spinal cord injury. Clin. Epidemiol. 6, 309–331. https://doi.org/10.2147/CLEP.S68889 (2014).
Merritt, C. H., Taylor, M. A., Yelton, C. J. & Ray, S. K. Economic impact of traumatic spinal cord injuries in the United States. Neuroimmunol. Neuroinflamm. 6 https://doi.org/10.20517/2347-8659.2019.15 (2019).
Wilson, J. R. et al. Early versus late surgery for traumatic spinal cord injury: The results of a prospective Canadian cohort study. Spinal Cord. 50, 840–843. https://doi.org/10.1038/sc.2012.59 (2012).
Li, C. et al. Temporal and spatial cellular and molecular pathological alterations with single-cell resolution in the adult spinal cord after injury. Signal. Transduct. Target. Ther. 7, 65. https://doi.org/10.1038/s41392-022-00885-4 (2022).
Plemel, J. R., Yong, W., Stirling, D. P. & V. & Immune modulatory therapies for spinal cord injury–past, present and future. Exp. Neurol. 258, 91–104. https://doi.org/10.1016/j.expneurol.2014.01.025 (2014).
Filipp, M. E. et al. Differences in neuroplasticity after spinal cord injury in varying animal models and humans. Neural Regen Res. 14, 7–19. https://doi.org/10.4103/1673-5374.243694 (2019).
Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 13, 89–102. https://doi.org/10.1038/nrm3270 (2012).
Chino, H. & Mizushima, N. ER-Phagy: Quality and quantity control of the endoplasmic reticulum by autophagy. Cold Spring Harb Perspect. Biol. 15 https://doi.org/10.1101/cshperspect.a041256 (2023).
Matsuyama, D., Watanabe, M., Suyama, K., Kuroiwa, M. & Mochida, J. Endoplasmic reticulum stress response in the rat contusive spinal cord injury model-susceptibility in specific cell types. Spinal Cord. 52, 9–16. https://doi.org/10.1038/sc.2013.118 (2014).
Beattie, M. S., Hermann, G. E., Rogers, R. C. & Bresnahan, J. C. Cell death in models of spinal cord injury. Prog Brain Res. 137, 37–47. https://doi.org/10.1016/s0079-6123(02)37006-7 (2002).
Szegezdi, E., Logue, S. E., Gorman, A. M. & Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885. https://doi.org/10.1038/sj.embor.7400779 (2006).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19, 432. https://doi.org/10.1186/s12859-018-2451-4 (2018).
Sticht, C., De La Torre, C., Parveen, A., Gretz, N. & miRWalk An online resource for prediction of microRNA binding sites. PLoS One. 13, e0206239. https://doi.org/10.1371/journal.pone.0206239 (2018).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–97. https://doi.org/10.1093/nar/gkt1248 (2014).
Han, H. et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386. https://doi.org/10.1093/nar/gkx1013 (2018).
Brunson, J. C. Ggalluvial: layered grammar for alluvial plots. J. Open. Source Softw. 5 https://doi.org/10.21105/joss.02017 (2020).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420. https://doi.org/10.1038/nbt.4096 (2018).
Narayan, A., Berger, B. & Cho, H. Assessing single-cell transcriptomic variability through density-preserving data visualization. Nat. Biotechnol. 39, 765–774. https://doi.org/10.1038/s41587-020-00801-7 (2021).
Aran, D. et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163–172. https://doi.org/10.1038/s41590-018-0276-y (2019).
Mehta, A. K., Gracias, D. T. & Croft, M. TNF activity and T cells. Cytokine 101, 14–18. https://doi.org/10.1016/j.cyto.2016.08.003 (2018).
Wang, N., Feng, Z., Zhao, W., Zhang, Z. & Zhang, L. Ultrashortwave radiation promotes the recovery of spinal cord injury by inhibiting inflammation via suppression of the MK2/TNF–alpha pathway. Int. J. Mol. Med. 42, 1909–1916. https://doi.org/10.3892/ijmm.2018.3786 (2018).
Chen, H. et al. Effect of hypoxia-inducible factor-1/vascular endothelial growth factor signaling pathway on spinal cord injury in rats. Exp. Ther. Med. 13, 861–866. https://doi.org/10.3892/etm.2017.4049 (2017).
Huang, J. H. et al. Exosomes derived from M2 macrophages improve angiogenesis and functional recovery after spinal cord injury through HIF-1alpha/VEGF Axis. Brain Sci. 12 https://doi.org/10.3390/brainsci12101322 (2022).
Xin, D. Q. et al. Schisandrin B attenuates the inflammatory response, oxidative stress and apoptosis induced by traumatic spinal cord injury via inhibition of p53 signaling in adult rats. Mol. Med. Rep. 16, 533–538. https://doi.org/10.3892/mmr.2017.6622 (2017).
Ji, X. et al. NIMA-related kinase 7 amplifies NLRP3 inflammasome pro-inflammatory signaling in microglia/macrophages and mice models of spinal cord injury. Exp. Cell. Res. 398, 112418. https://doi.org/10.1016/j.yexcr.2020.112418 (2021).
Zong, S. et al. The role of IL-17 promotes spinal cord neuroinflammation via activation of the transcription factor STAT3 after spinal cord injury in the rat. Mediators Inflamm. 2014 (786947). https://doi.org/10.1155/2014/786947 (2014).
Kim, H. et al. Polyamidoamine dendrimer-conjugated triamcinolone acetonide attenuates nerve injury-induced spinal cord microglia activation and mechanical allodynia. Mol. Pain. 13, 1744806917697006. https://doi.org/10.1177/1744806917697006 (2017).
Nie, X. et al. The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron 99, 464–479 e467 (2018). https://doi.org/10.1016/j.neuron.2018.06.035
Freria, C. M. et al. Impairment of toll-like receptors 2 and 4 leads to compensatory mechanisms after sciatic nerve axotomy. J. Neuroinflammation. 13, 118. https://doi.org/10.1186/s12974-016-0579-6 (2016).
Wasko, N. J. et al. Systemic TLR2 tolerance enhances central nervous system remyelination. J. Neuroinflammation. 16, 158. https://doi.org/10.1186/s12974-019-1540-2 (2019).
Ring, A. et al. MicroRNAs in peripheral artery disease: Potential biomarkers and pathophysiological mechanisms. Ther. Adv. Cardiovasc. Dis. 16, 17539447221096940. https://doi.org/10.1177/17539447221096940 (2022).
Kim, S. J. et al. Effective relief of neuropathic pain by adeno-associated virus-mediated expression of a small hairpin RNA against GTP cyclohydrolase 1. Mol. Pain. 5, 67. https://doi.org/10.1186/1744-8069-5-67 (2009).
Yuan, S., Wang, Y. X., Gong, P. H. & Meng, C. Y. MiR-124 inhibits spinal neuronal apoptosis through binding to GCH1. Eur. Rev. Med. Pharmacol. Sci. 23, 4564–4574. https://doi.org/10.26355/eurrev_201906_18032 (2019).
Lima, B. H. F. et al. Converging TLR9 and PI3Kgamma signaling induces sterile inflammation and organ damage. Sci. Rep. 9, 19085. https://doi.org/10.1038/s41598-019-55504-0 (2019).
Jin, R. et al. Phosphatidylinositol-3-kinase gamma plays a central role in blood-brain barrier dysfunction in acute experimental stroke. Stroke 42, 2033–2044. https://doi.org/10.1161/STROKEAHA.110.601369 (2011).
Liu, Q. et al. Electric field stimulation boosts neuronal differentiation of neural stem cells for spinal cord injury treatment via PI3K/Akt/GSK-3beta/beta-catenin activation. Cell. Biosci. 13, 4. https://doi.org/10.1186/s13578-023-00954-3 (2023).
Shichita, T. et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917. https://doi.org/10.1038/nm.2749 (2012).
Buonora, J. E. et al. Autoimmune profiling reveals peroxiredoxin 6 as a candidate traumatic brain Injury Biomarker. J. Neurotrauma. 32, 1805–1814. https://doi.org/10.1089/neu.2014.3736 (2015).
Daverey, A. & Agrawal, S. K. Pre and post treatment with curcumin and resveratrol protects astrocytes after oxidative stress. Brain Res. 1692, 45–55. https://doi.org/10.1016/j.brainres.2018.05.001 (2018).
Basu, S., Hellberg, A., Ulus, A. T., Westman, J. & Karacagil, S. Biomarkers of free radical injury during spinal cord ischemia. FEBS Lett. 508, 36–38. https://doi.org/10.1016/s0014-5793(01)02998-2 (2001).
Antezana, D. F. et al. High-dose ibuprofen for reduction of striatal infarcts during middle cerebral artery occlusion in rats. J. Neurosurg. 98, 860–866. https://doi.org/10.3171/jns.2003.98.4.0860 (2003).
Al Mamun, A. et al. Advances in immunotherapy for the treatment of spinal cord injury. Immunobiology 226, 152033. https://doi.org/10.1016/j.imbio.2020.152033 (2021).
Noble, B. T., Brennan, F. H. & Popovich, P. G. The spleen as a neuroimmune interface after spinal cord injury. J. Neuroimmunol. 321, 1–11. https://doi.org/10.1016/j.jneuroim.2018.05.007 (2018).
Laginha, I. et al. Natural killer (NK) cell functionality after human spinal cord Injury (SCI): protocol of a prospective, longitudinal study. BMC Neurol. 16, 170. https://doi.org/10.1186/s12883-016-0681-5 (2016).
Zeng, R. et al. Exploring the immunological role and prognostic potential of PPM1M in pan-cancer. Med. (Baltim). 102, e32758. https://doi.org/10.1097/MD.0000000000032758 (2023).
Wu, Y. et al. Temporal kinetics of CD8(+) CD28(+) and CD8(+) CD28(-) T lymphocytes in the injured rat spinal cord. J. Neurosci. Res. 95, 1666–1676. https://doi.org/10.1002/jnr.23993 (2017).
Zhao, X. et al. Different epidermal growth factor receptor signaling pathways in neurons and astrocytes activated by extracellular matrix after spinal cord injury. Neurochem Int. 129, 104500. https://doi.org/10.1016/j.neuint.2019.104500 (2019).
Li, J. H. et al. Bioinformatic identification of key candidate genes and pathways in axon regeneration after spinal cord injury in zebrafish. Neural Regen Res. 15, 103–111. https://doi.org/10.4103/1673-5374.264460 (2020).
Reinhold, S., Blankesteijn, W. M. & Foulquier, S. The interplay of WNT and PPARgamma signaling in vascular calcification. Cells 9 https://doi.org/10.3390/cells9122658 (2020).
Locati, M., Curtale, G., Mantovani, A. & Diversity Mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147. https://doi.org/10.1146/annurev-pathmechdis-012418-012718 (2020).
Ji, Z. et al. Neural stem cells induce M2 polarization of macrophages through the upregulation of interleukin-4. Exp. Ther. Med. 20, 148. https://doi.org/10.3892/etm.2020.9277 (2020).
Abumaree, M. H. et al. Decidua Basalis mesenchymal stem cells favor inflammatory M1 macrophage differentiation in Vitro. Cells 8 https://doi.org/10.3390/cells8020173 (2019).
Li, M. et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control Release. 321, 23–35. https://doi.org/10.1016/j.jconrel.2020.02.011 (2020).
Salzberger, W. et al. Tissue-resident NK cells differ in their expression profile of the nutrient transporters Glut1, CD98 and CD71. PLoS One. 13, e0201170. https://doi.org/10.1371/journal.pone.0201170 (2018).
Campagnolo, D. I., Dixon, D., Schwartz, J., Bartlett, J. A. & Keller, S. E. Altered innate immunity following spinal cord injury. Spinal Cord. 46, 477–481. https://doi.org/10.1038/sc.2008.4 (2008).
Author information
Authors and Affiliations
Contributions
YPZ and XMT contributed to the conception and design. XMT, YL and JD contributed to the collection and assembly of data. YPZ, XMT and JM analyzed and interpreted the data. All authors wrote and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The Ethics Committee of The Affiliated Huaian No.1 people’s Hospital of Nanjing Medical University deemed that this research is based on open-source data, so the need for ethics approval was waived.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Y., Tang, X., Dai, J. et al. Unveiling vital biomarkers and immune infiltration profiles in endoplasmic reticulum stress following spinal cord injury. Sci Rep 14, 29981 (2024). https://doi.org/10.1038/s41598-024-81844-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-81844-7
