
Abstract
Gastric cancer (GC) is characterized by notable heterogeneity and the impact of molecular subtypes on treatment and prognosis. The role of programmed cell death (PCD) in cellular processes is critical, yet its specific function in GC is underexplored. This study applied multiomics approaches, integrating transcriptomic, epigenetic, and somatic mutation data, with consensus clustering algorithms to classify GC molecular subtypes and assess their biological and immunological features. A machine learning model was developed to create the Gastric Cancer Multi-Omics Programmed Cell Death Signature (GMPS), targeting PCD-related genes. We verified the expression of the GMPS hub genes using the RT-qPCR method. The prognostic influence of GMPS on GC was then evaluated. Single-cell analysis was performed to examine the heterogeneity of PCD characteristics in GC. Findings indicate that GMPS notably correlates with patient survival rates, tumor mutational burden (TMB), and copy number variations (CNV), demonstrating substantial prognostic predictive power. Moreover, GMPS is closely associated with the tumor microenvironment (TME) and immune therapy response. This research elucidates the molecular subtypes of GC, highlighting PCD’s critical role in prognosis assessment. The relationship between GMPS and immune therapy response, alongside gastric cancer’s microenvironmental features, provides insights for personalized treatment.
Similar content being viewed by others


Introduction
Gastric cancer, ranking fourth globally in mortality among malignant tumors1, imposes a notable burden on public health systems. Its pronounced heterogeneity is not only a key biological characteristic but also a major challenge for current treatment strategies2. In clinical practice, the classification of gastric cancer is refined based on histopathological characteristics, including intestinal, diffuse, and indeterminate types, as well as molecular features, such as TCGA and ACRG subtypes3,4,5,6. Although treatment regimens based on these classifications, including surgery, chemotherapy, and radiotherapy, have led to some improvements in patient survival rates and quality of life, notable challenges remain in clinical practice due to considerable variations in treatment tolerance and the ongoing need for personalized treatment and prognosis assessment.
Recent studies have highlighted that biomarkers based on HER2 and PD-L1 can potentially improve the prognosis of gastric cancer and have spurred the development of corresponding clinical diagnostic and treatment strategies. Nevertheless, phase III clinical trials targeting these biomarkers have not consistently shown a notable survival advantage7,8. However, novel therapies such as immune checkpoint inhibitors, although they have shown efficacy in certain cases, are also accompanied by immune-related adverse events such as peripheral neuropathy and hearing loss9,10. At the same time, the tumor microenvironment has been proven to play a critical role in the progression and drug resistance of gastric cancer, suggesting the need to explore new therapeutic strategies targeting the microenvironment11. Although various prognostic tools are available for evaluating cancer patient outcomes, more precise biomarkers are still needed to better guide individualized treatment of gastric cancer12. A notable challenge in these trials is the selection of patients with specific molecular characteristics, which can limit the efficacy determination of targeted therapies. Consequently, the identification of new biomarkers and molecular subtypes becomes imperative to more accurately pinpoint patients who could benefit from specific targeted treatments.
PCD is a genetically regulated, actively coordinated cellular process, crucial for the growth and development of cellular organisms. PCD encompasses a diverse range of subtypes, including pyroptosis, ferroptosis, necroptosis, autophagy, and others, each characterized by distinct triggering stressors, morphological features, regulatory signaling pathways, and effector molecules13,14. PCD is pivotal in the pathogenesis, invasion, and metastasis of cancer, as demonstrated in various studies15,16. Notably, different PCD pathways, such as pyroptosis with its characteristic cell swelling and pro-inflammatory factor release, ferroptosis marked by iron-dependent lipid peroxide accumulation, cuproptosis driven by copper and lipoylated protein build-up, and apoptosis characterized by cell condensation and nuclear fragmentation, have been implicated in gastric cancer progression17,18,19,20. Additionally, emerging research is exploring other PCD pathways like Oxeiptosis, regulated by KEAP1’s reactive oxygen sensing, and parthanatos, associated with DNA damage and PARP-1 activation, for their potential roles in cancer21,22. However, the precise correlation between the various forms of PCD and the prognosis of gastric cancer remains under-explored, highlighting a gap in the current understanding of PCD’s specific functions in GC.
In this study, we conducted a comprehensive multiomics analysis of GC by utilizing mRNA, long non-coding RNA (lncRNA), and microRNA (miRNA) expression data, along with genomic mutation and DNA methylation epigenetic information. By integrating ten distinct clustering algorithms, we successfully identified consensus molecular subtypes of GC and extensively analyzed their PCD characteristics, epigenetic features, and immune characteristics. Utilizing 99 machine learning models, we developed the GMPS. GMPS showed notable prognostic value in independent training and validation cohorts and exhibited outstanding predictive performance for immune therapy responses and drug sensitivities. Moreover, single-cell analysis highlighted the cellular heterogeneity of PCD characteristics in GC, establishing a strong correlation between GMPS scoring and various biological processes in gastric cancer. The findings of our study provide essential insights into the molecular subtypes of gastric cancer and offer pivotal reference points for personalized treatment strategies in GC.
Methods
Multi-omics data collection and preprocessing of multicenter cohort data
In this study, we gathered multi-omics data from the gastric cancer cohort (STAD) of The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov), including patients with comprehensive transcriptomic expression, DNA methylation, and somatic mutation information. Patients lacking complete and relevant clinical data were excluded. Additionally, we sourced complete gastric cancer data from three other datasets in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), namely GSE15459, GSE38749, and GSE62254, encompassing gastric cancer tissue chip data and associated clinical information5,23,24. For single-cell analysis, we utilized three primary gastric cancer samples from GSE16355825. The datasets GSE78220 and GSE135222, along with clinical trial data from http://research-pub.gene.com/IMvigor210CoreBiologies, were employed as validation cohorts for immune analysis26,27,28. Following the conversion of transcriptome data to transcripts per million (TPM) format, we conducted background adjustment and quantile normalization on the merged data to create the final dataset. Batch effects, attributable to non-biological technical biases, were corrected using the ‘ComBat’ algorithm in the SVA package29. To assess the effectiveness of data merging, we applied principal component analysis (PCA) to visualize the data pre- and post-merging. We performed rigorous quality control on the data to eliminate low-quality samples and technical noise. Additionally, for identifying PCD-related genes, we curated a list of 2,173 genes associated with 15 types of PCD from the GeneCards, KEGG database, and pertinent literature30,31,32 (Supplementary Table 1).
Comprehensive multiomics consensus ensemble analysis
In this study, a comprehensive multi-omics analysis was undertaken by first collating information across five dimensions from a total of 346 TCGA samples, using their sample IDs. For DNA methylation data, we prioritized probes targeting CpG islands in promoter regions. The identification of mutated genes focused on those containing nonsynonymous variations, including frameshift insertions or deletions, in-frame insertions or deletions, and various types of mutations like nonsense, missense, nonstop, splice site, or translation start site mutations. We employed the ‘getElites’ function from the MOVICS R package for gene feature selection33. For continuous variables such as mRNA, lncRNA, miRNA, and methylation data, the ‘method’ parameter in ‘getElites’ was set to ‘mad’, enabling the selection of the top 1500 genes exhibiting the most pronounced variation. Subsequently, the ‘method’ parameter was adjusted to ‘cox’ for the identification of prognostic genes, integrating them with clinical data, and focusing on those with a significance level of p < 0.05 in each data dimension. For binary variables like gene mutation data, the ‘method’ parameter was set to ‘freq’, filtering for the top 10% of genes with the highest mutation frequency. The results from these five dimensions were then compiled for comprehensive further analysis.
After completing the initial feature selection, we proceeded to determine the optimal number of clusters for our data set. The goal was to identify the smallest number of clusters that would minimize noise yet retain essential information. We utilized the ‘getClustNum’ function in the MOVICS package for this purpose, which evaluates the optimal number of subtypes based on the Clustering Prediction Index (CPI), Gap statistics, and Silhouette Score34. Informed by previous gastric cancer research, we ultimately categorized the samples into two distinct subtypes. Subsequently, cluster analysis was conducted using the ‘getMOIC’ function. For preliminary clustering, ten clustering algorithms were chosen, including CIMLR, ConsensusClustering, SNF, iClusterBayes, PINSPlus, moCluster, NEMO, IntNMF, COCA, and LRA, as inputs for the ‘methodslist’ parameter. Following the computation of results from these ten methods, the ‘getConsensusMOIC’ function was employed to amalgamate the clustering outcomes of the various algorithms. This consensus clustering approach was adopted to enhance the stability and reliability of our clustering results. To validate the robustness of the clustering results, we performed multiple resampling and random seed settings to ensure the reproducibility of the outcomes.
Specific molecular features and stability in consensus molecular subtypes
In our analysis, the single-sample gene set enrichment analysis (ssGSEA) algorithm was employed to determine the molecular characteristics of PCD and treatment modalities within the molecular subtypes35. This included the evaluation of characteristic signatures comprising 15 PCD-related genes, gastric cancer TCGA molecular subtype-related signatures, and signatures associated with chemotherapy and targeted therapy36,37. Additionally, a transcriptional regulatory network was constructed using the RTN R package, focusing on 23 target transcription factors (TFs) that induce or inhibit, and 71 candidate regulatory factors related to cancer-specific chromatin remodeling. These factors served as key indicators for assessing the epigenetics of gastric cancer subtyping38,39. The distribution of immune checkpoints among these subtypes was also compared. The immune and stromal scores of tumor tissues were calculated using the ESTIMATE R package. Concurrently, the enrichment of 24 types of tumor immune microenvironment cells was assessed using ssGSEA. To evaluate the stability of the subtypes, the clustering results were first validated using subtype-specific biomarkers in the validation dataset. Subsequently, the consistency of the consensus clustering results was compared with the nearest template prediction (NTP) and partition around medoids (PAM) classifiers. To ensure the reliability of the results, we employed cross-validation in the validation set to evaluate the predictive performance of the model.
Development of a consensus-based machine learning prognostic signature
To ascertain the association of the GMPS with immune therapy response and prognosis, we utilized the TCGA STAD dataset as our training set and a composite of multiple datasets, termed the META-GC dataset, as our validation set. In developing a robust and generalizable GMPS, we integrated ten distinct machine learning algorithms, namely CoxBoost, stepwise Cox, Lasso, Ridge, elastic net (Enet), survival support vector machines (survival-SVMs), generalized boosted regression models (GBMs), supervised principal components (SuperPC), partial least squares Cox (plsRcox), and random survival forests (RSF).
The GMPS development process entailed the following steps:
Initially, from the intersection of the top 500 notably overexpressed genes in cancer subtypes (CS) and PCD genes, prognostically notable PCD-related genes (PRGs) were identified. This was achieved through univariate Cox regression analysis on the TCGA STAD and META-GC datasets.
Subsequently, ten machine learning algorithms were applied, and 99 algorithm combinations were utilized to construct a highly predictive GMPS, aiming for optimal C-index performance.
Upon establishing the model on the TCGA STAD training set, its efficacy was further assessed on the META-GC validation set. The model demonstrating the highest average C-index was selected as the optimal model.
Quantitative reverse transcription polymerase chain reaction
We validated the expression of GMPS hub genes using the RT-qPCR method, employing HGC-27 gastric cancer cell lines and GES-1 gastric mucosal epithelial cell lines for gene expression validation. After reverse transcription, RT-qPCR was performed using the Roche LightCycler96 PCR system and AceQ qRT-PCR SYBR Green Master Mix (Vazyme). GAPDH was used as the reference gene. The relative expression of GMPS hub genes in gastric cancer and normal gastric tissues was determined using the 2-ΔΔCt method. The primer sequences used in this study are listed in Supplementary Table 2.
Evaluating the prognostic impact and clinical application potential of GMPS in GC
Utilizing the developed model, we conducted multivariate Cox regression scoring for each sample in both the training and validation sets. Samples were stratified into high GMPS and low GMPS groups based on their scores. The GMPS score cutoff was determined using the ‘surv-cutpoint’ function of the survminer package. The prognostic significance of GMPS was evaluated using Kaplan–Meier survival curves. To augment GMPS’s clinical utility, we developed a nomogram incorporating factors identified through multivariate Cox regression analysis. The nomogram’s accuracy was assessed by plotting calibration curves and time-dependent C-index curves. Additionally, decision curve analysis were utilized to estimate the clinical benefit to patients.
Immune-omics profiles and immunotherapy responses linked to GMPS
In this segment of our study, we performed an extensive analysis of the immunological distinctions between high and low GMPS patient groups. This analysis focused on TME cell types and immune therapy response, utilizing the IOBR package40. We explored the differential distribution of TMB between these two groups and reclassified patients based on their GMPS categorization. For immune therapy response assessment, we initially examined patients’ delayed response survival following immune therapy. The TIP algorithm was employed to estimate immune therapy response41. Furthermore, the correlation between GMPS and immune therapy response was validated using data from the GSE78220 and GSE135222 immune datasets.
Screening potential therapy agents for patients
In this phase of the study, the gene set enrichment analysis (GSEA) was utilized to assess the activation status of oncogenic pathways in high GMPS patients42. We accessed expression data from human cancer cell lines (CCLs) via the Broad Institute Cancer Cell Line Encyclopedia (CCLE). Additionally, drug sensitivity data for these CCLs were obtained from both the Cancer Therapeutics Response Portal (CTRP) v.2.0 (https://portals.broadinstitute.org/ctrp) and the Profiling Relative Inhibition Simultaneously in Mixtures (PRISM) datasets (19Q4; https://depmap.org/portal/prism/).
Single-cell RNA-seq data processing and analysis
For this part of the study, single-cell RNA sequencing data from three primary GC patients were obtained from the GSE163558 dataset and analyzed utilizing the Seurat package43. Initial data processing involved stringent quality control measures, where cells with less than 15% mitochondrial gene content and genes expressed in at least 1000 cells, within an expression range of 100 to 7500, were retained. We then identified 2000 highly variable genes for in-depth analysis. To mitigate batch effects across the three sample datasets, the Harmony package was employed. The optimal resolution and number of clusters were determined using the clustree package, followed by the construction of cell clusters with the ‘FindClusters’ and ‘FindNeighbors’ functions. These clusters were visualized using the ‘t-SNE’ method. Cell annotation was based on marker genes for different cell types (Supplementary Table 3). The activity of specific gene sets in each cell was quantified using the ‘AddModuleScore’ function within Seurat. Differential gene expression analysis between groups was conducted using the ‘FindMarkers’ function. Additionally, the CellChat R package was applied for the analysis and computation of intercellular interactions44.
Statistical analysis
For comparative analysis between two groups, the unpaired Student’s t-test was utilized for variables adhering to a normal distribution. For variables not conforming to normal distribution, the Wilcoxon rank-sum test was employed. In cases involving more than two groups, parametric variables were analyzed using one-way ANOVA, while non-parametric variables were assessed with the Kruskal–Wallis test. Differential expression analysis was conducted employing the limma R package. All statistical procedures were performed using R software, version 4.1.3. Unless stated otherwise, the threshold for statistical significance was set at p < 0.05.
Result
Identification and prognostic correlation of multiomics-driven molecular subtypes in GC
The workflow of our study is depicted in Fig. 1. We initiated the process by rigorously preprocessing all data. PCA was applied to assess the data before and after batch effect processing, confirming the efficacy of the preprocessing (Supplementary Fig. 1A-B). Ten integrated clustering algorithms were employed, and the optimal clustering results were achieved with the dataset divided into two groups (Supplementary Fig. 2-3). Subsequently, we utilized a consensus clustering approach to merge these results with transcriptomic data, epigenetic methylation patterns, and somatic mutation expression profiles. The top 10 most notable data points in each group, pertaining to overall survival (OS), were displayed in a heatmap (Fig. 2A-C). Our molecular subtypes demonstrated a notable correlation with patient overall survival rates (p = 0.007; Fig. 2D), with CS1 showing a more favorable survival prognosis. Distinct differences in TMB were observed between these subtypes, especially in genes such as TTN, TP53, MUC16, LRP1B, and ARID1A (Fig. 2E-F). Furthermore, the genomic change proportions, including lost or gained genomes like CNV metrics, varied notably between the subtypes (Fig. 2G).

Overview of the study design.

GC integrated multimodal subtype analysis. (A) Extensive heatmap depicting consensus molecular subtypes, integrating mRNA, lncRNA, miRNA, DNA CpG methylation patterns, and gene mutations; (B) Stratification of GC cohorts via 10 diverse multimodal clustering methodologies; (C) Matrix representing consensus clustering of two emerging prognostic subtypes, derived from a synthesis of 10 distinct algorithms; (D) Kaplan–Meier survival plots delineating the prognostic trajectories of the identified subtypes; (E) TMB across the subtypes; (F) Mutated genes waterfall plot for each subtype; (G) Comprehensive evaluation of CNV within the subtypes.
Comprehensive stratification of GC using integrated consensus molecular subtypes
In this study, we recognized that the classification of most GC molecular subtypes is primarily based on molecular expression levels, which are likely to be associated with specific biological functions. Consequently, we delved deeper into the distinct molecular characteristics of these consensus molecular subtypes. Upon comparing our findings with the classical TCGA classification, a notable enrichment of the GS subtype was observed in CS2, while EBV and MSI subtypes were more prominent in CS1. Particularly, when assessing the 15 types of PCD, we found notable enhancements in parthanatos, necroptosis, necrosis-like morphology, pyroptosis, cuproptosis, and ferroptosis in CS1. On the other hand, anoikis, autophagy, lysosome-dependent cell death, extrinsic apoptosis, intrinsic apoptosis, immunogenic cell death, and necrosis were more pronounced in CS2. This suggests that differences in PCD types between different subtypes may be one of the key factors leading to differences in prognosis. These variations in cell death modalities underscore the pivotal role of PCD in influencing the prognosis of gastric cancer patients. Furthermore, our study revealed notable disparities in the response of different subtypes to specific treatments, indicating that CS2 might derive greater benefit from chemotherapy and targeted therapies (Fig. 3A).

Molecular characterization and validation of GC CSs. (A) Enrichment of subtypes in GC treatment-related signatures, PCD features, and within TCGA classifications; (B) Activity spectrum of 23 transcription factors and potential regulators associated with chromatin remodeling; (C) Immune profile in the TCGA STAD cohort with heatmap annotations showing tumor samples’ immune and stromal scores, expression of immune checkpoint genes, and enrichment levels of 22 immune cells in the TME; (D) Validation of GC CSs in the nearest template analysis of the META-GC cohort; (E) Survival analysis of GC CSs in the META-GC cohort; (F) Consistency of CSs with NTP in TCGA-STAD; (G) Consistency of CSs with PAM in TCGA-STAD; (H) Consistency between NTP and PAM in TCGA-STAD cohort.
In further analysis that integrated regulatory factors and transcription factors involved in cancer chromatin remodeling, notable differences were observed between the two subtypes. In CS2, regulatory factors such as AR, PGR, ESR1, STAT3, FGFR1, RARA, and RXRA demonstrated elevated expression levels. Conversely, in CS1, factors like GATA6, KLF4, FOXA1, and PPARG were found to be more highly expressed (Fig. 3B). These transcription factors play important roles in tumor occurrence and development, and their differences may explain the molecular mechanism specificity between different subtypes. Additionally, our investigation into immune infiltration revealed a higher stromal score in CS2. A notable differential enrichment in most immune checkpoints and immune cells was also detected between the two subtypes, highlighting distinct immunological landscapes (Fig. 3C).
Building on the differential expression analysis between the two subtypes, we identified 500 genes specifically upregulated in both subtypes and employed them as classifiers. These were further validated in an external dataset to confirm the stability of our subtype classification (Fig. 3D, Supplementary Table 4). Utilizing the NTP method, we classified samples in the external dataset into the appropriate CS. Within the META-GC dataset, which amalgamates multiple datasets, we observed notable prognostic differences between the two subtypes (P = 0.043; Fig. 3E). Additionally, the consistency of the PAM and NTP algorithms in differentiating these subtypes was evaluated, yielding notable results (p < 0.001; Fig. 3F-H).
Development and prognostic significance of GMPS
Recognizing the pivotal role of PCD in gastric cancer, we identified 31 prognostically relevant PCD-related genes (PRGs) notably associated with OS from the TCGA STAD and META-GC datasets. These PRGs were then integrated into a comprehensive framework to construct the GMPS. Utilizing 99 distinct algorithm combinations, we developed machine learning models for the TCGA STAD and META-GC datasets, calculating the average C-index for each model (Fig. 4A). Of these models, the Random Survival Forest (RSF) algorithm emerged with the highest average C-Index, leading to its selection for the final GMPS model, which was based on five hub genes (Fig. 4B-C). Subsequently, GMPS scores were computed for each sample in both datasets. Higher GMPS scores were associated with poorer clinical prognosis in both the TCGA STAD and META-GC datasets (Fig. 4D-E).

Generation and predictive value of GMPS. (A) Combined results of 99 machine learning algorithms based on an integrated computational framework, with C-index calculated for each model through TCGA STAD and META-GC cohorts and ranked by average C-index; (B) GMPS hub genes selected through the RSF algorithm; (C) Univariate Cox regression analysis results of hub genes in training and validation cohorts; (D-E) Survival analysis of high GMPS and low GMPS in TCGA STAD and META-GC cohorts; (F) The expression level differences of hub genes were determined by RT-qPCR between gastric cancer cell lines and normal gastric mucosal epithelial cell lines; (G) Protein expression levels of hub genes in stomach tissues and gastric cancer in the HPA database.
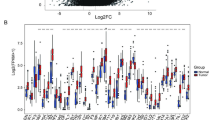
We aim to analyze the critical role of hub gene expression differences at the gene and protein levels in gastric cancer occurrence. First, we validated the expression of five GMPS hub genes in gastric cancer cell lines and normal gastric mucosal epithelial cell lines using RT-qPCR. The results showed that, consistent with our initial analysis, the expression levels of these hub genes were notably different in gastric cancer cells compared to normal gastric cells (Fig. 4F). This finding was corroborated by protein expression analysis of these hub genes in the Human Protein Atlas (HPA) database, highlighting notable disparities between gastric cancer and normal tissues45 (Fig. 4G).
During our examination of the hub genes composing the GMPS, Kaplan–Meier curves were employed to investigate the associations of these genes with OS, progression-free interval (PFI), and disease-specific survival (DSS) in gastric cancer patients. These analyses revealed notable correlations between the hub genes and patient prognosis (Supplementary Fig. 4). Further, we utilized the GSCALite public server (http://bioinfo.life.hust.edu.cn/web/GSCALite/) to systematically assess the multi-omics phenotypes of GMPS across 32 different cancer types (excluding STAD) within the TCGA database. The findings highlighted notable differential expression of these hub genes across various tumor types (Supplementary Fig. 5A). Moreover, a positive correlation was observed between the mRNA expression levels of these hub genes and CNV in most cancer types, particularly evident in KIF11 and MKI67 (Supplementary Fig. 5B). CNV frequency change analysis demonstrated that the CNV of these hub genes vary notably among different cancer types, with ROR2 predominantly exhibiting heterozygous deletion (Supplementary Fig. 5C-D). Additionally, methylation level comparisons between tumor and normal samples across multiple cancer types revealed notable differences in these hub genes (Supplementary Fig. 6A). In most cancers, there was an inverse correlation between methylation levels and mRNA expression of these genes (Supplementary Fig. 6B), suggesting that epigenetic changes might play a role in influencing patient prognosis. Further analysis indicated that these hub genes are involved in activating the cell cycle pathway and inhibiting the hormone ER pathway (Supplementary Fig. 6C-D).
Evaluating and comparing prognostic signatures in GC
Prognostic features and molecular subtyping based on transcriptome sequencing have garnered notable attention in gastric cancer research in recent years. To conduct a comprehensive comparison of GMPS with existing prognostic features, we systematically reviewed relevant literature from the past five years and selected 10 distinct gastric cancer prognostic features for our comparative analysis (Supplementary Table 5). These features encompass various biological processes, including metabolism, oxidative stress, and multiple types of PCD, such as pyroptosis, ferroptosis, and autophagy. Remarkably, GMPS outperformed all other prognostic models in terms of C-index in both the TCGA STAD and META-GC datasets (Fig. 5A-B). To explore GMPS’s potential in clinical applications, we conducted an independent prognostic analysis to identify key prognostic factors for gastric cancer (Supplementary Fig. 7). These factors were then integrated into a comprehensive prognostic nomogram, enhancing its clinical utility (Fig. 5C). The nomogram’s calibration curve displayed high accuracy in predicting 1-year, 2-year, and 3-year survival rates for gastric cancer patients (Fig. 5D). Decision curve analysis (DCA) demonstrated that the nomogram offered greater clinical benefit compared to using GMPS alone (Fig. 5E). Time-dependent C-index analysis further validated the superior predictive performance of the nomogram over other models (Fig. 5F).

Clinical application value of GMPS. (A-B) Comparison of GMPS with other 10 published models in TCGA STAD and META-GC cohorts; (C-D) Comprehensive nomogram based on GMPS with calibration curves; (E) Net decision curve analyses of the nomogram in comparison with other clinical features; (F) Time-dependent C-index curves for the nomogram and GMPS.
Exploring immune characteristics associated with GMPS
In light of the variation in immune cell composition within gastric cancer consensus subtypes, we embarked on an exhaustive analysis of the TME in relation to GMPS. Our findings revealed marked disparities in immune cell infiltration levels, including T cells, B cells, macrophages, and fibroblasts, between patients with high and low GMPS scores (Fig. 6A). Notably, patients with high GMPS exhibited increased features of immune suppression and rejection, such as epithelial-mesenchymal transition and upregulation of TGF-β family members, compared to the low GMPS group (Fig. 6B-C). Furthermore, traits associated with enhanced responsiveness to immune therapy were found to be notably more prevalent in patients with low GMPS (Fig. 6D). The TMB, a key biomarker for assessing patient response to immune therapy, was also examined. We discovered a higher enrichment of TMB in the low GMPS group, indicating a potentially stronger immunogenicity in these patients (Fig. 6E). The survival analysis results suggest that GMPS could be a valuable adjunct to TMB in evaluating the efficacy of immune therapy in gastric cancer patients (Fig. 6F).

TME-related molecular characteristics in high and low GMPS patients. (A) Distribution of TME immune cell type characteristics in high and low GMPS patients; (B) Distribution of immune suppression features in high and low GMPS patients; (C) Distribution of immune exclusion features in high and low GMPS patients; (D) Distribution of immunotherapy biomarkers in high and low GMPS patients; (E) Distribution of TMB in high and low GMPS patients; (F) Survival analysis of GMPS combined with TMB.
Evaluating the predictive efficiency of GMPS in gastric cancer immune therapy response
To thoroughly assess GMPS’s effectiveness in guiding immune therapy for gastric cancer, we carried out a systematic analysis. Initially, we delved into the IMvigor210 dataset, considering the comprehensive prognosis and treatment data of the patient cohort. By comparing the Restricted mean survival time (RMS) at 12 and 24 months between two patient groups, we illuminated the delayed effects of immune therapy and evaluated long-term survival (LTS) differences following 3 months of treatment (p < 0.05; Fig. 7A-B). The findings indicated that patients with lower GMPS scores had more favorable prognoses, suggesting enhanced benefits from immune therapy in this group. Analysis of treatment response degrees revealed that GMPS scores in the partial response (PR) group were notably lower than those in the non-responsive (progressive disease [PD]/stable disease [SD]) group (p < 0.05; Fig. 7C). This indicates that patients with lower GMPS scores respond better to immunotherapy and may benefit more from it. Additionally, we performed tracking tumor immunophenotype (TIP) analysis to investigate potential biological mechanisms linked to GMPS. In the fifth step of immune cell infiltration assessment, notable differences emerged between the high and low GMPS groups (Fig. 7D). Finally, we revalidated our model across several immune therapy validation datasets containing prognostic information. Consistently, patients in the low GMPS group exhibited better prognostic outcomes following immune therapy (GSE78220, p = 0.00014 [Fig. 7E]; GSE135222, p < 0.0001 [Fig. 7F]).

Value of GMPS in predicting immune therapy response in GC patients. (A) Difference curves of Restricted Mean Survival (RMS) time at 12 and 24 months after treatment between high and low GMPS groups; (B) Long-term Survival (LTS) difference curves after 3 months of treatment between high and low GMPS groups; (C) Distribution of GMPS across different immune therapy response groups; (D) Differences in activation levels between high and low GMPS groups at various stages of TIP; (E) Survival analysis for high and low GMPS groups in GSE78220; (F) Survival analysis for high and low GMPS groups in GSE135222.
Identification and assessment of potential therapeutic agents for GC
In our investigation, GSEA was employed to pinpoint pathways associated with the GMPS. GSEA results revealed that, in patients with high GMPS, biological pathways such as epithelial-mesenchymal transition (EMT), myogenesis, and coagulation were notably activated (Fig. 8A). Given the observed adverse reactions of high GMPS patients to immune therapy, we utilized the CTRP and PRISM databases to identify potential therapeutic drugs for this patient group. To validate the reliability of this approach, we compared our results with cisplatin’s known sensitivity, a standard treatment in gastric cancer. Aligning with previous studies46, our analysis indicated that gastric cancer patients with lower GDF15 expression levels responded more favorably to cisplatin-based chemotherapy, suggesting enhanced chemotherapy benefits (Fig. 8B). Consequently, drugs such as ML162, dasatinib, lorlatinib, and idronoxil were pinpointed as potentially more effective for high GMPS gastric cancer patients. Notably, both CTRP and PRISM databases underscored dasatinib as a promising therapeutic agent for these patients (Fig. 8C-D).

Potential therapeutics for high GMPS patients. (A) GSEA pathway enrichment analysis in the high CMLS group; (B) Sensitivity analysis of cisplatin; (C-D) Correlation and differential analysis of drug sensitivity for potential therapeutics screened from CTRP and PRISM datasets.
PCD characteristics in gastric cancer single-cell transcriptomics
In our investigation, we utilized single-cell RNA sequencing data from three primary gastric cancer patients. Following data filtering and batch effect elimination, we applied PCA and t-distributed stochastic neighbor embedding (t-SNE) techniques for dimension reduction of the top 2000 variable genes (Supplementary Fig. 8A-C). Further analysis using the Cluster tree enabled us to identify 14 distinct cell clusters at a resolution of 0.2, providing a stable and reasonable classification (Supplementary Fig. 8D). Each cell cluster exhibited unique gene expression characteristics, reflecting the cellular heterogeneity in gastric cancer tissues. We then categorized the cells into eight types based on marker genes and their expression profiles. These cell types included T cells, fibroblasts/pericytes, macrophages, monocytes, mast cells, endothelial cells, NK cells, and B cells (Fig. 9A and Supplementary Fig. 8E-F). A heatmap was generated to illustrate the top four most representative marker genes for each cell cluster, shedding light on their distinct profiles (Fig. 9B).

PCD characteristics in GC single-cell transcriptomics. (A) Identification of cell types based on marker genes; (B) Heatmap of the top four marker genes in each cell cluster; (C) PCD activity scores in different cells; (D) Distribution of PCD scores among different cell types; (E) Expression of GMPS hub genes in various cell types.
To quantify PCD activity across different cell types in gastric cancer, we assessed the expression levels of 2,172 PCD-related genes. This analysis spanned all identified cell types (Fig. 9C). Among the eight cell types examined, fibroblasts/pericytes, endothelial cells, and macrophages exhibited notably higher PCD activity compared to the other cell types (Fig. 9D). Additionally, our study delved into the expression patterns of hub genes within the TME. We discovered that these five hub genes were predominantly expressed in fibroblasts/pericytes, indicating their notable involvement in this cell type’s functions (Fig. 9E).
Correlation between GMPS and gastric cancer single-cell characteristics
To delve into GMPS’s attributes at the single-cell level, we computed GMPS risk scores for individual cells within the single-cell transcriptome data. Through GSEA, we determined that cells with high-risk scores predominantly participated in biological processes like epithelial-mesenchymal transition, IFN-α response, and intracellular signaling pathways (Fig. 10A). Given the notable role of fibroblasts/pericytes, we categorized them into high-risk and low-risk groups to analyze their interactions with other cell types in the TME. This led to the observation of varied intercellular communication patterns depending on the GMPS scores of fibroblasts/pericytes (Fig. 10B-C). Within the TME, different cell types assume roles such as sender, receiver, mediator, and influencer in cellular communication, orchestrating specific signal exchanges. Our findings revealed that fibroblasts/pericytes with high GMPS scores engage in more diverse interactions with other TME cells and assume more critical roles in mediating and influencing signaling pathways, specifically in FN1, COLLAGEN, and MIF (Fig. 10D–F). In terms of outgoing signaling, fibroblasts/pericytes with high GMPS scores were more prominent, while those with low GMPS scores played a more notable role in incoming signaling patterns (Fig. 10G).

Correlation of GMPS with single-cell characteristics. (A) GSEA analysis in the high-risk group; (B-C) Ligand-receptor interactions of high and low-risk Fibroblast/Pericyte cells; (D-F) Full network diagrams of FN1, COLLAGEN and MIF signaling pathways, with heatmap of pathway network interactions across different cell types; (G) Heatmap of incoming and outgoing signaling patterns for each cell type.
Discussion
Recent years have witnessed increasing focus on the molecular mechanisms and treatment strategies of gastric cancer47,48. While traditional treatment modalities like surgery, radiotherapy, and chemotherapy remain vital in improving patient prognosis, the inherent heterogeneity of gastric cancer presents notable challenges for many patients49,50,51. Recent studies have emphasized the importance of developing novel prognostic biomarkers and personalized therapeutic strategies that combine immune and inflammatory pathways52,53. A comprehensive understanding of molecular biomarkers and cell death pathways is crucial for improving the efficacy of gastric cancer treatments and patient outcomes. However, most current research on gastric cancer has primarily focused on single-omics approaches. The selection of omics clustering methods often relies on personal preference or limited comparative analyses. With growing research, it has become evident that gene expression is regulated by complex genetic and epigenetic processes, including DNA methylation, histone modification, and chromatin remodeling. Therefore, conducting a multi-omics analysis is essential to uncover the specific regulatory mechanisms underlying gastric cancer. In our study, we employed 10 clustering algorithms using mRNA, lncRNA, miRNA, DNA methylation, and somatic mutation data to classify gastric cancer, offering a detailed view of its molecular landscape. This classification not only correlates with patient prognosis but also shows strong associations with programmed cell death and responses to various treatment modalities. Furthermore, our analysis of chromatin remodeling regulators revealed distinct regulatory patterns among subtypes, suggesting that epigenetically-driven transcriptional networks may play a critical role in distinguishing these molecular subtypes. In line with the findings of Meng et al.54, which highlight the association of specific gastric cancer subtypes with immune activation status, TMB, and neoantigens based on DNA methylation differences, our comparison of TMB and CNV between two gastric cancer subtypes further underscores the notable impact of multi-omics variations on patient prognosis.
This study revealed the critical role of PCD features in the occurrence and development of gastric cancer. Specifically, the dysregulation of PCD processes, such as autophagy, pyroptosis, ferroptosis, cuproptosis, and necroptosis, plays a significant role in gastric cancer carcinogenesis. Alterations in the cuproptosis pathway enable cancer cells to evade death signals, promoting uncontrolled proliferation55. Abnormal autophagy allows tumor cells to survive under stress conditions, thereby accelerating tumor progression56. Activation of necroptosis can trigger inflammatory responses, alter the tumor microenvironment, affect immune cell functions, and promote tumor immune evasion and metastasis57,58. In-depth research into these mechanisms enhances our understanding of how PCD influences gastric cancer development by regulating the immune microenvironment and cell signaling pathways. This knowledge provides new insights for developing individualized therapeutic strategies targeting specific subtypes.
Furthermore, our multi-omics analysis found that PCD features exhibited differential dysregulation across the pathological subtypes classified by TCGA, suggesting that PCD may play distinct roles in different subtypes of gastric cancer. This finding aligns with previous studies59. Additionally, other research has confirmed that PCD features show differential dysregulation in Lauren’s pathological classification, further indicating that PCD functions vary among gastric cancer subtypes. For instance, the intestinal type may exhibit alterations in autophagy-related genes, while the diffuse type shows dysregulation in the cuproptosis pathway60,61. These findings support the development of new therapeutic approaches by integrating traditional pathological classifications with molecular subtypes. The involvement of these subtype-specific PCD features highlights the importance of individualized therapeutic strategies targeting these pathways.
Moreover, during tumor progression, PCD processes primarily regulate various immune cell types within the tumor microenvironment, including tumor-associated macrophages, T lymphocytes, and dendritic cells. Inflammatory forms of cell death, such as pyroptosis, can influence macrophage polarization by releasing inflammatory factors, thereby promoting tumor growth and immune evasion62,63. The absence or impairment of immunogenic cell death may lead to reduced release of damage-associated molecular patterns, weakening dendritic cell uptake and activation functions, subsequently affecting the activation of effector T cells and reducing anti-tumor immune responses64. Additionally, ferroptosis can release damage-associated molecular patterns such as HMGB1 and lipid peroxidation products, activating macrophages and triggering inflammatory responses, which affect immune cell recruitment and function65. B cells can inhibit tumor cell necrosis by secreting IL-10, reducing cancer cell death, impairing effector T cell activation, and promoting immune suppression, ultimately supporting tumor growth and progression66. These interactions between PCD processes and immune cells play a critical role in the immune microenvironment of gastric cancer, influencing tumor occurrence and development. An in-depth study of how PCD regulates immune cells will aid in developing new immunotherapeutic strategies and improving patient prognosis.
Epigenetic modifications and somatic mutations also play key roles in regulating PCD processes. Conversely, abnormalities in PCD may affect genomic stability and epigenetic states. In gastric cancer, epigenetic changes such as DNA methylation and histone modifications can influence the expression of PCD-related genes, hindering cancer cell apoptosis and promoting tumor growth67. Additionally, non-coding RNAs like miRNAs regulate PCD pathways through epigenetic mechanisms. For instance, miR-21 regulates the expression of PCD-related genes such as PDCD4, affecting gastric cancer cell survival68. Somatic mutations in key regulatory genes of PCD can disrupt cell death signals, allowing cancer cells to evade death and gain proliferative advantages69. Conversely, abnormalities in PCD processes may lead to the accumulation of DNA damage, inducing genomic instability and increasing the occurrence of somatic mutations70. Moreover, dysregulation of PCD can alter epigenetic states by influencing chromatin conformation and DNA repair mechanisms. These interactions suggest a complex bidirectional regulatory relationship between epigenetics, somatic mutations, and PCD. An in-depth exploration of these mechanisms will enhance our understanding of gastric cancer development and provide a foundation for new therapeutic strategies.
Machine learning offers notable potential for the effective integration of extensive medical data71. However, the use of a single machine learning method often faces limitations due to the complexity of diseases and the subjectivity involved in selecting the most suitable algorithm. To address this, we evaluated 99 combinations of 10 different machine learning methods to determine the most appropriate approach for constructing the GMPS model. Ultimately, the RSF algorithm was selected, and a model comprising five genes was developed, demonstrating strong predictive performance in both the training and validation cohorts. This model not only closely correlates with the prognosis of gastric cancer patients but also highlights the significant role of the immune microenvironment in the disease. Specifically, notable differences were observed in the tumor microenvironment between the high and low GMPS subtypes. The high GMPS subtype, characterized by activated immune suppression pathways, is associated with the ‘cold tumor’ phenotype and a poorer prognosis. In contrast, the low GMPS subtype, which features higher TMB and a stronger response to immune therapy, was validated using external datasets. These findings suggest that GMPS could be a valuable tool for identifying patients who are more likely to benefit from immune therapy. Previous studies by Hu et al. and Shen et al. have explored the correlation between single PCD pathways and gastric cancer prognosis, as well as the impact of crosstalk between two PCD genes on patient outcomes72,73. Compared to models based solely on single PCD or immune, metabolic pathways, GMPS demonstrated superior predictive accuracy across multiple datasets, underscoring the significance of analyzing multiple PCD pathways in conjunction and the value of machine learning in precision medicine.
Single-cell sequencing technology has revolutionized our ability to scrutinize diseases at a more granular level, offering insights into their heterogeneity74,75. Leveraging this advanced technology, our study investigated the relationships between PCD features in gastric cancer at the single-cell level and cellular communication in relation to the GMPS. We noted notable variations in PCD scores among different cell types in gastric cancer, with key GMPS-constructing genes showing marked expression in cells such as fibroblasts/pericytes. Cancer-associated fibroblasts (CAFs) are intricately involved in complex processes like tumor initiation, progression, and immune escape. However, the role of PCD in CAFs in the context of gastric cancer remains underexplored76,77,78. Dividing fibroblasts/pericytes based on GMPS scores into high and low groups, we discerned substantial differences in signaling targets and intensities between these groups. The high-score group predominantly contributed to outgoing signaling patterns, whereas the low-score group was more involved in incoming signals. Given GMPS’s relevance in immune therapy, developing treatment strategies that target CAFs based on GMPS scores could hold notable potential in enhancing the efficacy of immune therapy for gastric cancer.
The potential value of this study lies in demonstrating the importance of multi-omics integrative analysis in gastric cancer research and highlighting the critical role of PCD in the onset and progression of gastric cancer. By constructing the GMPS model, we offer a powerful tool for predicting the prognosis and immunotherapy response of gastric cancer patients, with significant clinical application prospects. Additionally, our research enriches the understanding of the interaction between CAFs and PCD in the tumor microenvironment, providing new avenues for developing therapeutic strategies. However, the specific molecular mechanisms of PCD in gastric cancer and its interactions with other biological processes remain insufficiently understood. Further functional studies and mechanistic explorations are necessary to elucidate the multiple interactions and specific pathways by which PCD influences gastric cancer progression. By combining technologies such as CRISPR gene editing, spatial transcriptomics, and spatial metabolomics, we can achieve a more comprehensive understanding of PCD’s role in gastric cancer development, which will help identify new therapeutic targets and improve patient prognosis. In the future, with the continuous advancement of high-throughput sequencing technologies and the integration of real-world data, we anticipate that the combined analysis of multi-omics data and electronic health records will be widely applied in disease research79. In-depth investigations into PCD and its related pathways will further unveil the molecular mechanisms underlying gastric cancer. Studies based on models like GMPS will enhance the diagnosis and treatment of gastric cancer, offer new perspectives on precision medicine and individualized therapy, and improve patient outcomes.
Overall, this study represents an advancement in understanding the molecular biology of gastric cancer. For the first time, we conducted an extensive investigation into the role of PCD pathways within a multi-omics framework for gastric cancer. Our analysis successfully links PCD variations with clinical features, prognosis, and the tumor microenvironment. The GMPS model we developed serves as a robust tool for guiding precision treatment strategies in gastric cancer. Theoretically, our findings offer a novel perspective, suggesting that gastric cancer pathogenesis is driven by the synergistic effects of multiple omics factors, with PCD playing a central role. Additionally, our research emphasizes the critical interaction between PCD and CAFs within the tumor microenvironment. We hope these insights will inspire further research in this area, ultimately leading to more effective and personalized therapeutic approaches for gastric cancer patients.
It is important to acknowledge certain limitations in our study. Firstly, while our data were sourced from multicenter datasets, the incidence of gastric cancer is intricately linked to a range of factors, including dietary habits, lifestyle, and genetic background80,81,82. Therefore, caution must be exercised when generalizing our findings to gastric cancer patients from different ethnic or regional backgrounds. Although we applied methods such as batch effect correction during data preprocessing, residual technical biases may still persist, and potential systematic errors in the multi-omics data integration process cannot be entirely eliminated. Furthermore, while our analysis comprehensively explores PCD variations in gastric cancer, it lacks in-depth investigation into specific molecular mechanisms, regulatory networks, and interactions with non-PCD pathways. Our study primarily relies on bioinformatics and machine learning methods, without extensive in vivo or in vitro experimental evidence or detailed correlation analyses with clinical data. Consequently, our conclusions require validation through larger-scale prospective studies and experimental investigations. Thirdly, our single-cell RNA sequencing data were derived from only three primary gastric cancer patients, representing a relatively small sample size that may not fully capture the heterogeneity of gastric cancer. Additionally, single-cell sequencing technology itself has certain technical limitations, such as insufficient sequencing depth and gene expression noise, which could affect the accuracy of the results. Fourthly, variations in treatment methods among patients in the different datasets used were not fully accounted for or corrected, potentially influencing our understanding of the relationship between cell death modalities and patient prognosis. Moreover, our model was trained and validated using retrospective data, and it lacks validation with prospective research data. Lastly, the PCD-related genes we selected may not encompass all cell death mechanisms associated with gastric cancer, and other potential factors—such as the microbiota, metabolic status, and environmental influences—were not considered in this study. These factors likely play critical roles in the occurrence and progression of gastric cancer. Therefore, future research should comprehensively incorporate additional biological processes and environmental factors to improve the accuracy and applicability of the model.
Conclusion
In this study, we successfully delineated two distinct subtypes of gastric cancer using a comprehensive multi-omics approach. These subtypes were intricately correlated with prognosis, PCD, and tumor microenvironment characteristics. The machine learning model we developed, based on PCD—the GMPS—exhibited notable clinical applicability and relevance to the immune landscape. This model lays a robust groundwork for personalized treatment approaches in gastric cancer. Additionally, our single-cell analysis underscored the pivotal role of PCD features within the gastric cancer microenvironment. This analysis not only offers fresh insights but also proposes potential biomarkers that could notably enhance the precision of treatment and prognosis assessment for gastric cancer patients.
Data availability
The data supporting this study’s findings are available in TCGA at [https://portal.gdc.cancer.gov], and the GEO at [https://www.ncbi.nlm.nih.gov/geo/], including datasets GSE15459, GSE38749, GSE62254, GSE78220, GSE135222 and GSE163558. Validation clinical trial data at [http://research-pub.gene.com/IMvigor210CoreBiologies] were also used. Detailed references for these resources are provided in the manuscript.
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249 (2021).
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet. 396, 635–648 (2020).
Berlth, F., Bollschweiler, E., Drebber, U., Hoelscher, A. H. & Moenig, S. Pathohistological classification systems in gastric cancer: diagnostic relevance and prognostic value. World J. Gastroenterol. 20, 5679–5684 (2014).
Nature CGARNJ Comprehensive molecular characterization of gastric adenocarcinoma. 513 (7517), 202 (2014).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21, 449–456 (2015).
Ricci, A. D., Rizzo, A. & Brandi, G. DNA damage response alterations in gastric cancer: knocking down a new wall. Future Oncol. (London, England). 17(8), 865–868 (2021).
Rodriquenz, M. G. et al. MSI and EBV positive gastric cancer’s subgroups and their link with novel immunotherapy. J. Clin. Med. 9(5), 1427 (2020).
Yan, H. H. N. et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell. 23, 882-897.e811 (2018).
Rizzo, A. et al. Peripheral neuropathy and headache in cancer patients treated with immunotherapy and immuno-oncology combinations: the MOUSEION-02 study. Exp. Opin. Drug Metab. Toxicol. 17(12), 1455–1466 (2021).
Guven, D. C. et al. Immune checkpoint inhibitor-related hearing loss: a systematic review and analysis of individual patient data. Support. Care Cancer. 31(12), 624 (2023).
Rihawi, K. et al. Tumor-associated macrophages inflammatory microenvironment in gastric cancer: novel translational implications. Int. J. Mol. Sci. 22(8), 3805 (2021).
Sahin, T. K., Rizzo, A., Aksoy, S. & Guven, D. C. Prognostic Significance of the Royal Marsden Hospital (RMH) Score in Patients with Cancer: A systematic review and meta-analysis. Cancers. 16(10), 1835 (2024).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Zou, Y. et al. Leveraging diverse cell-death patterns to predict the prognosis and drug sensitivity of triple-negative breast cancer patients after surgery. Int. J. Surg. 107, 106936 (2022).
Su, Z., Yang, Z., Xu, Y., Chen, Y. & Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 14, 48 (2015).
Zhou, C. B. & Fang, J. Y. The role of pyroptosis in gastrointestinal cancer and immune responses to intestinal microbial infection. Biochim. Biophys. Acta Rev. Cancer. 1872, 1–10 (2019).
Tu, R. H. et al. Neurotransmitter receptor HTR2B regulates lipid metabolism to inhibit ferroptosis in gastric cancer. Cancer Res. 83, 3868–3885 (2023).
Sun, L. et al. Lactylation of METTL16 promotes cuproptosis via m(6)A-modification on FDX1 mRNA in gastric cancer. Nat. Commun. 14, 6523 (2023).
Shen, J. et al. The E3 ubiquitin ligase TRIM17 promotes gastric cancer survival and progression via controlling BAX stability and antagonizing apoptosis. Cell Death Differ. 30, 2322–2335 (2023).
Strusi, G., Suelzu, C. M., Horwood, N., Münsterberg, A. E. & Bao, Y. Phenethyl isothiocyanate and dasatinib combination synergistically reduces hepatocellular carcinoma growth via cell cycle arrest and oxeiptosis. Front Pharmacol. 14, 1264032 (2023).
Maru, B. et al. PARP-1 improves leukemia outcomes by inducing parthanatos during chemotherapy. Cell Rep. Med. 4, 101191 (2023).
Ooi, C. H. et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 5, e1000676 (2009).
Pasini, F. S. et al. A gene expression profile related to immune dampening in the tumor microenvironment is associated with poor prognosis in gastric adenocarcinoma. J. Gastroenterol. 49, 1453–1466 (2014).
Jiang, H. et al. Revealing the transcriptional heterogeneity of organ-specific metastasis in human gastric cancer using single-cell RNA Sequencing. Clin. Transl. Med. 12, e730 (2022).
Hugo, W. et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165, 35–44 (2016).
Jung, H. et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 10, 4278 (2019).
Balar, A. V. et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 389, 67–76 (2017).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 28, 882–883 (2012).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353-d361 (2017).
Safran, M. et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010, baq020 (2010).
Zhou, Y. et al. Implications of different cell death patterns for prognosis and immunity in lung adenocarcinoma. NPJ Precis. Oncol. 7, 121 (2023).
Lu, X., Meng, J., Zhou, Y., Jiang, L. & Yan, F. MOVICS: an R package for multi-omics integration and visualization in cancer subtyping. Bioinformatics. 36, 5539–5541 (2021).
Chalise, P. & Fridley, B. L. Integrative clustering of multi-level ‘omic data based on non-negative matrix factorization algorithm. PLoS One. 12, e0176278 (2017).
Barbie, D. A. et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 462, 108–112 (2009).
Sohn, B. H. et al. Clinical significance of four molecular subtypes of gastric cancer identified by the cancer genome atlas project. Clin. Cancer Res. 23, 4441–4449 (2017).
Li, Y. et al. Proteomic characterization of gastric cancer response to chemotherapy and targeted therapy reveals new therapeutic strategies. Nat. Commun. 13, 5723 (2022).
Lu, X. et al. Multi-omics consensus ensemble refines the classification of muscle-invasive bladder cancer with stratified prognosis, tumour microenvironment and distinct sensitivity to frontline therapies. Clin. Transl. Med. 11, e601 (2021).
Audia, J. E. & Campbell, R. M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, a019521 (2016).
Zeng, D. et al. IOBR: Multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front. Immunol. 12, 687975 (2021).
Xu, L. et al. TIP: A web server for resolving tumor immunophenotype profiling. Cancer Res. 78, 6575–6580 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550 (2005).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell. 177, 1888-1902.e1821 (2019).
Jin, S. et al. Inference and analysis of cell-cell communication using Cell Chat. Nat. Commun. 12, 1088 (2021).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science. 347, 1260419 (2015).
Wang, S. F. et al. Growth differentiation factor 15 induces cisplatin resistance through upregulation of xCT expression and glutathione synthesis in gastric cancer. Cancer Sci. 114, 3301–3317 (2023).
Zhang, C. et al. The NR_109/FUBP1/c-Myc axis regulates TAM polarization and remodels the tumor microenvironment to promote cancer development. J. Immunother. Cancer. 11, e006230 (2023).
Lin, J. X. et al. Neoadjuvant camrelizumab and apatinib combined with chemotherapy versus chemotherapy alone for locally advanced gastric cancer: a multicenter randomized phase 2 trial. Nat. Commun. 15, 41 (2024).
Lei, Z. N. et al. Signaling pathways and therapeutic interventions in gastric cancer. Signal. Transduct. Target Ther. 7, 358 (2022).
Liu, J. Z. et al. BDH2 triggers ROS-induced cell death and autophagy by promoting Nrf2 ubiquitination in gastric cancer. J. Exp. Clin. Cancer Res. 39, 123 (2020).
Rizzo, A. et al. Third- and later-line treatment in advanced or metastatic gastric cancer: a systematic review and meta-analysis. Future Oncol. 16, 4409–4418 (2020).
Vitale, E., Rizzo, A., Santa, K. & Jirillo, E. Associations between “Cancer Risk”, “Inflammation” and “Metabolic Syndrome”: A Scoping Review. Biology (Basel). 13(5), 352 (2024).
Viscardi, G. et al. Comparative assessment of early mortality risk upon immune checkpoint inhibitors alone or in combination with other agents across solid malignancies: a systematic review and meta-analysis. Eur. J. Cancer. 177, 175–185 (2022).
Meng, Q. et al. DNA methylation regulator-mediated modification patterns and tumor microenvironment characterization in gastric cancer. Mol. Ther. Nucleic Acids. 24, 695–710 (2021).
Peng, Y., Shi, R., Yang, S. & Zhu, J. Cuproptosis-related gene DLAT is a biomarker of the prognosis and immune microenvironment of gastric cancer and affects the invasion and migration of cells. Cancer Med. 13, e70012 (2024).
Wang, S. et al. PTBP1 knockdown impairs autophagy flux and inhibits gastric cancer progression through TXNIP-mediated oxidative stress. Cell Mol. Biol. Lett. 29, 110 (2024).
Cheng, X. et al. Atlas of metastatic gastric cancer links ferroptosis to disease progression and immunotherapy response. Gastroenterology. https://doi.org/10.1053/j.gastro.2024.07.038 (2024).
Guo, D. et al. Celastrol induces necroptosis and ameliorates inflammation via targeting biglycan in human gastric carcinoma. Int. J. Mol. Sci. 20(22), 5716 (2019).
Wang, W. et al. CXCR4 induces cell autophagy and maintains EBV latent infection in EBVaGC. Theranostics. 10, 11549–11561 (2020).
Chen, Q., Xu, X. Y., Hou, X. X. & Chen, S. C. The upregulation of proteins light chain 3 and autophagy-related 5 and the occurence of intestinal-type gastric cancer. J. Physiol. Pharmacol. 72(6), 8 (2021).
Minikes, A. M. et al. E-cadherin is a biomarker for ferroptosis sensitivity in diffuse gastric cancer. Oncogene. 42, 848–857 (2023).
Yang, J. et al. FABP4 in macrophages facilitates obesity-associated pancreatic cancer progression via the NLRP3/IL-1β axis. Cancer Lett. 575, 216403 (2023).
Mónaco, A., Chilibroste, S., Yim, L., Chabalgoity, J. A. & Moreno, M. Inflammasome activation, NLRP3 engagement and macrophage recruitment to tumor microenvironment are all required for Salmonella antitumor effect. Cancer Immunol. Immunother. 71, 2141–2150 (2022).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125 (2021).
Jung, Y. J. et al. Effect of IL-10-producing B cells in peripheral blood and tumor tissue on gastric cancer. Cell Commun. Signal. 21, 320 (2023).
Yousefi, B. et al. Epigenetic changes in gastric cancer induction by Helicobacter pylori. J Cell Physiol. 234, 21770–21784 (2019).
Farasati Far, B. et al. The role of microRNA-21 (miR-21) in pathogenesis, diagnosis, and prognosis of gastrointestinal cancers: A review. Life Sci. 316, 121340 (2023).
Perillo, B. et al. ROS in cancer therapy: the bright side of the moon. Exp. Mol. Med. 52, 192–203 (2020).
Timmerman, D. M., Remmers, T. L., Hillenius, S. & Looijenga, L. H. J. Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells-Relevance for Understanding (Germ Cell) Tumorigenesis. Int. Mol. Sci. 22(10), 5377 (2021).
Nam, D., Chapiro, J., Paradis, V., Seraphin, T. P. & Kather, J. N. Artificial intelligence in liver diseases: Improving diagnostics, prognostics and response prediction. JHEP Rep. 4, 100443 (2022).
Hu, J. et al. A novel pyroptosis gene expression-based risk score for survival in gastric cancer. Front Endocrinol. (Lausanne). 14, 1120216 (2023).
Shen, Y. et al. Cross-talk between cuproptosis and ferroptosis regulators defines the tumor microenvironment for the prediction of prognosis and therapies in lung adenocarcinoma. Front Immunol. 13, 1029092 (2022).
Huang, X. Z. et al. Single-cell sequencing of ascites fluid illustrates heterogeneity and therapy-induced evolution during gastric cancer peritoneal metastasis. Nat. Commun. 14, 822 (2023).
Zulibiya, A. et al. Single-cell RNA sequencing reveals potential for endothelial-to-mesenchymal transition in tetralogy of fallot. Congenit. Heart Dis. 18, 611–625 (2023).
Qu, X. et al. Loss of cancer-associated fibroblast-derived exosomal DACT3-AS1 promotes malignant transformation and ferroptosis-mediated oxaliplatin resistance in gastric cancer. Drug Resist. Updat. 68, 100936 (2023).
Li, X. et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics. 12, 620–638 (2022).
Maeda, M. et al. Cancer cell niche factors secreted from cancer-associated fibroblast by loss of H3K27me3. Gut. 69, 243–251 (2020).
Foote, H.-P. et al. Using pharmacokinetic modeling and electronic health record data to predict clinical and safety outcomes after methylprednisolone exposure during cardiopulmonary bypass in neonates. Congenit. Heart Dis. 18, 295–313 (2023).
Hui, Y., Tu, C., Liu, D., Zhang, H. & Gong, X. Risk factors for gastric cancer: A comprehensive analysis of observational studies. Front. Public Health. 10, 892468 (2022).
Mohseni, S. et al. Effect of vitamins C and E on cancer survival; a systematic review. Daru. 30, 427–441 (2022).
Usui, Y. et al. Helicobacter pylori, homologous-recombination genes, and gastric cancer. N. Engl. J. Med. 388, 1181–1190 (2023).
Acknowledgements
We would like to express our gratitude to the maintainers of public databases for providing valuable data resources for this study, and to the developers in the R community who have offered robust algorithms and software packages. We also appreciate the assistance from our colleagues in data collection and analysis.
Author information
Authors and Affiliations
Contributions
ZHB, HW, and JRH conceptualized the research, downloaded, and analyzed the data. ZHB and JA produced the figures. ZHB, HW, and JRH wrote the manuscript. XMM and ZCY supervised and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bai, Z., Wang, H., Han, J. et al. Multiomics integration and machine learning reveal prognostic programmed cell death signatures in gastric cancer. Sci Rep 14, 31060 (2024). https://doi.org/10.1038/s41598-024-82233-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-82233-w
Keywords
This article is cited by
-
WHFDL: an explainable method based on World Hyper-heuristic and Fuzzy Deep Learning approaches for gastric cancer detection using metabolomics data
BioData Mining (2025)
-
CER1 as a manganese ion metabolism gene drives gastric cancer progression and therapeutic potential via oxidative stress and tumor microenvironment regulation
Discover Oncology (2025)
