
Abstract
Chronic kidney disease is defined as a progressive loss of kidney function associated with impaired recovery after acute kidney injury. Renal ischemia-reperfusion (IR) induces oxidative stress and inflammatory responses leading to severe tissue damage, where incomplete or maladaptive repair accelerates renal fibrosis and aging. To investigate the role of the purinergic P2Y2 receptor (P2Y2R) in these processes, we used P2Y2R knockout (KO) mice subjected to IR. KO mice showed severe kidney dysfunction and structural damage compared to WT mice. KO mice showed higher senescence-associated β-galactosidase expression and shorter telomere length than WT mice. Consistently, interstitial collagen accumulation and fibrogenic mediators were significantly upregulated in KO mice. Renal apoptosis and inflammation were highly elevated in KO mice. Interestingly, cell proliferation as shown by Ki-67 and PCNA expression, was increased for 3 days after IR in WT mice, whereas it maintained increased for 14 days in KO mice. Cell cycle inhibitors, p16 and p21, and regulators JunB and cyclin E were significantly increased after IR in KO mice, suggesting that cell cycle progression was impaired during recovery after IR. Proximal tubular cells treated with JunB siRNA showed a reduced expression of fibrogenic mediators and proinflammatory cytokines, consistent with the mice treated with MRS2768, a P2Y2 agonist that downregulated JunB levels. In conclusion, P2Y2R reduces kidney tissue damage after IR and repairs the tissue properly by regulating JunB-mediated signaling during the recovery process.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is a common condition in hospitalized patients, often resulting from ischemia-reperfusion (IR) injury, sepsis, or nephrotoxic medications1. AKI is a significant risk factor for the onset of chronic kidney disease (CKD). Complete recovery from AKI is uncommon, and impaired recovery can expedite the progression to CKD2,3. The repair process involves stress and metabolic adaptation (homeostasis), inflammatory responses, extracellular matrix production, cell proliferation, and dedifferentiation4,5. However, when these repair processes become excessive, uncontrolled, or sustained, they contribute to renal dysfunction and structural damage leading to renal fibrosis6,7. While considerable attention has been on pathology after ischemia or in the early stages after reperfusion8, there remains a lack of complete understanding regarding the recovery phase after IR and its underlying mechanisms.
During the recovery phase, adaptive responses are exerted to repair damaged tissue and restore renal function by tubular cell proliferation and differentiation9,10. However, severe or prolonged injury disrupts homeostasis and shifts toward maladaptive responses, resulting in tissue dysfunction and CKD progression3,8,11. Severe AKI causes DNA damage and cell cycle arrest, leading to impaired tissue regeneration and enhanced renal fibrosis and aging12,13.
Adenosine triphosphate (ATP) is released from cells in response to various stimuli, such as tissue injury, mechanical stress, inflammation, and hypoxia14. Extracellular ATP functions as a damage-associated molecular pattern molecule, triggering inflammatory and apoptotic responses. The ATP binds to purinergic P2 receptors, including ionotropic P2X and G-protein-coupled P2Y receptors15. Currently, eight mammalian P2Ys have been identified: P2Y1, 2, 4, 6, 11, 12, 13, and 14. P2Y2R plays a crucial role in regulating immune responses to resolve acute injury, and its expression is elevated in rats after spinal cord injury16, and in the duct-ligated rat submandibular gland17. Furthermore, P2Y2R mediates neuronal regeneration and vascular proliferation in rats after spinal cord injury18. P2Y2R promotes epithelial repair during wound healing and airway remodeling by inducing the proliferation and migration of pulmonary cells; its aberrant regulation results in the development of hyperplasia, fibrosis, and neovascularization, leading to the maladaptive repair of the pulmonary system19. The deletion of P2Y2R results in severe renal injury following subtotal nephrectomy20. P2Y2R recruits immune cells after acute injury, contributing to the clearance of apoptotic cells and promoting proliferation and differentiation of structural cells; however, if left uncontrolled, it can lead to chronic inflammation, fibrosis, and accelerated aging15. However, the specific role and underlying mechanisms of P2Y2R in the development of renal fibrosis and aging following AKI have not been fully elucidated.
In this study, we used P2Y2R knockout (KO) mice to investigate the role of P2Y2R during recovery after renal IR. P2Y2R KO mice exhibited significant increases in renal tubular damage, apoptosis, and inflammatory and fibrogenic responses, compared to WT mice. KO mice delayed cell proliferation during recovery after IR by inhibiting cell cycle progression through JunB signaling. We suggest that P2Y2R activation is required to reduce tubular apoptosis and inflammation after IR, regenerate damaged tissues properly, and thus prevent renal fibrosis and aging.
Materials and methods
Animals
Wild-type (WT) C57BL/6 mice were purchased from Koatech (Pyeongtaek, South Korea), and P2Y2R KO mice on C57BL/6 background (B6.129P2-P2ry2tm1Bhk/J) were obtained from Jackson Laboratory (Bar Harbor, Maine, USA). All mice were maintained in the animal facility of Gyeongsang National University. All animal experiments were approved by the Institutional Board of Animal Research at Gyeongsang National University (GNU-180615-M0028) and conducted in accordance with the National Institutes of Health guidelines for laboratory animal care. All authors complied with the ARRIVE guidelines. Mice were maintained with a 12-h light/dark cycle and provided freely with water and standard chow.
Animal model of renal IR injury
Male mice (7 weeks old) were habituated for 1 week. The mice were anesthetized with zoletil (0.5 mg/kg; Virbac Laboratories, Carros, France), and supine was placed on a heating pad under a heat lamp to maintain body temperature. After the abdominal incision, left and right renal pedicles were clamped with microvascular clips for 25 min. Mice remained hydrated with warm saline during the ischemic period, and the incision was sutured after removing the clamps. The sham mice were subjected to the same surgical procedure without clamping. MRS2768 (Tocris Bioscience, Bristol, UK) was dissolved in saline and administered intraperitoneally at a dose of 25 µg/kg. Treatment started 24 h after IR injury and was given daily until the mice were sacrificed. Mice were sacrificed 1, 3, 7, or 14 days after reperfusion (n = 6 for each group; total n = 24), and blood and kidney tissues were collected. The kidney tissues were rapidly frozen in liquid nitrogen for storage at -80 °C or fixed in 10% buffered formalin. Blood was from an inferior vena cava by using a heparinized syringe, centrifuged at 3,000 x g for 15 min, and the supernatants were stored at -80 °C for biochemical analysis. For survival analysis, 12 mice per group were subjected to the surgical procedure as described above, and the number of surviving mice was determined every morning (~ 9 am) for 14 days.
Biochemical assays
Plasma creatinine (Cr) was measured by a direct colorimetric Jaffe method and detected by using a spectrophotometer (Shimadzu UV-1800 spectrophotometer, Tokyo, Japan), as previously described21. Blood urea nitrogen (BUN) was measured using a commercial assay kit from IVDLab (Uiwang, Republic of Korea).
Periodic acid-Schiff (PAS) and Picro-Sirius Red staining
Kidney tissues were fixed in 10% formalin for 24 h, processed for paraffin embedding, and sectioned at 5-µm. Kidney sections were stained with PAS staining (Abcam, Cambridge, UK) for histological analysis, and histological scoring was performed as previously described21. Picro-Sirius Red staining (Abcam) was performed to visualize collagen deposition following a standard protocol. All images were captured by using a CKX41 light microscopy (Olympus, Tokyo, Japan).
Immunohistochemistry
The sections were deparaffinized, rehydrated, and antigen-retrieved in sodium citrate buffer (10 mM, pH 6.0) for 20 min. Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide, and nonspecific binding was blocked with 10% normal goat serum. The sections were incubated with a primary antibody against Ly-6B.2 (Bio-Rad, Hercules, CA, USA) or marker of proliferation Kiel 67 (Ki-67) (Abcam) overnight at 4 °C, and with a respective biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. The sections were incubated in an avidin-biotin-peroxidase complex solution (ABC solution; Vector Laboratories) for 30 min and developed using a 3,3ʹ-diaminobenzidine (DAB) Peroxidase Substrate Kit (Vector Laboratories). Then, the sections were counterstained with hematoxylin, and the images were obtained by a CKX41 light microscope (Olympus).
Immunofluorescence staining
The kidney sections were blocked in 2.5% normal horse serum and incubated with anti-proliferating cell nuclear antigen (PCNA) antibody (Abcam) for overnight at 4 °C. After washing, the sections were incubated with goat anti-rabbit secondary antibody conjugated with Alexa 488 (Vector Laboratories) for 1 h at RT and mounted with a ProLong Gold anti-fade mounting solution (Invitrogen, Waltham, MA, USA). The images were captured using a Fluoview 1000 (IX-81) microscope (Olympus) and analyzed by Image J (NIH, Bethesda, MD, USA).
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
TUNEL staining was performed by using an In situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer’s instruction. The images were captured using a Nikon Eclipse Ti-U microscope (Tokyo, Japan) and quantified by Image J (NIH). The number of TUNLE-positive cells was counted from five microscopic fields (200X) per section from each group (n = 3).
Western blot analysis
Kidney tissues were homogenized in an ice-cold RIPA buffer with protease inhibitors (Thermo Fisher Scientific, Waltham, MA, USA), sonicated, and incubated for 20 min on ice. The lysates were separated by electrophoresis, and transferred to polyvinylidene difluoride membranes (Bio-Rad). After blocking for 1 h at room temperature (RT), the membranes were incubated overnight at 4 °C in primary antibodies against caspase 3 (#9662), cyclin D1 (#2922), JunB (#3753), p21(#2947) (Cell Signaling Technology, Danvers, MA, USA); cyclin E (sc-247, Santa Cruz Biotechnology, Santa Cruz, CA, USA); PCNA (ab15497, Abcam); β-actin (A5441, Sigma-Aldrich, St. Louis, MI, USA) at 4 °C overnight. After washing, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) at RT for 1 h. Then, the membranes were developed using a Clarity™ Western ECL Substrate (Bio-Rad), and the protein band intensity was analyzed by the ChemiDoc XRS + System (Bio-Rad).
Silencing of P2Y2R and JunB by RNA interference
Small interfering RNA (siRNA) targets of P2Y2R and JunB were purchased from Bioneer (Daejeon, Korea). The siRNA oligonucleotides were synthesized to the following target sequences: P2Y2R; sense (5’-GAGGAAGGUGGCUUACCAA-3’) and antisense (5’-UUGGUAAGCCACCUUCCUC-3’), JunB; sense (5’-GAAAAGAAACACGCACUUA-3’) and antisense (5’-UAAGUGCGUGUUUCUUUUC-3’) and scramble control; 5’-GGAAAGACUGUUCCAAAAA-3’. The siRNAs were introduced into cells using Lipofectamine RNAiMAX (Invitrogen) by following the procedure recommended by the manufacturer.
Cell cycle analyses
Flow cytometry was used to analyze cell cycle distribution. HK2 cells (3 × 105 cells/well) were plated and transfected with scramble, P2Y2R, or JunB siRNA for 48 h. Cells were collected and fixed with cold 70% ethanol for 1 h at 4 °C. Cells were washed with PBS and incubated with RNase A (Qiagen, Hilden, Germany) and propidium iodide (5 µg/mL, Sigma-Aldrich) for 30 min at 4 °C in the dark. Cell cycle analysis was performed (FACS Calibur, BD Biosciences, Frankline Lakes, NJ, USA). The distribution of cells in the G0/G1, S, and G2/M phases was determined as the percentage of the total number of cells (FCS Express software, De Novo Software, Pasadena, CA, USA).
Quantitative reverse transcription PCR
Total RNA was extracted using Trizol (Invitrogen) and converted to cDNA using the RevertAid Reverser Transcription System (Thermo Fisher Scientific) according to the manufacturer’s instructions. Real-time PCR analysis was performed with a CFX Connect real-time PCR detection System using iQ SYBR Green Supermix (Bio-Rad). Relative mRNA levels were normalized to those of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences are shown in Table 1.
Telomere length analysis by quantitative real-time PCR
Genomic DNA (gDNA) was extracted from the kidney using a DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol. Telomere length was analyzed by a direct qPCR method as previously described22. The PCR reaction was optimized in 20 µL total reaction volume in a 96-well plate containing 20 ng gDNA, 10 µL TopReal 2x premix (Enzynomics, Daejeon, Korea), sense and antisense primers (900 nM each for telomere, 500 nM each for 36B4). The thermal program was one cycle at 95 °C for 10 min, two cycles at 95 °C for 15 s and 49 °C for 15 s for pre-amplification, followed by 45 cycles at 95 °C for 15 s, 62 °C for 10 s, and 72 °C for 15 s for target-amplification. Primers for telomere and a single-copy reference gene 36B4 were designed as previously reported and shown in Table 223,24.
Senescence-associated β-galactosidase (SA-β-gal) staining
The SA-β-gal assay was performed using an SA-β-gal staining kit (Roche, #11828673001) to determine the senescence state of cells according to the manufacturer’s instructions. Briefly, the tissues were embedded in OCT compound (Tissue-Tek OCT compound by Sakura, Torrance, CA), and flash-freezed in lipid nitrogen containing isopentane. Then the frozen tissues were sectioned at 5-µm and fixed in fixative solution for 10 min at RT. The tissues were washed with PBS, and incubated overnight at 37 °C with a fresh SA-β-gal staining solution without CO2. After mounting, all images were captured using a CKX41 light microscope (Olympus).
Statistical analysis
Statistical difference was determined using a two-tailed student t-test to compare two groups or by two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test for multiple groups. All statistical analyses were performed by GraphPad Prism 9 Software v.9.50 (GraphPad Software Inc., La Jolla, CA, USA). Data were expressed as means ± standard error of the mean (SEM). A P value < 0.05 was considered statistically significant.
Results
The deficiency of P2Y2R severely damages renal structure and function and accelerates aging after renal IR in mice
To investigate the role of P2Y2R during kidney damage and recovery after acute kidney injury, P2Y2R KO and WT mice were subjected to renal ischemia and collected the plasma and kidney tissues after perfusion of 1, 3, 7, and 14 days. The mRNA expression of P2Y2R was also confirmed in WT and KO mice (Fig. 1A). First, we measured plasma Cr and BUN levels and the mRNA expression of neutrophil gelatinase-associated lipocalin (NGAL), a renal injury marker (Fig. 1B-C). The increased levels of plasma Cr, BUN, and NGAL expression were attenuated along the recovery period (3 ~ 14 days) after reperfusion; however, the levels were significantly higher in KO mice than in WT mice. Then, histological analysis of kidney sections was performed by HE staining. KO mice showed higher levels of tubular injury than WT mice, accompanied by increased tubular necrosis and congestion, vacuolization, and simplification (Fig. 1D).

P2Y2R deficiency aggravates renal dysfunction and tubular damage during recovery after renal IR. Blood and kidney tissues were collected from WT and P2Y2R KO mice after sham operation or 1, 3, 7, and 14 days after renal IR. (A) Relative mRNA expression of P2Y2R was determined by real-time PCR analysis. (B,C) Plasma creatinine and BUN levels were measured and renal expression of NGAL was determined by real-time PCR analysis. (D) Representative HE-stained images of kidney sections are shown, and the extent of renal injury was scored as described. Arrows indicate the tubular necrosis. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT sham group, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. respective WT IR group. Scale bar, 100 μm.
The renal aging and fibrosis phenotypes were investigated after renal IR. The survival rate (%) at 2 days after renal IR was 90% for WT mice and 80% for KO mice, and was maintained until 14 days without significant difference (Fig. 2A). The senescence-associated β-galactosidase (SA-β-gal) staining showed a strong signal in KO mice compared to WT mice after IR (Fig. 2B). Telomere length was analyzed by a qPCR method optimized previously22 and the kidney age was estimated by a standard aging curve calculated from telomere length. The renal IR injury decreased average telomere length in WT mice, and the length was further decreased in KO mice after IR (Fig. 2C). The kidney aged faster in KO mice (~ 23-month old) than in WT mice (13.7-month old) after IR. Consistently, KO mice exhibited a higher accumulation of interstitial collagen than WT mice, as indicated by Sirius red staining (Fig. 2D). The mRNA expression of fibrogenic mediators, including fibronectin, collagen-I, transforming growth factor-β (TGF-β), and S100 calcium-binding protein A4 (S100A4) was significantly upregulated after IR, and KO mice showed higher levels than WT mice (Fig. 2E). These results suggest that P2Y2R is required during recovery after renal IR by reducing kidney structural and functional damage and delaying aging and fibrosis.

P2Y2R deficiency accelerates renal aging and fibrosis during recovery after IR. Renal IR was subjected to WT and KO mice, and kidney tissues obtained from the mice 14 days after IR were accessed for staining and telomere analysis. (A) Mouse survival percentages in mice after renal IR (n = 12). (B) Representative images of senescence-associated β-galactosidase (SA-β-gal) staining in the kidney sections. (C) The average telomere length was determined by qPCR analysis, and the kidney age was estimated according to telomere length. (D) Picro-sirius red staining was performed in kidney sections (arrows indicate collagen accumulation), and fibrotic area was presented as percentages (%) by using ImageJ software. (E) Relative mRNA expression of fibronectin, collagen-1, transforming growth factor (TGF)-β, and S100 calcium-binding protein A4 (S100A4) was determined by real-time PCR analysis. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT sham group, #P < 0.05, ###P < 0.001 vs. respective WT IR group. Scale bar, 100 μm.
The deficiency of P2Y2R increases renal inflammation and apoptosis after IR in mice
P2Y2R is shown to promote neutrophil infiltration as an essential chemotactic receptor to resolve damaged tissues and attenuate chronic inflammatory responses25,26. Thus, renal inflammation and apoptosis phenotypes were investigated in WT and KO mice after IR. First, the number of neutrophils recruited to the tubular cells was analyzed by immunohistochemical staining using an anti-Ly-6B.2 antibody. KO mice showed a severe neutrophil infiltration compared to WT mice after IR (Fig. 3A). Consistently, mRNA expression of tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and interleukin-6 (IL-6) was elevated in higher levels in KO than in WT mice (Fig. 3B). Second, TUNEL staining showed that the number of apoptotic cells was higher in KO than WT mice (Fig. 3C); the cleaved levels of caspases-3 was also higher in KO than in WT mice after IR (Fig. 3D). These results suggest that P2Y2R is required to resolve the excessive induction of neutrophils and proinflammatory cytokines and renal apoptosis after IR.

P2Y2R deficiency aggravates renal inflammation and apoptosis during recovery after IR. Kidney tissues were collected from WT and P2Y2R KO mice after sham operation or 1, 3, 7, and 14 days after renal IR. (A) Representative images of immunohistochemistry for neutrophil infiltration are shown by using an anti-Ly-6B.2 antibody. The number of stained neutrophils per x200 field image was counted. (B) Relative mRNA expression of tumor necrosis factor tumor necrosis factor (TNF-α), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and interleukin-6 (IL-6) was determined by real-time PCR analysis. (C) Representative TUNEL images and the quantification of apoptotic cells are shown. (D) Representative immunoblots of uncleaved- and cleaved caspase-3 and β-actin and the quantification of relative protein expression are shown. Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001 vs. WT sham group, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. respective WT IR group. Scale bar, 100 μm.
The deficiency of P2Y2R delays renal cell proliferation through JunB after IR in mice
To investigate the role of P2Y2R in renal cell proliferation during recovery after IR, the expression of proliferation markers, Ki-67 and PCNA, were analyzed (Fig. 4). There was a significant induction of Ki-67 expression for 1–7 days with a peak expression at 3 days after IR in WT mice, whereas the increased expression was maintained until 14 days after IR in KO mice, as shown in immunohistochemical staining (Fig. 4A) and qPCR analysis (Fig. 4B). Similarly, WT mice showed a significant PCNA induction for 3–7 days after IR; however, KO mice showed the increased levels until 14 days, as shown in immunofluorescence staining (Fig. 4C) and Western blot analysis (Fig. 4D).

P2Y2R deficiency delays renal tubular cell proliferation during recovery after IR. Kidney tissues were collected from WT and P2Y2R KO mice after sham operation or 1, 3, 7, and 14 days after renal IR. (A) Representative images of immunohistochemistry for proliferating cells are shown by using an anti-Ki-67 antibody and the number of stained cells was counted. (B) Relative mRNA expression of Ki-67 was determined by real-time PCR analysis. (C) Representative immunofluorescence images of proliferating cells are shown by using an anti-PCNA antibody. (D) Representative immunoblots of PCNA and β-actin and the quantification of relative protein expression are shown. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT sham group, #P < 0.05, ###P < 0.001 vs. respective WT IR group. Scale bar, 100 μm.
To investigate the role of P2Y2R in cell cycle progression during recovery after IR, the mRNA expression of p16 and p21, cyclin-dependent kinase inhibitors, was determined (Fig. 5A). WT mice showed a significant induction of p16 at 3 days after IR, whereas KO mice showed a delayed induction at 7–14 days. KO mice showed a higher increase in p21 than WT mice for 1–14 days after IR. These results suggest that P2Y2R is required for proper cell cycle control during recovery after IR. Further, the renal expression of JunB, a transcription factor and cell cycle regulators, cyclin D1 and cyclin E, was determined after IR (Fig. 5B). The levels of JunB and cyclin E in WT mice were increased at 1–3 days and decreased afterward, whereas the levels in KO mice were increased again at 14 days after IR. The levels of cyclin D1 were decreased for 1–7 days after IR compared to WT mice. These results suggest that P2Y2R is required for a proper cell cycle progression, possibly through Jun B, cyclin D1, and cyclin E signaling during recovery after renal IR.

P2Y2R deficiency alters the expression of cell cycle regulators during recovery after IR. Kidney tissues were collected from WT and P2Y2R KO mice after sham operation or 1, 3, 7, and 14 days after renal IR. (A) Relative mRNA expression of p16 and p21, cyclin-dependent kinase inhibitors, was determined by real-time PCR analysis. (B) Representative immunoblots for JunB, cyclin D1, cyclin E, and β-actin, and the quantification of relative protein expression are shown. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. WT sham group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. respective WT IR group.
The deficiency of JunB attenuates renal inflammation and fibrosis in proximal tubular cells exposed to TGF-β
To investigate the role of P2Y2R in cell cycle progression during fibrosis, TGF-β was treated in P2Y2R deficient (P2Y2R siRNA) cells, and the cell cycle stages and expression of cell cycle regulators, p21 and JunB were investigated. First, we confirmed the mRNA expression of P2Y2R in cells treated with scramble or P2Y2 siRNA (Fig. 6A). TGF-β treatment increased p21 and JunB levels, and the levels were further elevated by P2Y2R siRNA (Fig. 6A), suggesting that JunB is a critical regulator of P2Y2R in cell cycle progression during fibrogenesis. Consistently, the mRNA expression of JunB was further elevated by P2Y2R siRNA (Fig. 6B). Cell cycle progression was analyzed by flow cytometry, and the results showed that P2Y2R deficient cells inhibit G1/S transition, which was restored by JunB siRNA (Fig. 6B). Then, we investigated whether JunB deficient cells decrease renal fibrosis and inflammation, which were induced by TGF-β and enhanced further by P2Y2R siRNA. The results showed that mRNA expression of fibrogenic mediators (fibronectin and collagen I) and pro-inflammatory cytokines (TNF-α, MCP-1, MIP-2, and IL-6) were downregulated (Fig. 6C and D), suggesting that JunB-mediated signaling is important for regulation of P2Y2R on cell cycle progression.

JunB deficiency alters cell cycles and attenuates the expression of fibrogenic mediators and cytokines in proximal tubular HK2 cells. Cells were transfected with siRNA of scrambled, P2Y2R, or JunB for 24 h, and treated with TGF-β (5 ng/ml) for 24, 48, or 72 h. (A) After transfection with P2Y2R siRNA, relative P2Y2R mRNA expression was determined by real-time PCR analysis. Relative expression of JunB, p21, cyclin D1, cyclin E, and β-actin was analyzed by western blot and quantified. (B) After transfection with P2Y2R or JunB siRNA, relative JunB mRNA expression was determined by real-time PCR analysis. Percentage of cells in respective cell cycle stages after treatment of scrambled or siRNA of P2Y2R or JunB for 48 h. (C) Relative mRNA expression of TNF-α, MCP-1, MIP-2, and IL-6 was determined by real-time PCR analysis. (D) Relative mRNA expression of fibronectin and collagen I was determined by real-time PCR analysis. Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001 vs. vehicle group of scramble siRNA. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. respective TGFβ treatment group of scramble siRNA.
P2Y2R activation attenuates renal dysfunction, fibrosis, and inflammatory responses induced after IR by reducing JunB expression
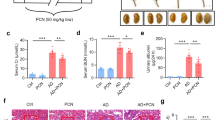
To investigate the effect of P2Y2R activation on the recovery phase after renal IR, we examined renal structure and function, and markers of fibrosis and inflammation by treating the mice with a selective P2Y2R agonist, MRS2768 (MRS, 25 µg/kg). MRS2768 was administered intraperitoneally starting at 24 h after IR injury and continued daily until sacrifice. Blood and kidney tissues were analyzed 3 days after renal IR. Plasma BUN levels and tubular injury increased after IR were significantly reduced by MRS treatment (Fig. 7A and B). In addition, fibrogenic mediators (TGF-β, fibronectin, and α-SMA) and proinflammatory cytokines (TNF-α, MCP-1, and MIP-2) were reduced by MRS treatment in mice subjected to renal IR, compared to vehicle treatment (Fig. 7C and D). JunB expression increased after IR was significantly reduced by MRS treatment, suggesting that JunB-mediated cell cycle progression was impaired during recovery after IR and restored by P2Y2 activation (Fig. 7E). After acute kidney injury, damaged proximal cells are cleared and replaced with newly proliferated and differentiated cells27; however, severe or repeated injury fails to complete the repair process, and P2YR deficiency alters cell cycle regulators p16, p21 through JunB signaling, resulting in increased maladaptive cells in cell cycle arrest, secreting fibrogenic cytokines, and accelerating aging.

P2Y2R activation by MRS treatment attenuates the renal dysfunction and expression of fibrogenic mediators and cytokines induced after IR. Blood and kidneys were collected from WT mice after a sham operation or 3 days after renal IR with vehicle or MRS treatment. (A) Plasma BUN levels were measured. (B) Representative PAS-stained images of kidney sections are shown. (C) Relative mRNA expression of TGF-β, fibronectin, and α-SMA was determined by real-time PCR analysis. (D) Relative mRNA expression of TNF-α, MCP-1, and MIP-2 was determined by real-time PCR analysis. (E) Representative immunoblots for JunB and β-actin and the quantification of relative protein expression are shown. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham group. ##P < 0.01, ###P < 0.001 vs. IR group. Scale bar, 100 μm.
Discussion
P2Y2R is involved in various diseases such as arterial hypertension28, cystic fibrosis29, dry eyes30, and neurodegeneration31. In the kidney, P2Y2R deficiency aggravates CKD progression after subtotal nephrectomy in mice20; however, its molecular mechanism is not clearly shown. In this study, we investigated the role of P2Y2R in renal fibrosis and aging after IR using P2Y2R KO mice. KO mice showed severe kidney injury and dysfunction, accelerated telomere shortening, and cellular senescence phenotypes. Consistently, KO mice exhibited enhanced collagen accumulation and expression of fibrogenic mediators and severe inflammatory and apoptotic responses, compared to WT. KO mice showed delayed and prolonged expression of cell proliferative markers and altered expression of cell cycle regulators, which indicates that P2Y2R is required for renal fibrosis and aging processes.
Cellular senescence is induced by several mechanisms, including telomere shortening, DNA damage, mitochondrial dysfunction, endoplasmic reticulum stress, metabolic imbalances, and epigenetic remodeling. Cellular senescence is an irreversible state of cell cycle arrest and exhibits senescence-associated secretory phenotype (SASP) and apoptotic resistance phenotype12. Renal tubular epithelial cells (RTEC) are susceptible to cellular senescence associated with AKI and can be transformed to function as inflammatory and fibrogenic cells during epithelial-mesenchymal transition, cell cycle arrest, and metabolic changes, contributing to the progression of CKD32. Patients with IgA nephropathy exhibit accelerated senescence of RTEC, showing increased p16, p21, and SA-β-gal expression, associated with high blood pressure and renal dysfunction33. Older donors are also more prone to IR injury in renal transplantation than younger donors34, suggesting RTEC senescence is a critical pathological feature35.
The pathways regulating cell cycle arrest are at cell cycle checkpoints, and cycle-dependent kinase inhibitors p53/p21 and p16/Rb pathways are the primary pathways driving RTEC senescence following AKI12. During severe and persistent AKI, senescent cells are accumulated and secrete SASP, causing inflammation and fibroblast proliferation, leading to maladaptive renal repair and fibrosis. Renal fibrosis is aggravated in senescent RTEC, where G2/M arrest activates JNK signaling and upregulates fibrogenic cytokine production36. In this study, KO mice showed upregulation of p21 following IR, suggesting that the accumulation of arrested cells secretes profibrogenic growth factors such as TGF-β1, and promotes interstitial fibrosis37. Additional research is needed to understand the exact mechanisms of P2Y2R regulating cell cycle progression during the recovery process after renal IR.
The cycle-dependent kinase inhibitors enhance checkpoint mechanisms, promoting cell cycle arrest at the G1/S or G2/M checkpoint and culminating senescence phenotypes. Loss of p16 increases epithelial cell proliferation and microvascular repair, protecting mice after renal IR38, while loss of p21 is susceptible to IR-induced renal failure and increased mortality associated with inappropriate cell cycle activity, indicating precise cell cycle control is required for repair processes39. In this study, mRNA levels of p16 and p21 were increased markedly in P2Y2R KO mice (p16 for 7–14 days; p21 for 1–14 days) after IR, indicating accumulation of senescent cells and accelerated SASP secretion and renal fibrosis. P2Y2R regulates cell cycle progression possibly through p16 and p21 in the G1/S transition, where the cyclin D and CDK4/6 complex and/or the cyclin E and CDK2 complex are involved13,40. Interestingly, the deficiency of P2Y2R reduced the protein expression of cyclin D at 1–7 days after IR and increased cyclin E at 14 days after IR, suggesting P2Y2R is required for appropriate cell cycle progression during the renal repair process after IR.
JunB is a transcription factor showing positive or negative transcriptional activity, depending on cell proliferation state and targeting genes41. JunB is shown to directly target p16 and induce premature senescence as a negative regulator of cell proliferation42. However, JunB inhibits cyclin D1 during the G1 phase and also stimulates cyclin A2 during the S phase, promoting cell cycle activity43. A recent study reports that JunB promotes cell cycle progression through cyclin E1 and TGF-β244. Interestingly, KO mice increased JunB expression, while P2Y2R activation by MRS decreased JunB expression after IR. In RTEC deficient of JunB, cell cycle arrest was restored, and the fibrogenic cytokine production was reduced. These results suggest that P2Y2R may regulate cell cycle progression through JunB in a cell cycle-specific manner; however, the precise mechanism must be elucidated to develop treatments.
In summary, P2Y2R is required for proper cell cycle progression of RTEC during recovery after renal IR (Fig. 8). P2Y2R activation regulates cyclin-dependent kinase inhibitors, p16 and p21, inhibits senescent cell accumulation, and consequently reduces fibrogenic cytokine production, which can be potential therapeutic potentials for patients with severe and/or frequent AKI by alleviating renal fibrosis and aging and delaying progression of CKD.

Schematic mechanism of P2Y2R on renal fibrosis and tubular senescence after renal IR injury. P2Y2R activation is required for proper cell cycle progression of RTEC during recovery and regulates cyclin-dependent kinase inhibitors, p16 and p21.
Data availability
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
References
Kellum, J. A. & Prowle, J. R. Paradigms of acute kidney injury in the intensive care setting. Nat. Rev. Nephrol. 14, 217–230 (2018).
Chawla, L. S. & Kimmel, P. L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 82, 516–524 (2012).
Ferenbach, D. A. & Bonventre, J. V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 11, 264–276 (2015).
Ruiz-Ortega, M., Rayego-Mateos, S., Lamas, S., Ortiz, A. & Rodrigues-Diez, R. R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 16, 269–288 (2020).
Strausser, S. A., Nakano, D. & Souma, T. Acute kidney injury to chronic kidney disease transition: Insufficient cellular stress response. Curr. Opin. Nephrol. Hypertens. 27, 314–322 (2018).
Rayego-Mateos, S. et al. Molecular mechanisms of kidney injury and repair. Int. J. Mol. Sci. 23 (2022).
Wynn, T. A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).
Zuk, A. & Bonventre, J. V. Acute Kidney Injury. Annu. Rev. Med. 67, 293–307 (2016).
Bonventre, J. V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 14 (Suppl 1), S55–61 (2003).
Humphreys, B. D. et al. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. U S A. 108, 9226–9231 (2011).
Yang, L., Humphreys, B. D. & Bonventre, J. V. Pathophysiology of acute kidney injury to chronic kidney disease: Maladaptive repair. Contrib. Nephrol. 174, 149–155 (2011).
Chen, J. et al. Cellular senescence of renal tubular epithelial cells in acute kidney injury. Cell. Death Discov. 10, 62 (2024).
Moonen, L., D’Haese, P. C. & Vervaet, B. A. Epithelial cell cycle behaviour in the injured kidney. Int. J. Mol. Sci. 19 (2018).
Lu, D. & Insel, P. A. Cellular mechanisms of tissue fibrosis. 6. Purinergic signaling and response in fibroblasts and tissue fibrosis. Am. J. Physiol. Cell. Physiol. 306, C779–788 (2014).
Idzko, M., Ferrari, D. & Eltzschig, H. K. Nucleotide signalling during inflammation. Nature 509, 310–317 (2014).
Rodriguez-Zayas, A. E., Torrado, A. I. & Miranda, J. D. P2Y2 receptor expression is altered in rats after spinal cord injury. Int. J. Dev. Neurosci. 28, 413–421 (2010).
Ahn, J. S., Camden, J. M., Schrader, A. M., Redman, R. S. & Turner, J. T. Reversible regulation of P2Y(2) nucleotide receptor expression in the duct-ligated rat submandibular gland. Am. J. Physiol. Cell. Physiol. 279, C286–294 (2000).
Cheng, R. et al. P2Y2 receptor mediated neuronal regeneration and angiogenesis to affect functional recovery in rats with spinal cord injury. Neural Plast. 2022, 2191011 (2022).
van der Vliet, A. & Bove, P. F. Purinergic signaling in wound healing and airway remodeling. Subcell. Biochem. 55, 139–157 (2011).
Potthoff, S. A. et al. P2Y2 receptor deficiency aggravates chronic kidney disease progression. Front. Physiol. 4, 234 (2013).
Dusabimana, T. et al. P2Y2R contributes to the development of diabetic nephropathy by inhibiting autophagy response. Mol. Metab. 42, 101089 (2020).
Baek, J. H., Son, H., Jeong, Y. H., Park, S. W. & Kim, H. J. Chronological aging standard curves of telomere length and mitochondrial DNA copy number in twelve tissues of C57BL/6 male mouse. Cells 8 (2019).
Callicott, R. J. & Womack, J. E. Real-time PCR assay for measurement of mouse telomeres. Comp. Med. 56, 17–22 (2006).
Cawthon, R. M. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 37, e21 (2009).
Chen, Y. et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314, 1792–1795 (2006).
Weisman, G. A. et al. Neuroprotective roles of the P2Y(2) receptor. Purinergic Signal. 8, 559–578 (2012).
Bonventre, J.V. Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int. Suppl. (2011). 4, 39–44 (2014).
Rieg, T. et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na + and water reabsorption. FASEB J. 21, 3717–3726 (2007).
Yerxa, B. R. et al. Pharmacology of INS37217 [P(1)-(uridine 5’)-P(4)- (2’-deoxycytidine 5’)tetraphosphate, tetrasodium salt], a next-generation P2Y(2) receptor agonist for the treatment of cystic fibrosis. J. Pharmacol. Exp. Ther. 302, 871–880 (2002).
Lau, O. C., Samarawickrama, C. & Skalicky, S. E. P2Y2 receptor agonists for the treatment of dry eye disease: A review. Clin. Ophthalmol. 8, 327–334 (2014).
Glaser, T. et al. Antagonistic roles of P2 × 7 and P2Y2 receptors in neurodegenerative diseases. Front. Pharmacol. 12, 659097 (2021).
Liu, B. C., Tang, T. T., Lv, L. L. & Lan, H. Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 93, 568–579 (2018).
Liu, J. et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl Res. 159, 454–463 (2012).
Melk, A. et al. Effects of donor age and cell senescence on kidney allograft survival. Am. J. Transpl. 9, 114–123 (2009).
Childs, B. G., Durik, M., Baker, D. J. & van Deursen, J. M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 21, 1424–1435 (2015).
Yang, L., Besschetnova, T. Y., Brooks, C. R., Shah, J. V. & Bonventre, J. V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 16, 535–543 (2010). 531p following 143.
Livingston, M. J. et al. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 19, 256–277 (2023).
Lee, D. H., Wolstein, J. M., Pudasaini, B. & Plotkin, M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 302, F183–191 (2012).
Megyesi, J., Andrade, L., Vieira, J. M. Jr., Safirstein, R. L. & Price, P. M. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 60, 2164–2172 (2001).
Silva Cascales, H., Mullers, E. & Lindqvist, A. How the cell cycle enforces senescence. Aging (Albany NY). 9, 2022–2023 (2017).
Piechaczyk, M. & Farras, R. Regulation and function of JunB in cell proliferation. Biochem. Soc. Trans. 36, 864–867 (2008).
Passegue, E. & Wagner, E. F. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 19, 2969–2979 (2000).
Andrecht, S., Kolbus, A., Hartenstein, B., Angel, P. & Schorpp-Kistner, M. Cell cycle promoting activity of JunB through cyclin A activation. J. Biol. Chem. 277, 35961–35968 (2002).
Perez-Benavente, B. et al. New roles for AP-1/JUNB in cell cycle control and tumorigenic cell invasion via regulation of cyclin E1 and TGF-beta2. Genome Biol. 23, 252 (2022).
Funding
This study was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea, funded by the Ministry of Science, ICT and Future Planning (RS-2023-00273563, 2021R1A5A8029490 and 2021R1A2C1010685).
Author information
Authors and Affiliations
Contributions
The manuscript and figures were generated by Kyuho Jeong and Jihyun Je, and revised by Hwajin Kim and Sang Won Park.Sang Won Park, a correspondence, contributed to funding, conceptualization, project administration, and supervision.Hwajin Kim, a co-correspondence, contributed to funding, conceptualization, and project administration.Kyuho Jeong and Jihyun Je contributed to conceptualization, data curation, formal analysis, project administration, and investigation.Theodomir Dusabimana, Jacques Karekezi, Tatang Aldi Nugroho, and Edvard Ndahigwa Ntambara supported formal analysis, and Hyun Joon Ki, Seung Pil Yun, and Hye Jung Kim supported the methodology.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jeong, K., Je, J., Dusabimana, T. et al. Deficiency of purinergic P2Y2 receptor impairs the recovery after renal ischemia-reperfusion injury and accelerates renal fibrosis and tubular senescence in mice. Sci Rep 14, 31932 (2024). https://doi.org/10.1038/s41598-024-83411-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-83411-6
