
Abstract
Allicin, a natural compound derived from garlic, protects against oxidative stress-mediated tissue inflammation and vascular remodeling. Although these are key processes in lung fibrosis, the effects of allicin on this disease have never been evaluated. In this study, we aimed to evaluate the effects of allicin on lung fibroblast-mediated lung fibrosis and its mechanisms. We assessed the effects of allicin on fibronectin-mediated lung fibroblast migration and the contraction of three-dimensional type I collagen gels, and evaluated its anti-fibrotic effects in mice models of bleomycin (BLM)-induced lung fibrosis. The results showed that allicin suppressed transforming growth factor beta 1 (TGFβ1)-stimulated gel contraction and migration, as well as α-smooth muscle actin (α-SMA) and fibronectin. Additionally, allicin upregulated AMP-activated protein kinase (AMPK) phosphorylation, while suppressing Smad3 phosphorylation. An AMPK inhibitor further stimulated TGFβ1-induced gel contraction and migration. Furthermore, allicin suppressed BLM-induced lung fibrosis with suppressed Smad3 phosphorylation and BLM-induced lung injury with suppressed inflammatory cell infiltration in the mouse models. These results suggest that allicin may be a candidate therapeutic agent for suppressing the fibrotic phase mediated by pulmonary fibroblasts through upregulated AMPK resulting in suppressed Smad3 pathway after reducing the acute inflammatory phase.
Similar content being viewed by others


Introduction
Pulmonary fibrosis is a progressive and debilitating respiratory disorder characterized by accumulation of fibrotic tissue in the lungs, resulting in impaired lung function and, ultimately, respiratory failure. Idiopathic pulmonary fibrosis (IPF) is a fatal disease that affects more than 150,000 patients annually in the United States and over 5 million patients worldwide1,2. Despite notable progress in understanding the pathological mechanisms underlying persistent fibrosis, effective treatment options for the disease are lacking3,4. Thus, it is critical to elucidate the basic molecular mechanisms underlying the development of IPF and identify effective therapeutic targets and drugs for its management.
Pulmonary fibrosis is characterized by excessive deposition of extracellular matrix components, such as collagen and fibronectin, which are primarily produced by activated myofibroblasts, resulting in fibrotic changes in the lung tissue. In response to oxidative stress-mediated lung injury, lung tissue remodeling and formation of fibrotic scars composed of contractile stress fibers (containing α-SMA produced by activated myofibroblasts) are initiated and maintained by a complex interplay of biochemical factors, including TGFβ15,6,7.
There has been growing recognition of natural products as viable alternatives for inhibiting tissue fibrosis8. Natural compounds in garlic extracts exert numerous protective effects, including anti-inflammatory, antiproliferative, antioxidative, and anti-vascular remodeling effects. Allicin, a key component of garlic, has been studied extensively owing to its ability to stimulate the production of several beneficial compounds, including sulfur dioxide, diallyl sulfide, and diallyl trisulfide9. This sulfur-containing compound has attracted considerable attention owing to its remarkable pharmacological properties10 and diverse biological activities, including modulation of cellular signaling pathways, immune system regulation, and antioxidant, anti-inflammatory, antimicrobial, and anticancer properties11,12,13,14,15,16. These multifaceted effects make allicin a highly versatile compound with promising therapeutic value17. However, the potential of allicin in the treatment of pulmonary fibrosis has not been explored to date. Although there is no direct evidence of the beneficial effects of allicin in the treatment of IPF, the role of oxidative stress-mediated inflammation and subsequent fibrotic processes in lung fibrosis disease suggest that allicin is a promising therapeutic agent for IPF. In this study, we investigated the effects of allicin on activated lung myofibroblast-mediated lung fibrosis using two established in vitro bioassays: fibroblast chemotaxis and fibroblast contraction of three-dimensional collagen gels, both of which are believed to model aspects of lung fibroblast-mediated fibrotic processes18 and the underlying signaling pathways.
Results
Effects of allicin on TGFβ1-stimulated lung fibroblast activity
The results of the bioassays indicated that allicin did not affect collagen gel contraction. However, 10–5 M allicin significantly inhibited the migration of HFL-1 cells toward fibronectin. We investigated whether allicin alters TGFβ1-induced increase in collagen gel contraction and chemotaxis in human fetal lung fibroblast (HFL-1) cells. We found that allicin significantly attenuated TGFβ1-stimulated collagen gel contraction and chemotaxis (P < 0.05 for 10–5 M allicin ± 0.25 ng/mL (10 pM) TGFβ1 versus control, Fig. 1a,b).

Effects of various concentrations of allicin with or without TGFβ1 on HFL-1-mediated collagen gel contraction and chemotaxis. Fibroblasts were grown in culture and mixed with three-dimensional collagen gels following treatment with various concentrations of allicin with or without TGFβ1 (a). The vertical axes show the collagen gel size on day 3 compared to that in the control. Fibroblasts were cultured and their response to fibronectin (20 μg/mL) was tested to evaluate chemotactic activity following treatment with various concentrations of allicin in the presence or absence of TGFβ1 (b). Vertical axes show the number of migrated cells in the five high-power fields. Data were evaluated by using one-way analysis of variance and are presented as mean ± SEM. *P < 0.05. 'v' represents the vehicle.
Effects of allicin on TGFβ1-mediated fibrotic regulators in lung fibroblasts
Immunoblotting showed that treatment with 10–5 M allicin significantly reduced TGFβ1-induced increase in the expression of fibronectin and α-SMA (P < 0.05; Fig. 2a–c). In addition, allicin pretreatment directly suppressed TGFβ1-induced increase in phosphorylated SMAD3 (Fig. 2d,e). Furthermore, allicin pretreatment increased AMPK levels by upregulating phosphorylated AMPK at Thr172 in the presence and absence of TGFβ1 (Fig. 2d,f).

Effects of allicin on TGFβ1-mediated fibrotic regulators via AMPK signaling in lung fibroblasts and effects of various concentrations of an AMPK inhibitor with or without TGFβ1 on collagen gel contraction and chemotaxis. Fibroblasts (HFL-1) were pretreated with allicin or the vehicle for 60 min and then treated with or without TGFβ1 for 24 h (a). HFL-1 cells were collected and evaluated using SDS-PAGE and immunoblotting for the detection of fibronectin (b) and αSMA (c). The vertical axes show the relative intensity of each target protein vs. β-actin. HFL-1 cells were pretreated with allicin or the vehicle for 60 min and then treated with or without TGFβ1 for 0–60 min (d). HFL-1 cells were evaluated through immunoblotting for phosphor(p)-SMAD3/ total(t)-SMAD3 (e) and phosphor(p)-AMPK/total (t)-AMPK (f). The vertical axis shows the relative intensities of the p-SMAD3/ t-SMAD3 and p-AMPK/t-AMPK signals. Data are presented as mean ± SEM and were assessed using one-way analysis of variance (ANOVA). HFL-1 cells were grown in culture and mixed with three-dimensional collagen gels following treatment with various concentrations of an AMPK inhibitor with or without TGFβ1 (g). The vertical axes show the collagen gel size on day 3 compared to that of the control. HFL-1 cells were cultured, and their responses to fibronectin (20 μg/mL) were tested to evaluate chemotactic activity following treatment with various concentrations of the AMPK inhibitor with or without TGFβ1 (h). Vertical axes show the number of migrated cells in the five high-power fields. Data were evaluated by using analysis of variance and are presented as mean ± SEM. *P < 0.05, ***P < 0.001, and ****P < 0.0001.
Effects of an AMPK inhibitor on TGFβ1-stimulated fibroblast activity
Our results revealed a correlation between allicin-induced AMPK signaling and canonical TGFβ1/Smad3 signaling; therefore, we investigated the effects of an AMPK inhibitor on TGFβ1-induced collagen gel contraction and migration toward fibronectin. The analysis showed that the AMPK inhibitor further stimulated TGFβ1-induced collagen gel contraction and chemotaxis (P < 0.05 for 107 M AMPK inhibitor ± 0.25 ng/mL (10 pM) TGFβ1 versus control, Fig. 2g,h).
Allicin attenuated BLM-induced lung fibrosis in mice
To examine the anti-fibrotic properties of allicin in vivo, we assessed a murine model of lung fibrosis induced through intratracheal injection of bleomycin (BLM) (Fig. 3a). All the mice survived until day 22. Oral administration of allicin significantly attenuated BLM-induced body weight loss on day 22 (P < 0.05, Fig. 3b). In addition, the allicin treatment reduced the BLM-induced increase in the total numbers of cells, macrophages, and lymphocytes in the bronchoalveolar lavage fluid (BAL) (P < 0.05, Fig. 3c). Furthermore, allicin attenuated lung fibrosis, determined using Masson’s Trichrome staining of lung specimens from the BLM-treated mice (Fig. 3d) and the Ashcroft score, which is used for quantitative histological analysis (P < 0.05, Fig. 3e). Immunostaining analysis revealed that Smad3 was positively expressed in the BLM-treated group and negatively expressed in the allicin-treated group, indicating suppression of Smad3 activation by allicin in this fibrosis model (Fig. 3d). The mice treated with BLM and allicin exhibited a lower number of lung cells positive for phospho-Smad3(p-Smad3) than the mice treated with BLM and the vehicle (P < 0.05, Fig. 3f).

Effect of allicin on a mouse model of BLM-induced lung fibrosis. Allicin was administrated from day 16 to 20 after intratracheal injection of BLM (a). Changes in body weight after BLM treatment. Body weight was defined as 0 on day 14 (after BLM treatment) (b). Concentrations of inflammatory cells (total cells, macrophages, lymphocytes, and neutrophils) in the bronchoalveolar lavage fluid of mice in each group. Cells were counted using a manual hemocytometer (c). Masson’s trichrome and hematoxylin and eosin staining of specimens from mice lungs harvested on day 22 at 40 × and 200× magnification. Scale bars represent 500 μm and 100 μm, respectively. Immunohistochemical analysis of phospho-SMAD3 in mouse lungs. Scale bar = 500 μm. (d) Ashcroft score for each group of five mice (e). Phospho-SMAD3-positive cells were counted in 10 fields at ×200 magnification. The average percentage of phospho-SMAD3-positive cells in each of the four groups was calculated by dividing the average for each group with that of the control group (f). Data were assessed using one-way analysis of variance and are presented as mean ± SEM. *P < 0.05.
Allicin eliminated BLM-induced lung injury in mice
To examine the anti-inflammatory properties of allicin in vivo, we created a murine model of lung injury induced through intratracheal injection of BLM (Fig. 4a). All the mice survived until day 8. Oral administration of allicin significantly restored BLM-induced body weight loss from day 4 (P < 0.05, Fig. 4b). In addition, allicin attenuated lung injury, as shown in the results of hematoxylin and eosin staining (Fig. 4c). Furthermore, infiltration of inflammatory cells, determined through Wright-Giemsa staining of BAL samples, was observed in the mice treated with BLM (Fig. 4d). Allicin treatment effectively reduced the BLM-induced increase in the total numbers of cells, macrophages, lymphocytes, and neutrophils in BAL (P < 0.05, Fig. 4e).

Effect of allicin on a mouse model of BLM-induced lung injury. Allicin was administered from days 3 to 7 after intratracheal injection of BLM (a). Changes in body weight after BLM treatment. Body weight was defined as 1.0 on day 0 (after BLM treatment) (b). Hematoxylin and eosin staining of specimens from mice lungs on day 8 at 40× and 200× magnification. Scale bars represent 500 μm and 100 μm, respectively (c). Wright–Giemsa staining of inflammatory cells in the bronchoalveolar lavage fluid of the mice on day 8. Scale bar = 100 μm (d). Concentrations of inflammatory cells (total cells, macrophages, lymphocytes, neutrophils) in the lavage fluid of the mice in each group. Cells were counted using a manual hemocytometer (e). Data were assessed using one-way analysis of variance and are presented as mean ± SEM. *P < 0.05.
Discussion
The progression of fibrosis is generally categorized into distinct phases, from the inflammatory phase to the organization and fibrosis phases. In the present in vitro study, we primarily focused on analyzing the effects of allicin on the mechanisms of lung fibroblast-mediated lung fibrosis in the presence of the profibrotic cytokine, TGF-β1. Our results demonstrated that allicin suppresses TGFβ1-stimulated lung fibroblast-mediated gel contraction and migration, as well as α-SMA and fibronectin expression via the upregulation and downregulation of the AMPK and Smad3 pathways, respectively. An AMPK inhibitor further stimulated the TGFβ1-induced collagen gel contraction and migration, supporting the reciprocal regulatory effect. Furthermore, allicin effectively attenuated BLM-induced lung fibrosis and eliminated lung injury in mice. Our data revealed that allicin-induced activation of AMPK could effectively alleviate lung fibroblast-mediated lung fibrosis via suppression of the canonical TGFβ1/Smad3 pathways.
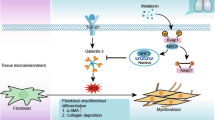
The BLM-induced lung fibrosis mice models demonstrated that allicin suppressed both the fibrotic and acute inflammatory phases of lung fibrosis. Previous studies have indicated that increased macrophage count is correlated with increased TGF-β production in lung fibrosis mice models19. In the present study, allicin strongly attenuated macrophage infiltration in BAL. Therefore, it is plausible that allicin indirectly suppresses TGF-β production by reducing the number of infiltrating macrophages in the acute inflammatory phase, resulting in suppression of the subsequent fibrosis phases induced by the lung fibroblast-mediated fibrotic processes (Fig. 5).

Allicin suppressed lung fibrosis via AMPK signaling. Allicin suppressed the number of infiltrating macrophages in the acute inflammatory phase, resulting in suppression of the subsequent fibrosis phases induced by the lung fibroblast-mediated fibrotic processes. The efficacy of Allicin in alleviating lung fibroblast-mediated fibrosis was attributed to its ability to enhance AMPK signaling, which in turn suppressed the TGFβ1-induced canonical Smad3 pathways.
Several studies have shown that allicin exhibits anti-inflammatory and anti-fibrotic effects in various organs. Allicin prevents inflammation and subsequent pulmonary vascular remodeling by suppressing proinflammatory and profibrotic markers, TNF-α, IL-6, and TGF-β20. A previous study demonstrated that allicin treatment prevents hepatic fibrosis by interfering with mitochondrial autophagy21. In addition, an animal study indicated that allicin improved kidney fibrosis in diabetes mice models by reducing the levels of pro-fibrotic markers, connective tissue growth factor, TGF-β1, and α-SMA through the inhibition of oxidative stress22. Another study demonstrated that allicin suppresses peritoneal fibrosis by decreasing the expression of TGF-β, α-SMA, and collagen I and alleviating epithelial-to-mesenchymal transition in human peritoneal mesothelial cells23. These findings suggest that allicin exerts its antifibrotic effects through the inhibition of profibrotic cytokines that target mesenchymal cell-mediated fibrotic processes following inflammation. Consistent with these reports, the present study further demonstrates the anti-fibrotic and anti-inflammation effects of allicin in lung fibroblast-mediated fibrosis.
AMPK, a serine-threonine protein kinase, plays a significant role in the regulation of cellular energy metabolism and may play a role in the activation of fibroblasts and pulmonary fibrosis23,24,25,26,27. A recent study revealed that several AMPK signal inducers could inhibit fibroblast-mediated tissue fibrosis through the regulation of the TGFβ/Smad pathway28,29,30,31,32. AMPK is activated when the AMP/ATP ratio decreases or when cells are exposed to prolonged cellular stress33. AMPK is involved in various physiological processes, including those that occur under the cellular stress induced by oxidation and inflammation34,35. Metformin-mediated activation of AMPK suppresses BLM-induced lung fibrosis and activation of lung myofibroblasts by inhibiting NADPH oxidase 4 (NOX4)-derived generation of reactive oxygen species, which activate the canonical TGFβ/Smad pathway25,36. Activation of AMPK also inhibits the fibroblast–myofibroblast transition that occurs in lung fibrosis. Recent evidence on the role of AMPK in the regulation of cell proliferation emphasizes the significance of exploring its impact on fibroblast proliferation and pulmonary fibrosis37,38. These findings suggest that AMPK activation can suppress fibroblast activities through the canonical TGFβ/Smad3-induced pulmonary fibrosis. The results of the present study provide new insights into the potential role of allicin-induced AMPK in the regulation of fibroblast activity and subsequent lung fibrosis. However, further research is needed to clarify the mechanisms underlying the functions of AMPK in pulmonary fibrosis and determine its suitability as a therapeutic target.
This study has some limitations. First, this study showed that allicin inhibited fibrosis through AMPK-mediated suppression of Smad3 phosphorylation in vitro but not in the murine model. The phosphorylation step of the AMPK activation pathway occurs early after the administration of allicin; therefore, immunostaining of the mice lung tissue was not feasible. However, the in vitro experiments demonstrated that allicin activates AMPK and inhibits phosphorylation of Smad3, supporting the hypothesis that allicin affects the AMPK pathway. Second, an appropriate in vitro experimental model designed to fully represent the inflammatory phase by incorporating the direct inhibitory effect of allicin on the production of TGF-β by macrophages has not yet been established. Thus, although our findings suggest that allicin may exhibit anti-fibrotic effects through the modulation of macrophages, further studies conducted using a suitable inflammatory model are warranted to validate this mechanism.
Broadly, our results indicate that allicin may have therapeutic effects on lung fibrosis. Analyses of the underlying mechanisms further demonstrated that allicin influences fibroblast–myofibroblast transition via the suppression of the AMPK-dependent canonical TGFβ/Smad3 pathway. Furthermore, our results provide proof-of-concept that AMPK activation by allicin or other pharmacological agents that target these pro-resolution pathways could serve as a promising therapeutic strategy for progressive fibrotic disorders.
Materials and methods
Materials
Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM), which was purchased from Wako (Osaka, Japan). Fetal calf serum (FCS) was procured from Sigma-Aldrich (St. Louis, MO, USA), TGFβ1 was obtained from R & D Systems (Minneapolis, MN, USA), and allicin was purchased from LKT Laboratories, Inc. (St. Paul, MN, USA). The allicin was dissolved in 100% dimethyl sulfoxide, which had no observed effect on the bioassay results owing to its low quantity.
Cell line and cell culture conditions
Human fetal lung fibroblasts (HFL-1) were obtained from the American Type Culture Collection (CCL-153, Manassas, VA, USA) and cultured in DMEM supplemented with 10% FCS and 1% antibiotics. The cells were maintained with 5% CO2 in a humidified incubator at 37 °C. Allicin and TGFβ1 were added to the culture medium for various experiments when the cells reached 60–70% confluence.
Collagen gel contraction assay
Type I collagen was extracted from rat-tail tendons, as described previously13. The effects of allicin on fibroblast-mediated gel contraction in the presence or absence of TGFβ1 were determined following previously described methods, with modifications16. Floating gels were cultured for a maximum of three days. To assess the ability of fibroblasts to contract the floating gels, the gel area was measured daily using an LAS 4000 image analyzer (GE Healthcare Bio-Science AB, Uppsala, Sweden). Data are presented as the percentage of the gel area relative to the initial gel size.
Chemotaxis assay
Cell migration was assessed using a Boyden blind-well chamber (Neuro Probe; Gaithersburg, MD, USA)16. Fibroblasts treated with TGFβ1 and/or allicin were added to the upper chamber, while fibronectin was used as a chemoattractant in the lower chamber. Chemotaxis was evaluated by counting the number of migrated cells in five high-power fields (i.e., one in the center and one in each quadrant). Negative controls were included in wells containing serum-free DMEM.
Western blot analysis
Western blot analysis was performed to evaluate HFL-1 cells after they were lysed with lysis buffer (RIPA; P0013C; Beyotime Institute of Biotechnology, Haimen, China) containing 100 mM phenylmethylsulphonyl fluoride (PMSF; ST506; Beyotime Institute of Biotechnology) as a proteinase inhibitor. After centrifugation at 13,000 RPM for 20 min at 4 °C, the supernatant was collected. Proteins were separated using SDS-PAGE and subsequently transferred to PVDF membranes (IPVH00010; Millipore, Billerica, MA, USA). After blocking the membrane with 5% non-fat milk at room temperature for 1 h, it was washed with phosphate-buffered saline containing Tween 20. The membrane was then incubated overnight at 4 °C with primary antibodies, including anti-AMPK (1:1000, Cell Signaling Technology, cat. no. 2532S, Danvers, MA, USA), anti-P-AMPK (1:1000, Cell Signaling Technology, cat. no. 2531. Danvers, MA, USA), anti-phospho-SMAD3 (1:1000 dilution; Cell Signaling Technology; cat. no. 9520), anti-SMAD3 (1:800 dilution; Cell Signaling Technology, Beverly, MA, USA; cat. no. 9513), anti-α-SMA (1:1000 dilution; Sigma-Aldrich; cat. no. A2547), anti-Fibronectin (1:1000 dilution; Enzo Life Sciences, Inc., Farmingdale, NY, USA; cat. no. BML-FG6010-0100), or anti-β-actin (1:5000 dilution; FUJIFILM Wako Pure Chemical Corporation; cat. no. 281-98721). On the second day of the experiment, the membranes were incubated in the dark with anti-mouse IgG (1:3000, Sigma-Aldrich, cat. no. NA931) or anti-rabbit IgG (1:2000, Sigma-Aldrich, cat. no. 934) for one hour. Imaging was performed using the Odyssey CLx (USA ), with H3 serving as an equivalent nucleoprotein control and β-actin as an equivalent protein control for quantitative analyses.
Bleomycin-induced lung injury and a fibrosis mouse model
Male C57BL/6J mice (8–10 weeks old) weighing approximately 25 g were obtained from Japan Oriental Kobo (Tokyo, Japan). The mice were housed in a specific pathogen-free room at the Laboratory Animal Center of Juntendo University under controlled temperature (25 °C), humidity, and lighting (12/12-h light–dark cycle) and provided water ad libitum and a standard diet. Animal disposal was performed in accordance with the regulations and requirements of the National Institutes of Health.
To create the lung fibrosis mouse model, the mice were randomly divided into three groups according to their body weight (groups A–C). Five mice were included in each group to reduce the number of experimental animals as much as possible. The mice in group A were intratracheally injected with normal saline, followed by oral treatment with 100 µL of normal saline. The mice in groups B and C were intratracheally injected with BLM 1.5 mg/kg/day (Nippon Kayaku, Tokyo, Japan) dissolved in 100 µL of normal saline for one day. Group B received oral treatment with normal saline only, whereas group C received allicin 50 mg/kg (LKT laboratories Inc., St Paul, MN, USA) dissolved in 100 µL of normal saline and administered orally between days 15 and 21 after administration of BLM. The weight of all the mice was measured daily. All the mice were sacrificed 22 days after BLM injection.
To create an experimental model of acute lung injury, the mice were randomly divided into three groups according to their body weight (n = 5 per group). Thereafter, the mice were intratracheally injected with BLM dissolved in normal saline for one day, followed by oral treatment with either normal saline only or allicin 50 mg/kg between 2 and 7 days after the initiation of BLM treatment. The dilution method used in this process was identical to that used in during the creation of the lung fibrosis models. All the mice were euthanized eight days after BLM injection through inhalation of 5% isoflurane.
The lung specimens were fixed in a buffered 10% formalin solution (Wako Pure Chemical Industries, Tokyo, Japan) for histological examination. Paraffin sections of 3 μm were cut and stained with hematoxylin and eosin or Masson trichrome to visualize the fibrotic lesions under a microscope. The lung specimens were obtained from all five lobes, and all the available specimens were reviewed. Fibrotic changes were graded using the Ashcroft score. Two physicians (MK and SN) independently reviewed the same samples to evaluate the interobserver agreement of our method. Subsequently, BAL samples were collected according to a previously described method39. The total number of cells was determined using Turk dye exclusion. Differential cell count was performed by staining cytospins with Diff-Quik (Sysmex International Reagents, Kobe, Japan).
Immunohistochemistry
Immunohistochemical staining of paraffin-embedded lung tissues was performed to evaluate the degree of fibrosis in the lungs. SMAD3 (Phospho-Ser425) antibody (Assay Biotech Inc., #A0031) was used for the detection of phosphorylated SMAD3 according to the manufacturer’s protocol.
Statistical analysis
Data are presented as the mean ± standard deviation. An independent samples t-test was used to analyze two groups of data, and one-way analysis of variance was used for comparisons among multiple groups. GraphPad Prism 6 was used to create column graphs. Statistical significance was set at P < 0.05.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Beers, M. F. & Morrisey, E. E. The three R’s of lung health and disease: Repair, remodeling, and regeneration. J. Clin. Investig. 121, 2065–2073 (2011).
Thannickal, V. J., Zhou, Y., Gaggar, A. & Duncan, S. R. Fibrosis: Ultimate and proximate causes. J. Clin. Investig. 124, 4673–4677 (2014).
Meyer, K. C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 11, 343–359 (2017).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012).
Duffield, J. S., Lupher, M., Thannickal, V. J. & Wynn, T. A. (host). Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 (2013).
Bueno, M. et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 125, 521–538 (2015).
Bernard, K. et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J. Biol. Chem. 290, 25427–25438 (2015).
Chen, D. Q., Feng, Y. L., Cao, G. & Zhao, Y. Y. Natural products as a source for antifibrosis therapy. Trends Pharmacol. Sci. 39, 937–952 (2018).
Reiter, J., Hübbers, A. M., Albrecht, F., Leichert, L. I. O. & Slusarenko, A. J. Allicin, a natural antimicrobial defence substance from garlic, inhibits DNA gyrase activity in bacteria. Int. J. Med. Microbiol. 310, 151359 (2020).
Borlinghaus, J., Albrecht, F., Gruhlke, M. C. H., Nwachukwu, I. D. & Slusarenko, A. J. Allicin: Chemistry and biological properties. Molecules 19, 12591–12618 (2014).
Hayat, S. et al. Garlic, from remedy to stimulant: Evaluation of antifungal potential reveals diversity in phytoalexin allicin content among garlic cultivars; allicin containing aqueous garlic extracts trigger antioxidants in cucumber. Front. Plant Sci. 7, 1235 (2016).
Liu, C. et al. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J. Nutr. Biochem. 21, 1238–1250 (2010).
Chan, J. Y., Yuen, A. C. Y., Chan, R. Y. K. & Chan, S. W. A review of the cardiovascular benefits and antioxidant properties of allicin. Phytother. Res. 27, 637–646 (2013).
Tedeschi, P. et al. Therapeutic potential of allicin and aged garlic extract in Alzheimer’s disease. Int. J. Mol. Sci. 23, 6950 (2022).
Ba, L. et al. Allicin attenuates pathological cardiac hypertrophy by inhibiting autophagy via activation of PI3K/Akt/mTOR and MAPK/ERK/mTOR signaling pathways. Phytomedicine 58, 152765 (2019).
Shan, Y. et al. Allicin ameliorates renal ischemia/reperfusion injury via inhibition of oxidative stress and inflammation in rats. Biomed. Pharmacother. 142, 112077 (2021).
El-Saber Batiha, G. et al. Chemical constituents and pharmacological activities of garlic (Allium sativum L.): A review. Nutrients 12, 872 (2020).
Kadoya, K. et al. Specific features of fibrotic lung fibroblasts highly sensitive to fibrotic processes mediated via TGF-β-ERK5 interaction. Cell. Physiol. Biochem. 52, 822–837 (2019).
Manoury, B. et al. Macrophage metalloelastase (MMP-12) deficiency does not alter bleomycin-induced pulmonary fibrosis in mice. J. Inflamm. (Lond) 3, 2 (2006).
Sánchez-Gloria, J. L. et al. Anti-inflammatory effect of allicin associated with fibrosis in pulmonary arterial hypertension. Int. J. Mol. Sci. 22, 8600 (2021).
Xiaoyan, C. & Bin, C. Mechanism of allicin for improvement of hepatic fibrosis by regulating autophagy. Indian J. Pharm. Educ. Res. 56, 1099–1105 (2022).
Arellano-Buendía, A. S. et al. Effects of allicin on pathophysiological mechanisms during the progression of nephropathy associated to diabetes. Antioxidants (Basel) 9, 1134 (2020).
Gan, L. et al. Allicin ameliorated high-glucose peritoneal dialysis solution-induced peritoneal fibrosis in rats via the JAK2/STAT3 signaling pathway. Cell Biochem. Biophys. (2024).
Mummidi, S. et al. Metformin inhibits aldosterone-induced cardiac fibroblast activation, migration and proliferation in vitro, and reverses aldosterone+salt-induced cardiac fibrosis in vivo. J. Mol. Cell. Cardiol. 98, 95–102 (2016).
Sato, N. et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 17, 107 (2016).
Herzig, S. & Shaw, R. J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018).
O’Neill, L. A. & Hardie, D. G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493, 346–355 (2013).
Efentakis, P. et al. Myocardial protection and current cancer therapy: Two opposite targets with inevitable cost. Int. J. Mol. Sci. 23, 14121 (2022).
Cieslik, K. A., Trial, J. & Entman, M. L. Aicar treatment reduces interstitial fibrosis in aging mice: Suppression of the inflammatory fibroblast. J. Mol. Cell. Cardiol. 111, 81–85 (2017).
Yang, J. Y. et al. Wedelolactone attenuates pulmonary fibrosis partly through activating AMPK and regulating Raf-MAPKs signaling pathway. Front. Pharmacol. 10, 151 (2019).
Xiao, Y. et al. Baicalin inhibits pressure overload-induced cardiac fibrosis through regulating AMPK/TGF-β/Smads signaling pathway. Arch. Biochem. Biophys. 640, 37–46 (2018).
Gao, L. et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 11, 44 (2020).
Angin, Y., Beauloye, C., Horman, S. & Bertrand, L. Regulation of carbohydrate metabolism, lipid metabolism, and protein metabolism by AMPK. Exp. Suppl. 107, 23–43 (2016).
Wong, K. A. & Lodish, H. F. A revised model for AMP-activated protein kinase structure: The alpha-subunit binds to both the beta- and gamma-subunits although there is no direct binding between the beta- and gamma-subunits. J. Biol. Chem. 281, 36434–36442 (2006).
Cheng, D. et al. Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J. Transl. Med. 19, 349 (2021).
Gamad, N. et al. Metformin alleviates bleomycin-induced pulmonary fibrosis in rats: Pharmacological effects and molecular mechanisms. Biomed. Pharmacother. 97, 1544–1553 (2018).
Mihaylova, M. M. & Shaw, R. J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 (2011).
Faubert, B. et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 17, 113–124 (2013).
Nie, Y. et al. S-allyl-l-cysteine attenuates bleomycin-induced pulmonary fibrosis and inflammation via AKT/NF-κB signaling pathway in mice. J. Pharmacol. Sci. 139, 377–384 (2019).
Acknowledgements
This study was supported by Japan China Sasakawa Medical Fellowship. This work was supported by a grant for cross-disciplinary collaboration from JSPS KAKENHI, grant number 22K08241.
Author information
Authors and Affiliations
Contributions
The authors confirm contribution to the paper as follows: study conception and design: S.N., J.H., M.K., S.T.; data collection: Y.A, H.M., K.K., T.S., H.H., Y.O., I.S.; statistical analysis: K.H.; analysis and interpretation of results: S.N., J.H., M.K., S.T., K.T.; draft manuscript preparation: S.N., J.H.; funding acquisition: J.W., H.I., K.K.; All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was performed in accordance with the ARRIVE guidelines. All the animal experiments were conducted in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology (Notice No. 71, 2006). All experiments were approved by the Committee for Animal Experimentation of Juntendo University (approval no. 1589). Patients or the public were not involved in the design, conduct, reporting, dissemination plans of this research.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nakazawa, S., Hou, J., Kato, M. et al. Allicin induced AMPK signaling attenuated Smad3 pathway mediated lung fibrosis. Sci Rep 15, 19060 (2025). https://doi.org/10.1038/s41598-025-01314-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-01314-6
