
Abstract
The future of hypertension management lies in distinguishing disease subtypes for precise control. The underlying drivers and pathology of non-obese hypertension (J-HTN) remain unclear. There is a lack of biomarkers for the early identification of J-HTN. The aim of this study was to identify circulating metabolomic profiles that facilitate the early detection of J-HTN patients, thereby providing valuable insights for more targeted and precision-based therapies. A non-targeted metabolomics approach was used to quantify serum metabolites in 120 patients with newly diagnosed hypertension, and to determine the metabolomic characteristics of J-HTN and two types of obese hypertension (fat-dominant and muscle-dominant). 4 metabolites unique to J-HTN were identified, with lysophosphatidylcholine 22:6 (LysoPC(22:6/0:0)) standing out as the marker showing the most pronounced difference. Using the serum metabolome alone, we were able to distinguish J-HTN from other hypertensive patients. In a secondary validation with an independent cohort of 60 medically treated J-HTN patients, 3 metabolites, including LysoPC(22:6/0:0), remained significantly altered. The serum metabolic profiles identified in this study enable the early detection of J-HTN, with LysoPC(22:6/0:0) emerging as a highly promising biomarker. This metabolite may also correlate with the clinical efficacy of J-HTN treatments.
Similar content being viewed by others

Introduction
Hypertension (HTN) is the leading cause of cardiovascular death and disability worldwide1. It is estimated that over 1 billion people are affected by HTN2, yet less than 25% achieve adequate blood pressure control3, contributing to approximately 8.5 million deaths annually4. The development of HTN is influenced by a complex interplay of genetic, lifestyle, and environmental factors, which complicates its treatment. One significant risk factor is elevated body mass index (BMI)5. While calorie restriction and exercise can effectively lower blood pressure in overweight individuals, these interventions are not suitable for most non-obese patients.
Metabolic disturbances are a hallmark of non-obese HTN (J-HTN) and often represent an early sign of pathological changes in the body, sometimes occurring before HTN itself develops6. It has been suggested that metabolic abnormalities in non-obese individuals can lead to a two- to five-fold increase in cardiovascular events7. Unlike obese individuals, J-HTN patients lack external indicators of metabolic dysfunction, such as increased body size, which can result in a certain degree of cardiovascular damage and vascular injury before HTN is clinically diagnosed. Our research revealed key alterations in energy metabolism, bile acid metabolism, and phospholipid metabolism in hypertensive animals with normal body weights8. However, the metabolic drivers and causative factors of elevated blood pressure in non-obese individuals remain poorly understood, limiting the development of targeted treatment strategies. There is a pressing need to discover specific and reliable disease markers and explore corresponding therapeutic measures.
Emerging metabolomics technologies offer promising tools for understanding the end products of complex interactions between various metabolic targets, signalling pathways, and the internal environment9,10,11,12. These approaches help identify disease-related metabolic disturbances, uncover biomarkers, and map disrupted metabolic pathways. To date, metabolomics has been applied in experimental studies to explore the diagnosis and pathogenesis of hypertension in both humans and animals, providing preliminary insights into the associated pathophysiology. However, the reproducibility of metabolic findings is highly dependent on the studied cohort, and only a limited number of studies have rigorously validated the diagnostic potential of specific metabolites.
In this study, we present, for the first time, an analysis of potential biomarkers for J-HTN based on body size classification. We compared serum metabolites in J-HTN patients with incipient HTN, as well as in two categories of obese patients (fat-dominant (T-HTN) and muscle-dominant (Y-HTN)), with those in healthy volunteers. Furthermore, we conducted a secondary validation using an independent cohort of 60 medically treated J-HTN patients. Our study aims to improve the understanding of the underlying pathophysiology of J-HTN and to guide more precise clinical management strategies for this condition.
Results
Characteristics of the study population
Our cross-sectional study included 120 participants, with 30 healthy volunteers serving as the control group (group N). The remaining 90 hypertensive patients were divided into three groups according to body type: J-HTN (group J, n = 30), T-HTN (group T, n = 30), and Y-HTN (group Y, n = 30). The BMI in group J was significantly lower than that in the other two groups (group J vs. T, adj.p = 0.001; group J vs. Y, adj.p = 0.001; group T vs. Y, adj.p = 0.093). Notably, patients in group J were more likely to be female and exhibited less severe metabolic abnormalities, such as lower triglyceride levels (group J vs. T, adj.p = 0.022; group J vs. Y, adj.p = 0.116; group T vs. Y, adj.p = 1.000), as well as lower uric acid levels (group J vs. T, adj.p = 0.042; group J vs. Y, adj.p = 0.015; group T vs. Y, adj.p = 1.000). Table 1 provides a comprehensive demographic, clinical, biochemical, and metabolic profile of the entire cohort.
Mass signal stability and anomalous data rejection
The stability of the ultra-high performance liquid chromatography quadrupole time-of-flight mass spectrometry (UHPLC-Q-TOF-MS) system was assessed via quality control (QC) samples. Supplementary Figure S1 displays stacked plots of the total ion flow in both positive and negative ion modes for the QC samples. The nearly complete overlap of these plots indicates that the instrumental method remained stable, and the data collected throughout the detection sequence were reliable and consistent.
To explore metabolic differences among the groups, principal component analysis (PCA) was first performed. PCA is an unsupervised analytical method that revealed a clear separation of the three types of HTN in both positive and negative ion modes (Fig. 1A,B). Within-group consistency was assessed using PCA-class, and samples falling outside the ‘2 standard deviation’ line were considered outliers and excluded from further analysis. As illustrated in Fig. 1C–J, the following samples were excluded due to inconsistencies: N6, T26 , and Y1 in negative ion mode, and N1, N2, N3, J1, J2, J3, and Y1 in positive ion mode.

Overall distribution of metabolome in all serum samples. PCA scores: (A) negative ion mode, (B) positive ion mode. N, normal group; J, non-obese patient group; T, obese patient (fat-dominant) group; Y, obese patient (muscle-dominant) group; QC, quality control group. Within-group consistency: Group N, (C) negative ion mode, (D) positive ion mode; Group J, (E) negative ion mode, (F) positive ion mode; Group T, (G) negative ion mode, (H) positive ion mode; Group Y, (I) negative ion mode, (J) positive ion mode. N6, T26, and Y1 in negative ion mode and N1, N2, N3, J1, J2, J3, and Y1 in positive ion mode were considered outliers and excluded from further analysis.
Overall changes in the serum metabolome of J-HTN
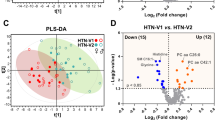
To characterize the metabolic profiles of J-HTN, orthogonal partial least squares discriminant analysis (OPLS-DA) was conducted to compare groups J, T, and Y with group N. OPLS-DA is a supervised analysis method that helps distinguish metabolic differences between groups and identifies potentially significant variables. In this analysis, the closer Q2 is to 1, the better the predictive power of the model, and the closer R2Y is to 1, the greater the difference between the two groups. The serum metabolic profile of group J significantly differed from that of group N (Fig. 2), and the self-cross-validation of the model yielded excellent fit and predictability (R2Y = 0.975, Q2 = 0.968). Further validation using permutation testing confirmed the model’s robustness, with R2Y intercepts less than 0.4 and Q2 intercepts less than 0 in both ion modes, indicating a well-constructed model (Supplementary Fig. S2).

Differential metabolic profiles of group J vs. group N based on non-targeted metabolomics. OPLS-DA scores: (A) negative ion model, (B) positive ion model. N, normal group; J, non-obese patient group. S-plots: (C) negative ion model, (D) positive ion model. The abscissa represents the co-correlation coefficient of principal component and metabolite, and the ordinate represents the correlation coefficient of principal component and metabolite. Points farther from the origin represent variables with a greater contribution to the observed variance. Volcano plots: (E) negative ion model, (F) positive ion model. The screening criteria were fold change (J/N) ≥ 1.50 or fold change (N/J) ≥ 0.67, and adjusted p-value ≤ 0.05. Blue represents a decrease, and red represents an increase.
Based on the criteria of variable importance in projection (VIP) ≥ 1, |partial correlation coefficient (Pcorr) ≥ 0.2, fold change (FC) ≥ 1.20 or ≤ 0.83, and adjusted p-value (adj.p) ≤ 0.05, a total of 40 differentially abundant metabolites were identified between groups J and N. The corresponding identifying information for these metabolites is provided in Table 2. Notably, the alterations in group J were predominantly related to lysophosphatidylcholine (LysoPC), lysophosphatidylethanolamine (LysoPE), amino acids, and metabolites such as carnitine and primary bile acids. Specifically, amino acid metabolites, including L-tryptophan and phenylalanine (PHE), as well as lipoylcarnitines (palmitoylcarnitine, oleoylcarnitine), and secondary bile acids (such as deoxycholic acid glycine conjugate and glycoursodeoxycholic acid 3-sulfate), were significantly elevated in group J. In contrast, LysoPE(18:2/0:0), LysoPE(20:4/0:0), and LysoPE(0:0/20:0) were decreased. Further details regarding the differentially abundant metabolites in groups T and Y can be found in Supplementary Tables S1 and S2.
Differentially abundant metabolites were orthogonalized via a Venn diagram (Fig. 3), revealing 25 metabolites common to all three types. Among these 25 metabolites, 10 exhibited significant reductions in hypertensive patients, with myristic acid showing an increase in group Y. The remaining 15 metabolites demonstrated a marked trend toward elevation in patients, with LysoPC(16:0/0:0), LysoPC(18:1/0:0), LysoPC(0:0/18:0), and LysoPC(20:4/0:0) decreasing in group Y. Detailed information on these 25 metabolites is provided in Supplementary Table S3.

Venn diagram of all differential metabolites.
J-HTN exhibited 4 unique characteristic metabolites: glycoursodeoxycholic acid 3-sulfate, deoxycholic acid glycine conjugate, LysoPE (0:0/24:0), and LysoPC(22:6/0:0). All of these metabolites were significantly increased compared with those in group N, as illustrated in Fig. 4. The characteristic metabolites for groups T and Y are shown in Supplementary Figures S3 and S4. Observing variations in sex distribution among the three patient groups, we conducted supplementary analyses to evaluate potential sex-specific differences. The results demonstrated no statistically significant variations in HTN-related metabolites between male and female participants within our study cohort (refer to the Results Appendix 2).

Scatter plots of peak area of J-HTN characteristic metabolites. The ordinate indicates the ratio of the signal value measured by the sample and the quality control sample. The p value was adjusted via the Benjamini‒Hochberg method (with a target FDR of 0.05). *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001.
Potential for early identification of J-HTN through serum metabolites
The random forest algorithm, an ensemble learning method combining multiple decision tree classifiers, was employed to build a diagnostic model using the differentially abundant metabolites from Table 2 and Supplementary Tables S1 and S2. This model aimed to predict the subtype of HTN based on serum metabolomic data.
As shown in Fig. 5A, the random forest model with 500 decision trees effectively separated the three groups. The prediction accuracy for the test set was 0.211 (i.e., a prediction rate of 79.90%), with an ROC value of 81.90% (Fig. 5B), demonstrating good predictive efficacy. The top five significant parameters identified by the model were LysoPC(22:6/0:0), LysoPC(20:2/0:0), LysoPE(0:0/22:6), LysoPC(20:3/0:0), and indoxyl sulfate (Fig. 5C). The intensity of LysoPC(22:6/0:0), the most significant metabolite, across the respective groups is shown in Fig. 5D.

Random forest model for J-HTN. (A) categorical prediction results; (B) ROC curves; (C) importance assessment; (D) relative intensity of LysoPC(22:6/0:0), ****, p ≤ 0.0001.
Potential association of circulating metabolic profiles with the effectiveness of J-HTN treatment
Currently, there are no specific drugs that target metabolic disorders in HTN. There is a lack of evidence that existing antihypertensive medications influence the differentially abundant metabolites mentioned earlier. In a previous study, the Chinese herbal compound Chai-Gui Decoction (CGD) was found to modulate the serum intensity of LysoPC(22:6/0:0) in spontaneously hypertensive rats (SHRs)8. In this study, we analysed the changes in the serum metabolome of 60 J-HTN patients before and after two treatment regimens: 30 patients in the control group and 30 in the CGD group. The demographics of the participants are presented in Supplementary Table S4. The control group (treated with a dihydropyridine calcium antagonist-based regimen) and the CGD group (receiving CGD in addition to the control regimen) were evaluated after 4 weeks of treatment. The antihypertensive effects in both groups are shown in Fig. 6A,B.

Effect of CGD on J-HTN (A) systolic blood pressure, (B) diastolic blood pressure, ***, p ≤ 0.001, (C) key metabolites (the CGD group), (D) key metabolites (the control group).
Using the criteria of VIP ≥ 1, |Pcorr|≥ 0.2, FC ≥ 1.50 or ≤ 0.67, and adj.p ≤ 0.05, 13 differentially abundant metabolites in the control group and 25 differentially abundant metabolites in the CGD group were identified (Supplementary Tab. S7). Compared with the pretreatment groups, the control group presented elevated levels of fatty acids (linoleic acid, docosahexaenoic acid, myristic acid), steroids (desoxycorticosterone acetate, flumethasone, pregnenolone), triglycerides (MG(18:1/0:0), and glycerol 1-myristate). Similarly, the CGD group presented increases in metabolites related to fatty acids (stearic acid, heptadecanoic acid), steroids (cortisol), and glycerol esters (MG(20:4/0:0)), along with a broader spectrum of changes across metabolites, including organic acids (pyruvic acid, 3-hydroxymethylglutaric acid, 4-hydroxybenzoic acid), sugars (ADP-glucose, L-fucose), amino acids (PHE), phospholipids (LysoPC(14:0/0:0), LysoPC(22:6/0:0)), and bile acids (cholic acid), all of which were significantly increased.
Compared with the circulating metabolic profiles of J-HTN, three key metabolites—myristic acid in the control group and PHE and LysoPC(22:6/0:0) in the CGD group—emerged as important recurring features (Fig. 6C,D). Notably, myristic acid and LysoPC(22:6/0:0) presented biphasic alterations: myristic acid was downregulated in the early phase of J-HTN but upregulated following treatment, whereas LysoPC(22:6/0:0) initially increased but then decreased after the addition of CGD. PHE consistently increased.
These findings align with our previous animal studies, where LysoPC(22:6/0:0) decreased after treatment, reinforcing the notion that this metabolite may serve as a distinctive marker for J-HTN, providing a potential metabolic target for diagnosis and treatment. The screening and identification of different metabolites in each group via UHPLC-Q-TOF-MS are detailed in the Results Appendix 1.
Discussions
In recent decades, breakthroughs in high-throughput sequencing and large-scale histological analyses have revolutionized our understanding of biological systems. The advent of new technologies has enabled researchers to capture biological information at an unprecedented resolution, spanning genomics, epigenomics, proteomics, metabolomics, and lipidomics13. HTN, a multifactorial disease, is influenced by genetic factors, environmental conditions, and their interactions, with the human metabolome offering a measurable representation of these gene-environment interactions14. The integration of clinical, health, social, behavioural, and personal data has the potential to create powerful predictive models for personalized treatment, providing deeper insights into the genetic, environmental, and social determinants of HTN.
The aim of this study was to explore metabolic markers specific to J-HTN and provide a theoretical foundation for the development of more precise diagnostic and therapeutic strategies for this group. Through serum metabolomic analysis, we characterized the circulating metabolic profiles of J-HTN and identified specific markers for its early identification. In particular, glycoursodeoxycholic acid 3-sulfate, deoxycholic acid glycine conjugate, LysoPE (0:0/24:0), and LysoPC(22:6/0:0) emerged as key markers, with LysoPC(22:6/0:0) being the most significant. Previous studies have highlighted the potential of certain LysoPC isoforms in HTN classification. For example, LysoPC(16:0), LysoPC(20:3), and sphingosine C24:1 are independently associated with reduced systolic blood pressure (SBP), and LysoPC(20:3) is linked with lower diastolic blood pressure (DBP)15. Additionally, four specific LysoPCs (LysoPCs 18:2, 16:0, 18:0, and 18:1) were negatively correlated with vascular ageing16. Furthermore, serum bile acid profiling in SHRs revealed significant increases in glycoursodeoxycholic acid17, which aligns with our findings. Moreover, recent research has identified deoxycholic acid as an early diagnostic biomarker for HTN18.
In an independent cohort of J-HTN patients treated with two different drug regimens, the addition of the herbal compound CGD led to more pronounced changes in the serum metabolome. The major metabolite, LysoPC(22:6/0:0), was again highlighted as a key marker for J-HTN. LysoPC(22:6/0:0), an isoform of LysoPC carrying docosahexaenoic acid (DHA), is known to release DHA in response to phospholipases (PLA1/PLA2), which lowers triglyceride levels and reduces cardiovascular disease risk19; it also mediates vasodilation to help lower blood pressure20. However, LysoPC has adverse cardiovascular effects, including inhibition of nitric oxide production, enhanced oxidative stress21, and disruption of cardiac structure and function22,23. Nevertheless, previous animal experiments have shown that LysoPC(22:6/0:0) levels decrease in SHRs after CGD treatment8. In line with these findings, our study demonstrated downregulation of LysoPC(22:6/0:0) in J-HTN patients, with a further decrease in levels as blood pressure decreased after CGD treatment. This strengthens the potential of LysoPC(22:6/0:0) as a biomarker for distinguishing between superior and inferior antihypertensive effects in J-HTN patients.
Another key metabolite identified in our cohort was PHE, an aromatic amino acid closely associated with protein synthesis and enzymatic reactions. PHE exists primarily in its L-configuration (L-PHE), which is not synthesized by the human body. L-PHE has been shown to reduce aortic thickness and myocardial collagen content24. In SHR models, L-PHE has been shown to increase vasodilation and lower blood pressure25. Conversely, retrospective population-based studies have reported that PHE intake is associated with an increased risk of HTN26. A recent secondary analysis of the SABPA study also suggested a positive association between PHE levels and SBP dynamics in individuals with masked HTN27. Our findings align with these studies, as PHE levels were elevated in the early stages of J-HTN and increased further after treatment. At present, there are also relevant descriptions of PHE variation for several subtypes of HTN, including masked HTN27, H-type HTN28, and gestational HTN29. We have underscored the importance of considering PHE metabolism in the pathophysiology of J-HTN. Additionally, the other recurring metabolite myristic acid has been identified in several studies analysing HTN metabolomics30,31. It reflects an imbalance between saturated and unsaturated fatty acids due to fatty acid metabolism disorders, whereas deeper investigation into their role is still warranted.
Globally, only approximately 20% of hypertensive patients are adequately controlled32. The future of HTN management lies in distinguishing disease subtypes for precise control. There remains an urgent clinical need for early identification and targeted treatment of hypertensive patients with normal or lean body sizes. In this study, we first analysed the circulating metabolic profiles of hypertensive patients with different body sizes, using an orthogonal approach to identify metabolites strongly correlated with J-HTN. Then, we validated the importance of each metabolic component through the random forest algorithm, which produced more robust results. Additionally, we performed secondary validation in an independent cohort receiving pharmacological treatment and identified 3 metabolic markers associated with both diagnosis and prognosis. Among these, LysoPC(22:6/0:0) stood out as the most significant biomarker for early recognition of J-HTN.
Our study enhances the understanding of the pathological aspects of metabolic disorders in J-HTN, with LysoPC(22:6/0:0) emerging as a promising biomarker. Although the current work was validated in an independent cohort receiving pharmacological treatment, it has not yet been fully explored through functional studies, and the molecular mechanisms linking metabolites to J-HTN remain unclear. Preliminary exploration of the mechanism revealed that serum levels of total LysoPC, calcium-independent PLA2, and RGS2 were altered in both healthy individuals and J-HTN patients. Additionally, in J-HTN patients, these changes were consistent before and after treatment (Supplementary Fig S5). Furthermore, the subjective nature of abdominal palpation for distinguishing T-HTN from Y-HTN by specialists was a limitation. Other limitations included variables such as comorbidities, medications, and the duration of sample freezing, all of which could influence serum metabolites. Despite these challenges, our study provides valuable insights into the pathophysiology of J-HTN and lays the groundwork for future research into new targets and therapies for precision medicine in HTN.
In brief, this work, on the basis of non-targeted metabolomics data, identified specific metabolic profiles of J-HTN, with LysoPC(22:6/0:0) emerging as the most notable biomarker. This finding is valuable for the early prevention and prognosis of this HTN subtype and offers new insights into HTN classification. Nonetheless, further preclinical and clinical studies are needed to explore new therapeutic targets and drugs for precision therapy in J-HTN.
Methods
Study population
All the subjects were recruited and evaluated by the Department of Cardiovascular Medicine, Wuxi Affiliated Hospital of Nanjing University of Chinese Medicine from March 17, 2023, to January 10, 2024. The study protocol was approved by the Institutional Review Board of Wuxi Affiliated Hospital of Nanjing University of Chinese Medicine (No. YJS2023031307, March 17, 2023). All study participants, including patients and healthy volunteers, provided informed consent and were grouped by cardiovascular specialists for assessment after a thorough medical examination.
Patients who met the diagnostic criteria for HTN (SBP ≥ 140 mm Hg and/or DBP ≥ 90 mm Hg) according to three separate measurements without previous use of antihypertensive medication were eligible for screening. Patients who had used antihypertensive medication or had a confirmed diagnosis of secondary HTN were excluded. Hypertensive patients whose weight exceeded 10% above the standard weight were categorized into the “obese” group, based on the standard weight formula (standard weight [kg] = height [cm]—105). The remaining patients were classified as having J-HTN. Among the obese patients, abdominal muscle tone was assessed by physical examination, and those with flaccid abdominal muscles were classified as T-HTN (fat-dominant HTN), whereas those with tight abdomens were classified as Y-HTN (muscle-dominant HTN).
Sample preparation
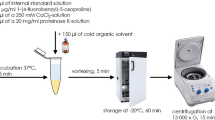
A total of 30 healthy volunteers and 90 hypertensive patients (30 each of J-HTN, T-HTN, and Y-HTN) were recruited for this study. Fasting venous blood was collected from all subjects in the morning. The venous blood samples were centrifuged at 1500 rpm for 10 min to obtain the serum. The serum was collected into 1.5 ml EP tubes and stored at − 80 °C in a refrigerator. 600 μl of acetonitrile was added to 200 μL of pre-treated serum, vortexed for 60 s, and centrifuged for 10 min (13,000 rpm, 4 °C). A 480 μL aliquot of the supernatant was pipetted into a 1.5 ml EP tube and blown dry under nitrogen. Before non-target metabolomics experiments, the blow-dried samples were reconstituted with 100 μL of 50% acetonitrile, vortexed again for 60 s, and centrifuged for 10 min (13,000 rpm, 4 °C), after which the supernatant was aspirated into the sample. Another appropriate amount of each sample was taken, mixed in equal parts, and prepared according to the above steps for the QC samples. One injection of the QC sample was inserted into every 10 samples during the assay to monitor the stability of the instrument and method.
Liquid chromatography mass spectrometry
A SCIEX Exion LC instrument combined with an X500B mass spectrometer (AB Sciex, Foster City, CA, USA) was used for sample analysis. The chromatographic and mass spectrometric conditions were as follows: chromatographic column, Waters Acquity BEH C18 column (2.1 × 100 mm, 1.7 μm); column temperature, 35 °C; mobile phase, A-water (containing 0.1% formic acid); B-acetonitrile; flow rate, 0.3 ml/min; and injection volume, 2 μl. The elution gradient and scanning mode are provided in Supplementary Tables S5 and S6.
Independent treatment cohort validation
To validate the potential of differentially abundant metabolites as markers of J-HTN efficacy, we performed an additional untargeted metabolomics analysis on an independent cohort of 60 pharmacologically treated J-HTN patients who were not part of the population with initial HTN described above. In this cohort, we compared the dynamic changes in metabolites before and after the two drug treatment regimens, with a focus on classification markers. All procedures were performed in accordance with the Declaration of Helsinki and registered with the Chinese Clinical Trial Registry (https://www.chictr.org.cn/, ChiCTR2400085731, 17/06/2024). The full study protocol for this section is provided in the Methods Appendix.
Data analysis
Numerical variables for clinical characteristics are expressed as the means ± SDs, and differences in clinical and biochemical parameters between groups were detected via independent samples t-tests or analysis of variance (ANOVA). The multigroup comparison p values were adjusted via the Bonferroni method. Raw plots were extracted and transformed into data matrices via SCIEX OS Analytics, and all the data were analysed after normalization to the total peak area. SIMCA 14.1 (Umetrics, Umeå, Sweden) software was used for multivariate mathematical statistics. First, the overall situation and the consistency of the samples within groups were analysed via PCA and PCA-Class to exclude anomalous data. The grouping variables were subsequently included in the modelling two by two for OPLS-DA, and the replacement test was used to test the validity of the model. On the basis of this model, the variables that had a greater impact on the establishment of this model were screened according to the conditions of a VIP value ≥ 1, |Pcorr| ≥ 0.2, and FC ≥ 1.20 or ≤ 0.83, and these variables were further subjected to the Mann‒Whitney test. The p value was adjusted via the Benjamini‒Hochberg method (with a target FDR of 0.05), and a p value ≤ 0.05 was found to be significantly different. The screened differential variables were identified based on accurate molecular mass (within 10 ppm mass error) and tandem mass spectrometry secondary information, and the identified candidate differentially abundant metabolites were searched and compared with the HMDB online database and annotated to obtain the differentially abundant metabolites. The dataset was randomly divided into a training set and a test set (2:1) by machine algorithm, the training set was used to construct a random forest model, and the test set was used to predict the classification results to evaluate the diagnostic efficacy of the metabolic markers.
Data availability
The individual participant data that underlie the results reported in this article will be shared in the communal repository after deidentification (Xu, Yifan (2024), “Clinical efficacy evaluation and serum metabolomics study of Chai-Gui Decoction on patients with Jueyin-type hypertension”, Mendeley Data, V1, doi: 10.17632/d3y5zxcnty.1). Anyone who wishes to access the data is allowed to view and use it for any purpose.
References
Tran, K. B. et al. The global burden of cancer attributable to risk factors, 2010–19: A systematic analysis for the Global Burden of Disease Study 2019. The Lancet 400(10352), 563–591. https://doi.org/10.1016/S0140-6736(22)01438-6 (2022).
Xue, B. & Johnson, A. K. Sensitization of hypertension: the impact of earlier life challenges: Excellence award for hypertension research 2021. Hypertension 80, 1–12. https://doi.org/10.1161/hypertensionaha.122.18550 (2023).
Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 398, 957–980, https://doi.org/10.1016/s0140-6736(21)01330-1 (2021).
Zhou, B., Perel, P., Mensah, G. A. & Ezzati, M. Global epidemiology, health burden and effective interventions for elevated blood pressure and hypertension. Nat. Rev. Cardiol. 18, 785–802. https://doi.org/10.1038/s41569-021-00559-8 (2021).
Foti, K. et al. BMI and blood pressure control among United States adults with hypertension. J. Hypertens. 40, 741–748. https://doi.org/10.1097/hjh.0000000000003072 (2022).
Deng, Y., Huang, C., Su, J., Pan, C. W. & Ke, C. Identification of biomarkers for essential hypertension based on metabolomics. Nutr. Metab. Cardiovasc. Dis. 31, 382–395. https://doi.org/10.1016/j.numecd.2020.11.023 (2021).
Cirulli, E. T. et al. Profound perturbation of the metabolome in obesity is associated with health risk. Cell Metab. 29, 488–500. https://doi.org/10.1016/j.cmet.2018.09.022 (2019).
Zhu, H., Xu, C., Dong, Y., Lu, S. & Guo, L. Chai-Gui Decoction and its representative components ameliorate spontaneous hypertension rats by modulating lipid metabolism and gut microbiota. J. Ethnopharmacol. 305, 116116. https://doi.org/10.1016/j.jep.2022.116116 (2023).
Au, A., Cheng, K. K. & Wei, L. K. Metabolomics, lipidomics and pharmacometabolomics of human hypertension. Adv. Exp. Med. Biol. 956, 599–613. https://doi.org/10.1007/5584_2016_79 (2017).
Louca, P., Menni, C. & Padmanabhan, S. Genomic determinants of hypertension with a focus on metabolomics and the gut microbiome. Am. J. Hypertens. 33, 473–481. https://doi.org/10.1093/ajh/hpaa022 (2020).
Nikolic, S. B., Sharman, J. E., Adams, M. J. & Edwards, L. M. Metabolomics in hypertension. J. Hypertens. 32, 1159–1169. https://doi.org/10.1097/hjh.0000000000000168 (2014).
Xie, H. et al. Gut microbiota and metabonomics used to explore the mechanism of Qing’e Pills in alleviating osteoporosis. Pharm. Biol. 60, 785–800. https://doi.org/10.1080/13880209.2022.2056208 (2022).
Dzau, V. J. & Hodgkinson, C. P. Precision Hypertension. Hypertension 81, 702–708. https://doi.org/10.1161/hypertensionaha.123.21710 (2024).
Zheng, Y. et al. Metabolomics and incident hypertension among blacks: The atherosclerosis risk in communities study. Hypertension 62, 398–403. https://doi.org/10.1161/hypertensionaha.113.01166 (2013).
Huang, C. C., Huang, Y. L., Lin, C. H. & Chen, J. W. Plasma metabolomic profiles associated with hypertension and blood pressure in response to thiazide diuretics. Hypertens. Res. 45, 464–473. https://doi.org/10.1038/s41440-021-00825-9 (2022).
Polonis, K. et al. Metabolomic signature of early vascular aging (EVA) in hypertension. Front. Mol. Biosci. 7, 12. https://doi.org/10.3389/fmolb.2020.00012 (2020).
Zhou, R. et al. Effect of gut microbiota on the pharmacokinetics of nifedipine in spontaneously hypertensive rats. Pharmaceutics 15(8), 2085. https://doi.org/10.3390/pharmaceutics15082085 (2023).
Yaren, Y. et al. Unique intestinal microflora and metabolic profile in different stages of hypertension reveal potential biomarkers for early diagnosis and prognosis. J. Med. Microbiol. https://doi.org/10.1099/jmm.0.001839 (2024).
Retterstøl, K. & Rosqvist, F. Fat and fatty acids - a scoping review for nordic nutrition recommendations 2023. Food Nutr. Res. https://doi.org/10.29219/fnr.v68.9980 (2024).
Bercea, C. I., Cottrell, G. S., Tamagnini, F. & McNeish, A. J. Omega-3 polyunsaturated fatty acids and hypertension: A review of vasodilatory mechanisms of docosahexaenoic acid and eicosapentaenoic acid. Br. J. Pharmacol. 178, 860–877. https://doi.org/10.1111/bph.15336 (2021).
Watanabe, T. et al. Human urotensin-II potentiates the mitogenic effect of mildly oxidized low-density lipoprotein on vascular smooth muscle cells: Comparison with other vasoactive agents and hydrogen peroxide. Hypertens. Res. 29, 821–831. https://doi.org/10.1291/hypres.29.821 (2006).
Sargent, C. A., Vesterqvist, O., Ogletree, M. L. & Grover, G. J. Effects of endogenous and exogenous lysophosphatidylcholine in isolated perfused rat hearts. J. Mol. Cell Cardiol. 25, 905–913. https://doi.org/10.1006/jmcc.1993.1103 (1993).
Hoque, A. N., Hoque, N., Hashizume, H. & Abiko, Y. A study on dilazep: II. Dilazep attenuates lysophosphatidylcholine-induced mechanical and metabolic derangements in the isolated, working rat heart. Jpn. J. Pharmacol. 67, 233–241. https://doi.org/10.1254/jjp.67.233 (1995).
Zhao, G., Li, Z. & Gu, T. Antihypertension and anti-cardiovascular remodeling by phenylalanine in spontaneously hypertensive rats: Effectiveness and mechanisms. Chin. Med. J. (Engl.) 114, 270–274 (2001).
Mitchell, B. M., Dorrance, A. M. & Webb, R. C. Phenylalanine improves dilation and blood pressure in GTP cyclohydrolase inhibition-induced hypertensive rats. J. Cardiovasc. Pharmacol. 43, 758–763. https://doi.org/10.1097/00005344-200406000-00004 (2004).
Teymoori, F., Asghari, G., Mirmiran, P. & Azizi, F. High dietary intake of aromatic amino acids increases risk of hypertension. J. Am. Soc. Hypertens. 12, 25–33. https://doi.org/10.1016/j.jash.2017.11.004 (2018).
Strauss-Kruger, M. et al. Identifying a metabolomics profile associated with masked hypertension in two independent cohorts: Data from the African-PREDICT and SABPA studies. Hypertens. Res. 45, 1781–1793. https://doi.org/10.1038/s41440-022-01010-2 (2022).
Gao, S., Zhao, J., Liu, X., Liu, L. & Chen, R. Metabolomics reveals serum metabolic signatures in H-type hypertension based on mass spectrometry multi-platform. Eur. J. Clin. Invest. 53, e14063. https://doi.org/10.1111/eci.14063 (2023).
Dasgupta, S. et al. Discovery of novel metabolic signatures for early identification of women at risk of developing gestational hypertension. Metabolomics 19, 50. https://doi.org/10.1007/s11306-023-02012-y (2023).
Arghavani, H., Bilodeau, J. F. & Rudkowska, I. Association between circulating fatty acids and blood pressure: A review. Curr. Nutr. Rep. 14, 15. https://doi.org/10.1007/s13668-024-00602-3 (2025).
Castillo, E. C. et al. Integrative analysis of lipid profiles in plasma allows cardiometabolic risk factor clustering in children with metabolically unhealthy obesity. Oxid. Med. Cell Longev. 2020, 2935278. https://doi.org/10.1155/2020/2935278 (2020).
Losurdo, G. et al. Prevalence and associated factors of obesity in inflammatory bowel disease: A case-control study. World J. Gastroenterol. 26, 7528–7537. https://doi.org/10.3748/wjg.v26.i47.7528 (2020).
Acknowledgements
This work was funded by Jiangsu Province Traditional Chinese Medicine leading talent training project, grant number Su Zhong Yi Ke Jiao [2023] No. 17. We appreciate all our participants and staff from Wuxi Hospital of Traditional Chinese Medicine. The technical assistance provided by the laboratory is also acknowledged.
Author information
Authors and Affiliations
Contributions
Conceptualization, Hongjun Zhu, Yifan Xu, Yilin Jiang and Jia Lu; methodology, Hongjun Zhu, Chen Xu and Qing Li; software, Jia Lu; validation, Jia Lu; formal analysis, Jia Lu; investigation, Yilin Jiang and Jia Lu; resources, Hongjun Zhu; data curation, Chen Xu and Qing Li; writing—original draft preparation, Yifan Xu and Yilin Jiang; writing—review and editing, Hongjun Zhu and Yifan Xu; visualization, Yifan Xu; supervision, Yifan Xu; project administration, Hongjun Zhu and Yilin Jiang; funding acquisition, Hongjun Zhu. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Y., Jiang, Y., Lu, J. et al. Identification of serum metabolic markers in non-obese hypertensive patients using non-targeted metabolomics. Sci Rep 15, 18320 (2025). https://doi.org/10.1038/s41598-025-02162-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02162-0

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}