
Abstract
Rhynchosporium commune is a fungal pathogen responsible for causing scald disease in barley, leading to significant yield losses and reduced grain quality in susceptible cultivars. Effector proteins secreted by R. commune play crucial roles in manipulating host defenses and facilitating infection. Hence, this study aimed to identify and characterize effector candidates (ECs) in R. commune using a comprehensive bioinformatics approach combined with experimental validation. Initially, a dataset of 12,211 genes from the R. commune strain UK7 genome was analyzed to identify potential ECs, resulting in the selection of 48 candidate proteins. These candidates were further validated using RNA-Seq analysis, which confirmed significant expression of 27 ECs during infection. Our analysis re-identified key effectors, including CZT06923 and CZT13833, with 100% identity to NIP3 and NIP2, respectively, in R. commune. Novel ECs, such as CZT07600, CZT13755, and CZT13375, were identified with lower identity to NIP2, suggesting potential variants. Additionally, structural analysis revealed that CZT07873 EC indicates significant structural similarity to known fungal effector. qRT-PCR validation confirmed the differential expression of CZS93219 and CZT13755, with peak expression at 9 and 12 dpi, respectively. This comprehensive approach enhances our understanding of R. commune’s pathogenic mechanisms and provides insights into potential targets for developing disease management strategies in barley cultivation.
Similar content being viewed by others

Introduction
Barley (Hordeum vulgare), one of the most crucial cereal crops worldwide, was cultivated on 52 million hectares in 2020 1,2. Among the first domesticated grains, barley is a vital food source in developing countries, valued for its nutritional benefits and adaptability to diverse environmental conditions3. However, barley production faces severe threats from scald, a major disease caused by the hemibiotrophic fungal pathogen R. commune4. Infected barley crops often experience reduced yields and lower seed quality, with global losses averaging 10% due to pathogen infection4,5,6. In the United Kingdom, yield losses from R. commune infections are estimated to cost £7.2 million annually, despite the use of treatments7. However, research indicates that yield losses due to R. commune can vary between 10% and 40%, with severe instances resulting in reductions of up to 35% across different regions8,9. Fungal plant pathogens like R. commune thrive in cold, wet conditions and spread primarily through infected seeds or straw, with further dispersal facilitated by water splashes. This pathogen is known for its high genetic variability, with 58% of global genetic diversity represented within individual fields4,10. This genetic diversity complicates disease management and significantly contributes to yield losses.
However, plants have developed a variety of defense mechanisms against pathogens in order to prevent loss of yield, but pathogens have also evolved strategies to counter these defenses by producing effectors and metabolites. During interactions between plants and pathogens, plants utilize plasma membrane-localized pattern recognition receptors (PRRs) to identify pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), leading to the activation of PAMP-triggered immunity (PTI). Pathogens secrete effectors that can disrupt PTI or increase virulence11,12. In response, plants have evolved resistance proteins encoded by R genes that can detect these effectors, initiating a strong defense mechanism known as effector-triggered immunity (ETI), often resulting in localized tissue necrosis13,14. Fungal effector proteins exhibit significant diversity and can be categorized based on their structure, function, and the specific host processes they target. Certain effectors are recognized for their capability to suppress the immune responses of the host, whereas others are renowned for their ability to manipulate the physiological functions of host cells in favor of the pathogen. Moreover, these effectors can be differentiated by the mechanisms through which they are delivered: some are secreted into the extracellular space, while others are directly injected into host cells via specialized infection structures15,16,−17.
The identification of fungal effector proteins is crucial for advancing our understanding of plant-pathogen interactions and developing effective crop resistance strategies. Effector proteins help pathogens invade host plants, suppress immune responses, and manipulate cellular functions. Understanding these mechanisms allows researchers to uncover plant vulnerabilities and develop resistance genes that recognize and counteract these effectors. Additionally, effector identification aids in creating targeted disease management strategies, reducing the reliance on broad-spectrum pesticides and enhancing crop resilience18. Despite the significant impact of R. commune on barley production, relatively few studies have been conducted to gain comprehensive insights into the repertoire of effectors utilized by this pathogen. Kristen et al. demonstrated that the necrosis-inducing proteins NIP1, NIP2, and NIP3 in R. commune play varying roles in disease development, with their impact differing based on host genotype19. The expression analysis conducted in this study revealed that NIP1 transcripts are present in spores, whereas NIP2 and NIP3 transcripts are synthesized following host plant inoculation19. Several studies on different isolates consistently demonstrated the widespread presence of NIP2 and NIP3 genes in R. commune populations, emphasizing their significance for the pathogen20,21. In a study by Mohd-Assaad et al. (2019), two distinct NIP1 paralogs, namely, NIP1A and NIP1B, were identified in almost all Rhynchosporium species22. NIP1A, which has been extensively studied, exhibited a greater impact on virulence compared to NIP1B. Furthermore, NIP1A was found to be under significant positive selection, indicating its dominant role as an effector that co-evolves with host receptors. Variations in the copy number of NIP1A and NIP1B were also observed across Rhynchosporium species22. Similarly, Penselin et al. (2016) identified NIP2 families consisting of 7–10 members in R. commune isolates23. During the subsequent investigation, the examination of gene expression revealed the presence of three previously unidentified genes, namely Rc_10934, Rc_2091, and Rc_2835, which emerged as potential effectors. These genes exhibited the greatest level of abundance throughout the infection progression24.
Despite previous research and the development of various effector-prediction pipelines25,26,27,28, identifying reliable effector candidates in R. commune remains challenging for three key reasons: (1) the lack of pathogen-specific prediction tools, (2) insufficient integration of multi-omics validation, and most critically, (3) our limited understanding of effector biology in this system. While effector research has advanced significantly in model fungal pathogens, current knowledge in R. commune remains restricted to the well-characterized NIP family (NIP1-3), and few other candidates, leaving major gaps in understanding: first, the complete effector repertoire; second, their stage-specific regulation during infection; and third, the structural basis of host manipulation.
To overcome these limitations, our study implemented an integrated pipeline combining three complementary approaches: (1) rigorous bioinformatics prediction using modified criteria optimized for R. commune, (2) temporal expression profiling via RNA-Seq across infection stages, and (3) experimental validation through qRT-PCR. This systematic strategy not only identified novel effector candidates beyond the NIP family but also revealed their dynamic regulation patterns, thereby providing unprecedented insights into effector-mediated pathogenicity in R. commune.
Results
Pathogenicity test
The Yusuf variety of barley showed the first signs of barley scald disease 12 days (Fig. 1) after being infected with R. commune. These signs presented as small, oval or spindle-shaped lesions with gray-white centers and reddish-brown margins. As time passed, these lesions grew larger and merged together, creating irregularly shaped spots. The disease symptoms progressed until 21 days after infection, when the affected barley plants experienced widespread tissue death and total leaf collapse.

The progression of disease stages and the development of barley scald caused by R. commune strain 1 observed (from left to right) on the 12, 15, 18, and 21 days after inoculation.
Identification of ECs
To identify ECs, an initial dataset of 12,211 genes from R. commune strain UK7 genome was analyzed. Proteins exceeding 300 amino acids in length were excluded, resulting in a subset of 4,628 proteins. SignalP v.6.0 software was then used to identify 509 secreted proteins. Subsequent filtering involved the use of TargetP v1.01 to remove proteins with N-terminal targeting signals, Big-PI to exclude cell wall-associated proteins, and TMHMM v.2.0 to eliminate membrane proteins. Additionally, proteins containing six or more cysteine residues were manually selected, resulting in a final set of 91 secreted proteins. Proteins with an EffectorP score above 80% were considered candidate effector proteins, leading to the selection of 48 proteins as the final ECs. From these 48 ECs, 45 were further validated and predicted as fungal effectors by the POOE database29. The cellular localization of these candidate effectors was further evaluated using LOCALIZER (Table S1).
Expression profile of ECs
The RNA-Seq data analysis revealed a total of 27 unique DE_ECs (differentially expressed effector candidates) across five pairwise comparisons from 48 ECs identified in this study (count matrix in Table S2). Specifically, the comparison between LBA and 13 dpi identified 12 DE_ECs (Fig S1). The comparison between LBA and 9 dpi resulted in the identification of 20 DE_ECs. The higher number of DE_ECs at 9 dpi suggests an early, broad effector response that may facilitate initial host colonization. In the comparison between PDB media and 13 dpi, 13 DE_ECs were identified. The comparison between PDB and 9 dpi yielded 7 DE_ECs. The nutrient-rich PDB environment appears to suppress effector diversity compared to host-like (LBA) conditions. Lastly, the direct comparison between 13 dpi and 9 dpi identified 13 DE_ECs. This distinct set of temporally-regulated effectors likely governs stage-specific virulence strategies.
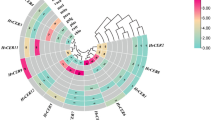
The RNA-Seq data analysis also revealed distinct patterns of DE_ECs across various comparisons, with each class exhibiting unique ranges of log2 fold change values and numbers of upregulated genes (Fig. 2, Table S3). In the comparison of 13dpi samples versus CLBA as control (CLBAvs13dpi), the log2 fold change ranged from 11.50 to 996.82, with 7 genes upregulated in 13dpi. For 9dpi samples versus CLBA as control (CLBAvs9dpi), the log2 fold change spanned 17.08 to 1615.81, with 6 genes upregulated in 9dpi. The higher maximal fold-change at 9 dpi may reflect aggressive early effector deployment. The 13dpi samples versus PDB as control (CPDBvs13dpi) showed a log2 fold change from 45.46 to 11140.71, and 11 genes were upregulated in 13dpi. The 9dpi samples versus PDB as control (CPDBvs9dpi) exhibited log2 fold changes from 27.34 to 14166.39, with 3 genes upregulated in 9dpi. The extreme upregulation in PDB comparisons (log2FC > 10,000) suggests nutrient stress may hyperactivate specific virulence factors. Lastly, in the comparison of 13dpi versus 9dpi samples (13dpivs9dpi), the log2 fold change ranged from 12.22 to 762.60, with 12 genes upregulated in 13dpi. This strong late-stage upregulation implies a shift toward effectors specialized for host tissue exploitation.

Heatmap illustrates the expression levels of ECs identified as DE_ECs through RNA-Seq data analysis. The heatmap displays scaled log2 fold-change values to manage the broad range of expression levels and highlight patterns, with red indicating positive values (upregulated genes) and blue indicating negative values (downregulated genes). Clustering of both rows and columns was performed based on correlation and average linkage methods to group similar expression profiles, facilitating a clearer visualization of trends and relationships within the data. CPDB refers to in vitro culture on Potato Dextrose Broth (PDB) medium, while CLBA indicates culture on Luria-Bertani Agar (LB) medium. 9 days post-inoculation (9 dpi) and 13 days post-inoculation (13 dpi) refer to samples collected from infected barley leaves.
Structural analysis and similarity search
Utilizing the hmmsearch algorithm, our analysis revealed that 11 out of the 27 DE_ECs contained at least one PFAM domain. This subset included proteins with known motifs, such as the cyanovirin-N domain (PF08881), frequently implicated as candidate fungal effectors in other fungal species. Notably, the DE_EC CZS94242, exhibiting high expression levels at 9 dpi, contained this domain. Additionally, CZS90720 was found to possess a Hydrophobin 2 domain (PF06766), with elevated expression in infected samples at 13 dpi, and CZT06923 contained a whey acidic protein (WAP) domain (PF00095). Further investigation of other ECs, which did not show significant expression in RNA-seq data, identified four ECs, CZS97401, CZS97401, CZS89959, and CZT05762, based on the presence of the CFEM domain.
Moreover, MEME analysis identified three novel conserved regions within the DE_ECs (Fig. 3). One of these motifs, “M[KQR]F[SL][IS][LA][FL]L[LA]AV,” was highly conserved across all 27 DE_ECs, with the multilevel consensus sequence “MKFSILFLLAV.” This motif was predominantly located between positions 1 and 11. Two other motifs were identified in 4 (CZT13755, CZT13833, CZT13375, CZT07600) and 3 (CZT13755, CZT13833, CZT13375) DE_ECs, respectively. An almost identical motif with the consensus sequence “MKLSTLLLALL” was found in ECs with no differential expression pattern, occupying the same position as the first motif identified in DE_ECs.

Conserved motif analysis of differentially expressed effector candidates (DE_ECs) identified from Rhynchosporium commune genome v1 by the MEME Suite program v5.5.7. The conserved motifs have been highlighted in specific regions of the EC sequences.
To further elucidate the characteristics of ECs (included DE_ECs), we utilized PHI-base database30, which contains expertly curated molecular and biological information on genes that influence host-pathogen interactions, as documented in peer-reviewed research articles. Our BLAST search of ECs against the PHI-base database identified significant homology in 11 ECs, including seven DE_ECs (Table S4). Among these DE_ECs, CZT06923 and CZT13833 showed 100% identity with the NIP3 and NIP2 effectors from R. commune, respectively, thereby confirming their re-identification as previously characterized effectors. In contrast, three additional DE_ECs, including CZT07600, CZT13375, and CZT13755, exhibited notable similarity with NIP2 from R. commune, although with lower identity percentages. However, it is important to note that these ECs have not been previously identified as effectors (Ec), nor have they been experimentally validated through expression analysis or structural characterization. The observed partial homology suggests that these candidates may represent either novel effectors distantly related to NIP2 or potential variants arising from copy number variation. Further investigation is warranted to elucidate their functional and evolutionary significance. Additionally, two ECs (not differentially expressed), CZT07873 and CZT10411, were found to be homologous to NIP1 and NIP2 from R. commune, respectively, further supporting their potential role as effectors. CZS89216 demonstrated similarity to genes across various fungi, including Alternaria brassicicola, Fusarium solani, and Phytophthora capsici, suggesting a broader functional relevance. Similarly, CZT08080 exhibited homology with the Fghyd5 gene from Fusarium graminearum and MHP1 from Magnaporthe oryzae, indicating possible functional parallels with known effectors in other fungal pathogens. These findings suggest that some effectors are conserved across various pathogens. However, the lack of upregulation under the tested conditions implies that these genes may not play a significant role in pathogenicity at these specific times or conditions To elucidate the further potential roles of ECs, we employed the Swiss-model to predict their three-dimensional structures. We then compared these predicted structures with known proteins that have characterized tertiary structures and functions using the Dali server. This study specifically concentrated on the most abundantly expressed ECs. Notably, only one EC demonstrated structural similarities to other ECs or Avr effector proteins from various plant-pathogenic fungi, whose tertiary structures have been previously determined. Our structural analysis revealed a significant resemblance between CZT07873 and the NIP1 elicitor protein from Rhynchosporium secalis (1kg1), with a z-score of 7.8, an RMSD of 1.54, and a remarkable sequence identity of 100% in 60 aa alignment length (Fig. 4).

Sequence and structural alignment. (A) Sequence alignment of CZT07873 (001 A) and 1 kg1 highlighting conserved regions. (B) Structural alignment of CZT07873 (blue) and 1kg1 (gold) reveals conserved folding in C-terminal domain, suggesting shared functional roles in substrate binding. The proteins are color-coded to differentiate between structures, and aligned regions are highlighted to emphasize structural conservation.
Gene expression profile using qRT-PCR
Gene expression analysis was conducted using qRT-PCR to validate four potential ECs. The analysis included three biological and three technical replicates to ensure robustness and accuracy. Samples were divided into two distinct groups for analysis (Fig. 5). The first group of barley leaves was initially collected at 1, 3, 6, 9, 12, 15, 18, and 21 dpi with R. commune. However, RNA extraction and quantitative analysis from samples at 1 and 3 dpi did not reveal any detectable gene expression (Fig. S2, 3). Consequently, these early time points were excluded from further qRT-PCR analysis, and subsequent assays focused on samples collected from 6 to 21 days post-inoculation. The second group comprised samples subjected to a mycelium starvation test. The temporal expression pattern of the CZS98606 gene demonstrated a dynamic profile. At 6 and 9 dpi, expression levels were notably elevated, reaching a peak at 9 days. This was followed by a marked decline at 12 dpi, after which expression remained consistently low at 15, 18, and 21 dpi. Interestingly, RNA-Seq analysis did not reveal significant differential expression for CZS98606; however, it was selected for further investigation based on its relatively high expression levels in the count matrix (Table S2). Moreover, homology searches identified significant similarity to the XadA3 protein from Xylella fastidiosa, a well-characterized virulence factor. This combination of elevated baseline expression and potential functional importance led to its inclusion in qRT-PCR validation. The expression pattern of CZT13755 showed an initial low expression at 6 dpi, followed by a significant increase peaking at 9 dpi. Subsequently, the expression level decreased at 12 dpi and continued to decline further by 15 dpi. A slight increase was observed at 18 dpi, followed by a stable and consistently low expression level at 21 dpi. This pattern suggested that CZT13755 exhibited peak expression early in the infection process, with a subsequent decrease as the infection progressed. The gene expression analysis of CZT13833 post-inoculation with the pathogen also revealed a similar dynamic pattern. Initially, expression was low at 6 dpi, followed by a notable increase in expression. This peak was observed at 12 dpi, indicating a potential role in the infection process. Subsequently, expression decreased progressively, remaining relatively low towards later time points (15 dpi onwards). The gene expression pattern of CZS93219 following inoculation showed a low expression level at 6dpi. This was followed by a noticeable increase at 9 dpi, indicating a response to the pathogen. Subsequently, the expression level declined sharply by 12 dpi, reaching a minimum. From 15 dpi onwards, there were slight fluctuations with no clear trend towards increasing or decreasing expression levels. In conclusion, genes CZT13755, CZT13833, and CZS93219 exhibited distinct yet overlapping expression patterns in response to R. commune inoculation. CZT13755 showed an initial low expression followed by a peak at 9 dpi and subsequent decline, CZT13833 displayed a similar trend with increased expression at 12 dpi, followed by a decrease, and CZS93219 demonstrated a responsive pattern peaking at 9 dpi with subsequent decline.

Relative expression of four ECs at intervals of 6, 9, 12, 15, 18, and 21 days’ post inoculation with R. commune and samples subjected to the mycelium starvation test. The samples were subjected to various nutrient conditions: B5-R (nutrient-rich source: complete B5 medium), B5-N (absence of nitrogen: potassium nitrate, ammonium sulfate), B5-C (absence of carbon), and B5-NC (absence of both nitrogen and carbon). In the presented data, error bars represent the standard error. Statistical significant was performed one-way ANOVA. * p < 0.05 and ** p < 0.01.
In the course of further investigation, the expression profiles of four genes were evaluated under different nutrient conditions, which included a nutrient-rich source (B5-R), the absence of nitrogen (B5-N), the absence of carbon (B5-C), and the absence of both nitrogen and carbon (B5-NC). All four genes demonstrated a significant increase in expression under the nutrient-rich conditions, suggesting that their regulatory roles are influenced by the availability of nutrients. Conversely, the genes exhibited noticeable downregulation when nitrogen or carbon was absent individually. However, the genes CZT13833 and CZS93219 exhibited enhanced expression when both nitrogen and carbon were absent, indicating a distinct regulatory response to these specific nutrient deficiencies. Correlation analysis between RNA-seq and qRT-PCR results for these four genes indicated a highly reliable correlation (R2 = 0.94), confirming the accuracy and reproducibility of the RNA-seq data (Figure S6).
Discussion
The importance of R. commune in barley cultivation is considerable due to its role in causing scald, a severe disease impacting barley worldwide8,31,32. Scald primarily affects regions with cool, humid climates, manifesting as elongated pale blotches with distinctive brown margins on leaves and leaf sheaths4. This disease can lead to yield losses of up to 30–40% in susceptible barley cultivars and degrade grain quality33.
While previous studies have identified a limited number of putative effectors, proteins critical for pathogen virulence through host defenses manipulation, our understanding still remains incomplete. To bridge this gap, robust bioinformatics methodologies and rigorous experimental validations are essential for the thorough identification and characterization of ECs in R. commune. Several computational pipelines have been developed for predicting fungal secretomes, including Secretool34, Fungal Secretome Database (FSD)35, FunSecKB36, FunSecKB237, and Aspertome38. However, these tools have not provided a comprehensive and reliable framework specifically for identifying ECs. Despite recent advancements in bioinformatics that have introduced more sophisticated tools and methodologies for effector identification, improving prediction accuracy39,40, gaps remain in existing pipelines. To address these limitations, we utilized a refined approach based on the methodology of Mueller et al.41, incorporating several modifications to enhance its effectiveness42. This advanced pipeline not only improves the accuracy of EC identification but also enables rapid and reliable predictions, representing a significant advancement over current methodologies and offering deeper insights into the secretome of R. commune.
Using our comprehensive pipeline, we have identified a total of 48 ECs. Out of these, 13 ECs align with the 22 ECs previously reported by Gamble from nine different strains24. It is worth noting that our approach differs significantly from that of Gamble et al.24. In their study, they used a criterion based on the presence of more than four cysteines, whereas we imposed a more stringent threshold, requiring the presence of more than six cysteines. This higher cysteine count is supported by research indicating that proteins with six or more cysteines are more reliable as predicted effectors. However, we also recognize that effector proteins with fewer than four cysteines have been identified in some pathogens, suggesting that cysteine content alone is not always a definitive predictor of effector functionality43,44,45,46. Furthermore, it is important to mention that our methodology incorporates the use of the EffectorP 2.0 tool for predicting fungal effectors within secretory proteins, which was not utilized by Gamble et al.24. Upon conducting further analysis of the 48 ECs, we made significant findings. By performing a BLASTp search against the fungal secretome database35 and PHI-base, we obtained significant hits for 36 ECs in the fungal secretome database. Additionally, we identified 11 unique potential effectors in PHI-base. These findings highlight the relevance of these ECs as secreted proteins within the fungal kingdom.
In addition to robust and precise bioinformatics analysis, validating expression data is essential for confirming bioinformatics findings. In our study, we employed two methods, RNA-Seq and qRT-PCR, to verify the bioinformatics results. Notably, 27 ECs with distinct temporal expression profiles during barley infection (9 vs. 13 dpi) were identified through RNA-Seq analysis as highly expressed genes during R. commune infection of barley leaves. These patterns reveal a likely biphasic virulence strategy: early-stage effectors (9 dpi) are enriched for host penetration and immune suppression functions, while late-stage effectors (13 dpi) dominate tissue colonization and nutrient acquisition. Among these, seven ECs demonstrated structural similarity to previously identified NIP effector proteins from R. commune, including one copy each of NIP1 and NIP3 (CZT07873 and CZT06923, respectively) and five copies of NIP2. The structural conservation strongly suggests functional parallels, particularly for CZT07873 which shares NIP1’s characteristic β-barrel fold and cysteine spacing, features critical for host membrane interaction and apoplastic stability. Besides, these findings are consistent with prior research indicating that NIP1 and NIP3 exist as single genes in R. commune genome8,20,23. However, previous studies have indicated that NIP2 contains 7 to 10 members, highlighting the potential variability and complexity within this effector family8,23. NIP1, NIP2, and NIP3 are crucial for pathogenicity of R. commune.
Initial characterization of NIP1, NIP2, and NIP3 demonstrated their capacity to induce necrosis in barley leaves, with subsequent studies revealing their role in modulating host physiology. Specifically, NIP1 and NIP3 have been shown to stimulate plasma membrane H+-ATPase activity, potentially promoting apoplastic acidification to facilitate fungal colonization. Notably, NIP1 also functions as a virulence factor, recognized by barley resistance gene Rrs1, thereby eliciting host defenses. The discovery of paralogs such as NIP1A, NIP1B, and multiple NIP2 variants (NIP2.1–NIP2.11), along with NIP2-like proteins (NLPs), underscores the genetic diversity within this effector family19,47,48.
Studies indicate the near-universal presence of NIP2 and NIP3 in various isolates, emphasizing their importance20,21. NIP1, existing in paralogs NIP1A and NIP1B, is under positive selection, showing co-evolution with host receptors and significant impact on virulence22. These proteins play a vital role during the early infection stages, aiding fungal spread and virulence20. NIP1 functions as both an effector and elicitor, inducing necrosis and resistance responses through the Rrs1 gene. Deletion or mutation of NIP1 enhances virulence, allowing the pathogen to bypass resistance mechanisms20,49. Conversely, NIP2 and NIP3 primarily induce necrosis without elicitor activity50. Understanding these effector proteins is crucial for developing effective disease resistance strategies in barley. Transcriptional analyses of the NIP effector family reveal distinct temporal dynamics, suggesting stage-specific contributions to pathogenesis. The previous studies indicate NIP expression peaks during early infection in susceptible barley cultivars but exhibits delayed yet more pronounced induction in moderately susceptible genotypes. In contrast, NIP2 and NIP3 are upregulated post-penetration, with their expression levels varying significantly depending on host genotype. For instance, NIP2 transcripts accumulate more abundantly in moderately susceptible cultivars than in susceptible ones, while NIP3 expression displays delayed but sustained peaks. These differential expression profiles suggest that NIP effectors may fulfill specialized roles at distinct infection stages, with their regulation finely tuned by host genetic factors. The early induction of NIP1 may facilitate initial colonization, whereas the later expression of NIP2 and NIP3 could contribute to sustained fungal growth or immune evasion. Such host-dependent modulation of effector expression underscores the dynamic interplay between pathogen virulence strategies and plant defense responses19.
Beyond the NIP family, additional effector candidates have been implicated in R. commune pathogenicity. RcCDI1, a fungal PAMP, induces cell death in Solanaceae species, while PFP1, containing an Epc-N domain, appears critical for infection, possibly through epigenetic regulation. Transcriptomic and proteomic analyses have identified other potential effectors, including Rc_10934, Rc_2091, Rc_2835, Rc07_03591, and Rc07_02334, which exhibit elevated expression during the biotrophic phase. Furthermore, RcLysM3 may contribute to immune evasion by binding chitin and suppressing host defenses, analogous to related fungal proteins. Secondary metabolites, such as the necrotic-inducing meroterpenoid Rhynchospene B, have also been implicated in symptom development51,52.
The high expression of CZS93219 at 9 dpi according to our expression data analysis, it is plausible that CZS93219 functions as a potential EC. Additionally, the identification of the YafQ toxin (PF15738) as a conserved domain within CZS93219 provides further evidence for its role as an effector. These findings collectively suggest that CZS93219 may play a significant role in the pathogenicity of R. commune. Further investigation using our integrated bioinformatics and expression analysis identified CZS94242 as a significant EC. This gene exhibited distinct expression dynamics during infection, particularly showing heightened responsiveness during the early stages of the infection process. Additionally, a motif search revealed that CZS94242 contains a CVNH domain. The CVNH protein is frequently identified as a candidate effector in various phytopathogenic ascomycete fungi, such as Sclerotinia sclerotiorum53,54 and Colletotrichum higginsianum55. CZS90720, another DE_EC identified in this study, contains a Hydrophobin 2 domain. Hydrophobin 2 is a type of hydrophobin, which is a small protein class found in filamentous fungi. Hydrophobins are known for their ability to modify the surface properties of fungal spores and hyphae, allowing them to interact with hydrophobic and hydrophilic surfaces. In the context of fungal effectors, hydrophobins have been implicated in the interaction between fungi and their hosts26.
This study establishes a framework for effector candidate identification through integrated computational and transcriptomic approaches. While our multi-tiered pipeline demonstrates improved predictive accuracy over existing methods, we recognize two fundamental considerations: (1) in silico predictions ultimately require empirical validation through targeted mutagenesis and complementation assays to confirm virulence functions, and (2) expression patterns, while temporally resolved, necessitate biochemical characterization of host-pathogen interactions to establish mechanistic causality. Looking forward, the identified effector candidates present immediate opportunities for translational applications, including HIGS targets and marker-assisted breeding strategies. Specifically, conserved effectors showing stage-specific expression (e.g.,CZT07600, CZT13755, and CZT13375) could serve as priority targets for developing durable resistance in barley cultivars.
Conclusion
Our study employed a comprehensive bioinformatics pipeline to identify and characterize 48 ECs in R. commune, the causal agent of barley scald disease. Through integrated RNA-Seq and qRT-PCR analyses, we validated the expression patterns of 27 ECs during host infection, including known virulence factors (NIP2/NIP3 homologs) and novel candidates such as CZT07600, CZT13755, CZT13375, CZT07600, CZT13755, and CZS93219 containing conserved effector domains. These findings provide multiple actionable pathways for improving barley resistance: (1) The identified ECs serve as molecular markers for screening barley germplasm and can guide marker-assisted breeding programs to pyramid resistance genes; (2) Highly expressed effectors like CZS94242 (CVNH domain) and CZS90720 (Hydrophobin 2 domain) represent prime targets for host-induced gene silencing (HIGS) strategies; (3) Structural characterization of ECs (e.g., CZT07873’s similarity to NIP1) enables the rational design of protein inhibitors or decoy receptors to disrupt effector function. The multi-copy nature of NIP2 homologs and sequence variation among identified ECs highlight the pathogen’s evolutionary adaptability while simultaneously revealing vulnerable points for intervention. By bridging fundamental knowledge of R. commune pathogenicity with practical disease management applications, this work lays the foundation for developing durable, effector-informed solutions to combat barley scald.
Materials and methods
Isolation and purification of R. commune
The barley leaves exhibiting typical scald disease symptoms were collected from barley fields (Table 1). To obtain these samples the permissions were not necessary. The formal identification of the plant material was undertaken by the herbarium of Agricultural and Natural Resources College, University of Tehran, and no voucher specimens were collected and deposited in the collection (it is not necessary as we don’t describe a novel species). We also stated that the field studies were in compliance with local legislation of Iran in the experimental greenhouse of Isfahan University of Technology, Isfahan, and no specific licenses were required.
The leaf segments containing scald lesions underwent fungal isolation. To this aim, the leaf segments were surface-disinfected using 1% sodium hypochlorite and 70% ethanol, followed by rinsing with sterile water and drying on filter paper. Subsequently, the leaf segments were transferred on a 2% water-agar medium and placed in an incubator at 17 °C under continuous darkness for 10 days. The single-spore isolation procedure was employed to obtain pure cultures. Isolates 1 and 2 were obtained from leaves infected with barley scald pathogen, sourced from Korzan, Kermanshah (isolate 1), and Harsin village (isolate 2). The geographical coordinates for these locations are provided in Table 1. Besides, microscopic examination of the isolates was conducted (Fig. S4) to assess the colony morphology, sporulation, and spore characteristics. These observations were consistent with the known morphological traits of R. commune.
Pathogenicity test, inoculation, and sampling
To facilitate further growth and proliferation of the fungus, the isolated strain 1 was (due to its consistent behavior in infection development) transferred to an enriched white bean-agar medium (80 g of white bean extract, 4 g of yeast extract, and 4 g of sucrose per liter). The cultures were maintained in a refrigerated incubator at 17 °C under continuous darkness for 4 weeks to promote optimal fungal growth and sporulation. The spore suspension was adjusted to 2 × 106 spores/mL and amended with 0.1% Tween 20. The susceptible Yusuf cultivar, with fully unfolded second leaves, was inoculated using a hand sprayer. The inoculation was performed until runoff, ensuring that all leaves were uniformly coated with the spore suspension for consistent pathogen exposure and reliable experimental results. The pots were enclosed in plastic bags and placed in a growth chamber at 15 °C, to keep high humidity (> 95%), and continuous darkness for 48 h. Subsequently, the barley plants were transferred to a greenhouse with a day/night temperature regime of 20 °C/17° C, under a photoperiod of 16 h of light and 8 h of darkness. Symptom observation and leaf sampling was performed on the day post-inoculation (dpi), 1, 3, 6, 9, 12, 15, 18 and 21 dpi. Leaves were sampled in triplicate and stored in sterile aluminum foil at − 70 °C until RNA extraction.
Starvation test
A controlled experimental setup was utilized to carry out the starvation test, mimicking the harsh conditions that pathogens encounter at the onset of plant infection when nutrient levels are scarce. These conditions create an environment conducive to the activation of effector and pathogenicity genes. Nutrient stress, such as carbon or nitrogen limitation, is highly relevant to effector expression during infection because it mirrors the conditions fungal pathogens naturally encounter in the early stages of host colonization. When a pathogen first contacts the host surface, nutrient availability is typically low, creating a starvation-like environment. Under these conditions, many fungi respond by upregulating effector genes, which help suppress host defenses and facilitate nutrient acquisition. Initially, the fungi were cultured on solid white bean-agar medium for a period of two weeks to establish growth, after which they were transferred to the liquid Yeast Synthetic Medium (YSM, consisting of 10 gr of yeast extract and 20 gr of sucrose per liter) to encourage further mycelial development. Subsequently, the cultures were placed in a shaker at 17 °C for a duration of 10 days. To conduct the mycelium starvation test, the isolates were then shifted to three different formulations of the B5 medium: B5 medium lacking a carbon source (sucrose), B5 medium devoid of nitrogen sources (potassium nitrate, ammonium sulfate), and B5 medium without both carbon and nitrogen sources (sucrose, potassium nitrate, ammonium sulfate). The cultures were incubated in a shaker at 17 °C for a period of 24 h56,57,58,59.
Identification and selection of ECs
Given the accessibility of R. commune v1 genome (GCA_900074885: Technische Universitat Munchen - WZW (2016). Strain: UK7) on the NCBI portal (https://www.ncbi.nlm.nih.gov/datasets/genome/?taxon=914237), protein sequences corresponding to the genes were retrieved. Subsequently, a systematic process was conducted to filter and select candidate genes42. This process included selecting proteins smaller than 300 amino acids, as smaller proteins are often associated with effector functions in fungal pathogens due to their role in manipulating host plant processes40,60,61, identify secretory proteins using SignalP v.6.062 software (http://www.cbs.dtu.dk/services/SignalP-6.0/), and employing TargetP v1.0163 software (http://www.cbs.dtu.dk/services/TargetP-1.0/) to remove proteins with N-terminal sequences such as mitochondrial targeting peptides, chloroplast transit peptides, or secretory pathway signal peptides. Additionally, membrane proteins were eliminated using TMHMM v.2.064 software (http://www.cbs.dtu.dk/services/TMHMM/), and proteins attached to the cell wall were excluded using Big_PI65 software (http://mendel.imp.ac.at/gpi/fungi_server.html). Proteins containing six or more cysteine amino acids were manually selected, and their presence in other fungi was assessed through homology searches. The six-cysteine threshold was chosen based on prior research indicating that cysteine-rich proteins often function as effectors in fungal pathogens, with most experimentally validated effectors containing six or more cysteines. Although this criterion may overlook some true effectors with fewer cysteines, it minimizes false positives, as proteins lacking these conserved residues are less likely to demonstrate effector activity in functional assays. Functional analysis was performed using Pfam (http://pfam.xfam.org/) and InterPro (https://www.ebi.ac.uk/interpro/)66,67. Fungal effectors in secretory proteins were initially predicted using the EffectorP 3.0 platform (http://effectorp.csiro.au/)68 with score above 80%. To enhance the reliability of identified ECs, these predictions were further validated using the POOE database (http://zzdlab.com/pooe/index.php)29, thereby increasing the likelihood of accurately identifying ECs. Subsequently, the subcellular localization of these effector proteins within plant cells was predicted utilizing the LOCALIZER tool (http://localizer.csiro.au/)69.
Identification of expressed ECs through RNA-seq
The expression of identified ECs was assessed by analyzing RNA-Seq data obtained from the European Nucleotide Archive (ENA) through the BioProject accession number PRJNA804666 (https://www.ebi.ac.uk/ena). Stalder et al. (2023) previously provided a comprehensive description of the sampling, RNA extraction, and sequencing methodologies70. In brief, RNA-Seq experiments were conducted under four different conditions, including two in vitro conditions and two in planta conditions. The in vitro conditions involved growth on Luria-Bertani (LB) and Potato Dextrose Broth (PDB) media. In planta experiments, the spring barley cultivar Beatrix (Viskosa 9 × Pasadena, Saaten Union, breeder’s Reference NS01/2449), was infected and leaf samples were collected at 9 and 13 days post-infection (dpi). Each experiment was performed with two replicates. Total RNA extraction was carried out using TRIzol (Invitrogen), and libraries were prepared using the TruSeq stranded mRNA sample prep kit (Illumina). The selected dataset was subsequently analyzed using the Galaxy platform (https://usegalaxy.eu)71. Initial quality checks on the reads were conducted with FastQC (v.0.11.8; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality bases and adapter sequences were then removed using Trimmomatic v.0.3872. High-quality clean reads were aligned to R. commune reference genome using STAR v2.7.10b73. Transcript assembly was performed with StringTie v.2.1.174. Differential expression analysis was conducted on the gene read count matrices with the Python script prepDE.py. The resulting matrices were uploaded to the IDEAMEX platform for further analysis75. DESeq276 was used to identify differentially expressed (DE) ECs (DE_ECs), applying screening criteria of FDR ≤ 0.05, log2 fold change (logFC) ≥ 2, and CPM (count per million) = 1.
Structural analysis and homology search
For further investigation, the inferred DE_ECs were analyzed through hmmsearch77, regex, and homology analysis. Conserved domains in each effector candidate were predicted using Pfam v37 (http://pfam.xfam.org/)66. Additionally, novel motifs in the ECs were detected using MEME v5.3.2 software. The Pathogen–Host Interactions database (PHI-base) was utilized to gain further insights into the identified DE_Ecs30. The tertiary structures of DE_EC proteins were predicted using the Swiss-Model, focusing on the most highly expressed EC members. These predicted tertiary structures were then screened against the RCSB PDB database using the Dali server78 to identify proteins with similar folds, considering hits with a Z-score of ≥ 2 as significantly similar. The alignment and visualization of protein tertiary structures were conducted using PyMol v2.5, employing the CEalign plugin tool79. Furthermore, the TM-align method80 was used to calculate the root-mean-square deviation (RMSD) value to explore structural similarities in greater detail.
Primer design
After identifying the candidate genes, gene-specific primers were meticulously designed to assess gene expression (Table 2). This process utilized several software tools, including Oligo, Primer3 (http://primer3.ut.ee/), and IDT-OligoAnalyzer (https://eu.idtdna.com/calc/analyzer). To ensure primer specificity, validation was carried out using NCBI Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primers were designed to have lengths between 18 and 23 nucleotides, with a GC content ranging from 40 to 60%. Additionally, the desired PCR product length for qRT-PCR was set between 155 and 195 bp to ensure optimal amplification efficiency. To further validate the primers, genomic DNA was extracted using the CTAB method81, and PCR was performed for each primer pair. The PCR products were subsequently subjected to agarose gel electrophoresis, which confirmed the presence of specific amplicons for each primer set (Fig. S5).
RNA extraction, cDNA synthesis and qRT-PCR analysis
RNA extraction from the leaf samples followed the protocol utilizing a 2% CTAB buffer81. The quantity and quality of the extracted DNA and RNA were assessed using a Nanodrop spectrophotometer and electrophoresis on a 1% agarose gel prepared with 0.5X TBE buffer.
To initiate cDNA synthesis, the RNA samples were initially subjected to DNase I treatment as per the guidelines provided by the manufacturer (Biofact) to eliminate any genomic DNA contamination. Specifically, 2 µg of total RNA was exposed to 1 U of DNase I (Thermo Fisher Scientific) at 37 °C for 30 min, followed by heat inactivation at 75 °C for 10 min. Subsequently, the treated RNA underwent reverse transcription utilizing Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT). The reaction mixture comprised 2 µg of DNase I-treated RNA, 200 U M-MLV RT, 500 µM dNTPs, 5 µM random hexamer primers, 10 mM DTT, and 20 U RNase inhibitor, incubated at 37 °C for 1 h and then inactivated at 70 °C for 15 min. To further validate the cDNA synthesis and ensure the quantitative assay’s reliability, a PCR test was performed using the designed primers on the synthesized cDNA. The PCR products were then analyzed through agarose gel electrophoresis to confirm successful cDNA synthesis and amplification specificity.
For the quantitative assessment of gene expression, qRT-PCR was carried out using a StepOne Real-Time PCR system in a 15 µL reaction volume. The reaction mixture consisted of 7.5 µL of SYBR Green Master Mix (BioFACT, Korea), 2 µL of diluted cDNA, and 0.1 µL of each primer (10 pM), with PCR-grade water added to complete the volume. The qRT-PCR process began with an initial denaturation step at 95 °C for 5 min. This was followed by 40 cycles of 10 s at 95 °C, 20 s at the primer-specific annealing temperature, and 20 s at 72 °C. The process concluded with a melting curve analysis. Gene expression levels were quantified using the 2−∆∆Ct method, as described by82, with glyceraldehyde-3-phosphate dehydrogenase (GPD : JQ349030) used as the internal reference gene19,23. In the gene expression analysis, control samples were incorporated for both the pathogenicity and starvation tests to ensure accurate calculation of relative expression levels. For the pathogenicity test, control samples without fungal inoculation were collected at each time point and used as the baseline for comparison. For the starvation test, nutrient-rich medium (R) was used as the control for relative expression calculations across different nutrient-deprivation conditions. Due to variability in Ct values across biological replicates, the geometric mean was used to determine the average expression level. The geometric mean was calculated by multiplying the Ct values of biological replicates and then taking the nth root, where n represents the number of replicates (e.g., ∆Ct Control geometric average = \(\sqrt[3]{{{\text{(Ct1}} \times {\text{(Ct2}} \times {\text{(Ct3))}}}}\). Finally, statistical analysis of the qRT-PCR expression data was performed using one-way analysis of variance (ANOVA) followed by Tukey’s test to assess the significance of differences in gene expression. We also performed a correlation analysis between RNA-seq and qRT-PCR results using R.
Data availability
All RNA-Seq data were deposited in the NCBI SRA database under the project PRJNA804666 (https://www.ncbi.nlm.nih.gov/bioproject/ PRJNA804666).
References
Bouchetat, F. An analytical overview of the multiple uses of barley in the development of agro-food industries. YMER Digit. 22, 1131–1145 (2023).
Maanju, S. et al. Genetic diversity and population structure analyses in barley (Hordeum vulgare) against corn-leaf aphid, Rhopalosiphum maidis (Fitch). Front. Plant. Sci. 14, 1188627 (2023).
Saisho, D., Takeda, K. & Barley Emergence as a new research material of crop science. Plant. Cell. Physiol. 52, 724–727 (2011).
Zhan, J., Fitt, B. D. L., Pinnschmidt, H. O., Oxley, S. J. P. & Newton, A. C. Resistance, epidemiology and sustainable management of rhynchosporium secalis populations on barley. Plant. Pathol. 57, 1–14 (2008).
Newton, A. C., Searle, J., Guy, D. C., Hackett, C. A. & Cooke, D. E. L. Variability in pathotype, aggressiveness, RAPD profile, and rDNA ITS1 sequences of UK isolates of rhynchosporium Secalis/Veränderlichkeit der pathogenität, aggressivität, RAPD-Profile und der rDNA ITS1 sequenz für UK-Isolate von rhynchosporium secalis. Zeitschrift Für Pflanzenkrankheiten Und Pflanzenschutz/Journal Plant. Dis. Prot 446–458 (2001).
Avrova, A. & Knogge, W. Rhynchosporium commune: A persistent threat to barley cultivation. Mol. Plant. Pathol. 13, 986–997 (2012).
Newman, C. W. & Newman, R. K. A brief history of barley foods. Cereal Foods World 51, 4–7 (2006).
Zhang, X., Ovenden, B. & Milgate, A. Recent insights into barley and rhynchosporium commune interactions. Mol. Plant. Pathol. 21, 1111–1128 (2020).
Novakazi, F., Göransson, M., Stefánsson, T. S., Jalli, M. & Hallsson, J. H. Virulence of rhynchosporium commune isolates collected in Iceland. J. Plant. Pathol. 103, 935–942 (2021).
Zaffarano, P. L., McDonald, B. A., Zala, M. & Linde, C. C. Global hierarchical gene diversity analysis suggests the fertile crescent is not the center of origin of the barley scald pathogen rhynchosporium secalis. Phytopathology 96, 941–950 (2006).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Bigeard, J., Colcombet, J. & Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant. 8, 521–539 (2015).
Jones, J. D. G., Vance, R. E. & Dangl, J. L. Intracellular innate immune surveillance devices in plants and animals. Science 354 (2016).
Liang, W., Tong, M. & Li, X. SUSA2 is an F-box protein required for autoimmunity mediated by paired NLRs SOC3-CHS1 and SOC3-TN2. Nat. Commun. 11, 5190 (2020).
Stergiopoulos, I. & de Wit, P. J. Fungal effector proteins. Annu. Rev. Phytopathol. 47, 233–263 (2009).
Rocafort, M., Fudal, I. & Mesarich, C. H. Apoplastic effector proteins of plant-associated fungi and oomycetes. Curr. Opin. Plant. Biol. 56, 9–19 (2020).
Vleeshouwers, V. G. A. A. & Oliver, R. P. Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and necrotrophic plant pathogens. Mol. Plant. Microbe Interact. 27, 196–206 (2014).
Van de Wouw, A. P. & Idnurm, A. Biotechnological potential of engineering pathogen effector proteins for use in plant disease management. Biotechnol. Adv. 37, 107387 (2019).
Kirsten, S. et al. Necrosis-Inducing proteins of rhynchosporium commune, effectors in quantitative disease resistance. Mol. Plant. Microbe Interact. 25, 1314–1325 (2012).
Schürch, S., Linde, C. C., Knogge, W., Jackson, L. F. & McDonald, B. A. Molecular population genetic analysis differentiates two virulence mechanisms of the fungal avirulence gene NIP1. Mol. Plant. Microbe Interact. 17, 1114–1125 (2004).
Stefansson, T. S., Willi, Y., Croll, D. & McDonald, B. A. An assay for quantitative virulence in hynchosporium commune reveals an association between effector genotype and virulence. Plant. Pathol. 63, 405–414 (2014).
Mohd-Assaad, N., McDonald, B. A. & Croll, D. The emergence of the multi-species NIP1 effector in rhynchosporium was accompanied by high rates of gene duplications and losses. Environ. Microbiol. 21, 2677–2695 (2019).
Penselin, D. et al. Comparative genomics to explore phylogenetic relationship, cryptic sexual potential and host specificity of rhynchosporium species on grasses. BMC Genom. 17, 953 (2016).
Gamble, M. Molecular characterisation of the Rhynchosporium commune interaction with barley (2016).
Li, J. et al. Fungtion: A server for predicting and visualizing fungal effector proteins. J. Mol. Biol. 436, 168613 (2024).
Seong, K. & Krasileva, K. V. Prediction of effector protein structures from fungal phytopathogens enables evolutionary analyses. Nat. Microbiol. 8, 174–187 (2023).
Jones, D. A. B. et al. An automated and combinative method for the predictive ranking of candidate effector proteins of fungal plant pathogens. Sci. Rep. 11, 19731 (2021).
Carreón-Anguiano, K. G., Islas-Flores, I., Vega-Arreguín, J. & Sáenz-Carbonell, L. & Canto-Canché, B. EffHunter: A tool for prediction of effector protein candidates in fungal proteomic databases. Biomolecules 10 (2020).
Miao, Z. et al. POOE: Predicting oomycete effectors based on a pre-trained large protein Language model. mSystems 9, e01004–e01023 (2023).
Urban, M. et al. PHI-base: The pathogen-host interactions database. Nucleic Acids Res. 48, D613–D620 (2020).
Brunner, P. C., Schürch, S. & McDonald, B. A. The origin and colonization history of the barley scald pathogen rhynchosporium secalis. J. Evol. Biol. 20, 1311–1321 (2007).
Brunner, P. C., Stefansson, T. S., Fountaine, J., Richina, V. & McDonald, B. A. A global analysis of CYP51 diversity and Azole sensitivity in rhynchosporium commune. Phytopathology 106, 355–361 (2016).
Paulitz, T. & Steffenson, B. Biotic stress in barley: Disease problems and solutions. Barley Prod. Improv. Uses 307–354. https://doi.org/10.1002/9780470958636.ch11 (2011).
Cortázar, A. R., Aransay, A. M., Alfaro, M., Oguiza, J. A. & Lavín, J. L. SECRETOOL: Integrated secretome analysis tool for fungi. Amino Acids. 46, 471–473 (2014).
Choi, J. et al. Fungal secretome database: Integrated platform for annotation of fungal secretomes. BMC Genom. 11, 105 (2010).
Lum, G. & Min, X. J. FunSecKB: The fungal secretome knowledge base. Database (Oxford) bar001 (2011).
Meinken, J. et al. FunSecKB2: A fungal protein subcellular location knowledgebase. Comput. Mol. Biol. 4 (2014).
Vivek-Ananth, R. P. et al. Comparative systems analysis of the secretome of the opportunistic pathogen Aspergillus fumigatus and other Aspergillus species. Sci. Rep. 8, 6617 (2018).
Sonah, H., Deshmukh, R. K. & Bélanger, R. R. Computational prediction of effector proteins in fungi: Opportunities and challenges. Front. Plant. Sci. 7, 126 (2016).
Lovelace, A. H. et al. Effector identification in plant pathogens. Phytopathology 113, 637–650 (2023).
Mueller, O. et al. The secretome of the maize pathogen Ustilago maydis. Fungal Genet. Biol. 45(Suppl 1), S63–70 (2008).
Kamajian, M., Soorni, A. & Mehrabi, R. Identification of effector candidates in Bipolaris sorokiniana and their expression profile analysis during pathogen-wheat interactions. Physiol. Mol. Plant. Pathol. 133, 102343 (2024).
Rep, M. et al. A small, cysteine-rich protein secreted by fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383 (2004).
Qian, Y. et al. Systematic identification and functional characterization of the CFEM proteins in Poplar fungus Marssonina brunnea. Front. Cell. Infect. Microbiol. 12, 1045615 (2022).
Blondel, C. J., Amaya, F. A., Bustamante, P., Santiviago, C. A. & Pezoa, D. Identification and distribution of new candidate T6SS effectors encoded in Salmonella pathogenicity Island 6. Front. Microbiol. 14, 1252344 (2023).
Du, Z. et al. Characterization of a small cysteine-rich secreted effector, TcSCP_9014, in Tilletia controversa. Plants 13. https://doi.org/10.3390/plants13111523 (2024).
Jeffress, S. et al. Genome mining of the citrus pathogen Elsinoë fawcettii; prediction and prioritisation of candidate effectors, cell wall degrading enzymes and secondary metabolite gene clusters. PLoS One 15, e0227396 (2020).
Darma, R. et al. Revisiting the evolution and function of NIP2 parologs in the Rhynchosporium spp. complex. https://doi.org/10.1101/2024.10.15.618441 (2024).
Stukenbrock, E. H. & McDonald, B. A. The origins of plant pathogens in agro-ecosystems. Annu. Rev. Phytopathol. 46, 75–100 (2008).
Hahn, M., Jüngling, S. & Knogge, W. Cultivar-specific elicitation of barley defense reactions by the phytotoxic peptide NIP1 from rhynchosporium secalis. Mol. Plant. Microbe Interact. 6, 745–754 (1993).
Siersleben, S., Penselin, D., Wenzel, C., Albert, S. & Knogge, W. PFP1, a gene encoding an Epc-N domain-containing protein, is essential for pathogenicity of the barley pathogen rhynchosporium commune. Eukaryot. Cell 13, 1026–1035 (2014).
Franco-Orozco, B. et al. A new proteinaceous pathogen-associated molecular pattern (PAMP) identified in ascomycete fungi induces cell death in Solanaceae. New. Phytol. 214, 1657–1672 (2017).
Guyon, K., Balagué, C., Roby, D. & Raffaele, S. Secretome analysis reveals effector candidates associated with broad host range necrotrophy in the fungal plant pathogen Sclerotinia sclerotiorum. BMC Genom. 15, 336 (2014).
Heard, S., Brown, N. A. & Hammond-Kosack, K. An interspecies comparative analysis of the predicted secretomes of the necrotrophic plant pathogens sclerotinia sclerotiorum and Botrytis cinerea. PLoS One 10, e0130534 (2015).
Kleemann, J. et al. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathog. 8, e1002643 (2012).
Ries, L. N. A., Beattie, S., Cramer, R. A. & Goldman, G. H. Overview of carbon and nitrogen catabolite metabolism in the virulence of human pathogenic fungi. Mol. Microbiol. 107, 277–297 (2018).
Kahmann, R. & Basse, C. Fungal gene expression during pathogenesis-related development and host plant colonization. Curr. Opin. Microbiol. 4, 374–380 (2001).
Tudzynski, B. Nitrogen regulation of fungal secondary metabolism in fungi. Front. Microbiol. 5, 656 (2014).
Coleman, M., Henricot, B., Arnau, J. & Oliver, R. P. Starvation-induced genes of the tomato pathogen Cladosporium fulvum are also induced during growth in planta. Mol. Plant. Microbe Interact. 10, 1106–1109 (1997).
Feldman, D., Yarden, O. & Hadar, Y. Seeking the roles for fungal small-secreted proteins in affecting saprophytic lifestyles. Front. Microbiol. 11, 455 (2020).
Shen, Q., Liu, Y. & Naqvi, N. I. Fungal effectors at the crossroads of phytohormone signaling. Curr. Opin. Microbiol. 46, 1–6 (2018).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Emanuelsson, O., Nielsen, H., Brunak, S. & von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 (2000).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Pierleoni, A., Martelli, P. L. & Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 9, 392 (2008).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Hunter, S. et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Singh, K. B. & Taylor, J. M. Improved prediction of fungal effector proteins from secretomes with effectorp 2.0. Mol. Plant. Pathol. 19, 2094–2110 (2018).
Sperschneider, J. et al. Localizer: Subcellular localization prediction of both plant and effector proteins in the plant cell. Sci. Rep. 7, 44598 (2017).
Stalder, L., Oggenfuss, U., Mohd-Assaad, N. & Croll, D. The population genetics of adaptation through copy number variation in a fungal plant pathogen. Mol. Ecol. 32, 2443–2460 (2023).
Jalili, V. et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 48, W395–W402 (2020).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Jiménez-Jacinto, V., Sanchez-Flores, A. & Vega-Alvarado, L. Integrative differential expression analysis for multiple experiments (IDEAMEX): A web server tool for integrated RNA-Seq data analysis. Front. Genet. 10, 279 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Yoon, B. J. Hidden Markov models and their applications in biological sequence analysis. Curr. Genomics 10, 402–415 (2009).
Holm, L. Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42 (2020).
Shindyalov, I. N. & Bourne, P. E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11, 739–747 (1998).
Zhang, Y. & Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Wang, L. & Stegemann, J. P. Extraction of high quality RNA from polysaccharide matrices using cetyltrimethylammonium bromide. Biomaterials 31, 1612–1618 (2010).
Rao, X., Huang, X., Zhou, Z. & Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma Biomath. 3, 71–85 (2013).
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
S.A: investigation; methodology; formal analysis, writing-review and editing. A.S: Conceptualization; investigation; project administration; methodology; formal analysis; validation; visualization; writing-original draft; writing-review and editing. R.M: Investigation; methodology; funding acquisition; project administration; writing-review and editing. M.T: writing-review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Asadi, S., Soorni, A., Mehrabi, R. et al. Exploring effector candidates in Rhynchosporium commune: insights into their expression dynamics during barley infection. Sci Rep 15, 17667 (2025). https://doi.org/10.1038/s41598-025-02572-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02572-0
