
Abstract
This study aimed to explore the molecular mechanisms underlying the therapeutic effects of Qihuang JianPi ZiShen granule (QJZG) in treating Sjögren’s syndrome (SS) using a combination of network pharmacology, transcriptomics, and experimental validation. We conducted differential expression analysis, weighted gene co-expression network analysis, and utilized GeneCards along with other databases to identify potential therapeutic targets for SS. Concurrently, the active ingredients of QJZG and their associated targets were extracted from the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform. Network pharmacology was then employed to predict the therapeutic targets and biological pathways through which QJZG may exert its effects on SS. To validate these predictions, we performed immune infiltration analysis and molecular docking studies, followed by transcriptomic sequencing and in vitro experiments for further confirmation. A total of 1,570 SS-related targets were identified, along with 70 active ingredients of QJZG and 253 associated targets, 99 of which overlapped with the SS-related targets. Protein-protein interaction network analysis revealed hub targets, including IL-1β, TNF, and IL-6. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis highlighted significant pathways such as the “TNF signaling pathway” and “Apoptosis”. Immune infiltration analysis suggested that IL-1β, TNF, and IL-6 play critical roles in the pathogenesis and progression of SS. Molecular docking studies demonstrated strong binding affinities between the core ingredients of QJZG and hub targets. Transcriptomic sequencing confirmed that QJZG modulates SS through the TNF signaling pathway. In vitro experiments showed that QJZG, at a concentration of 4 mg/mL, exerted the most pronounced effects on human salivary gland (HSG) cells. Furthermore, treatment of interferon-γ-induced HSG cells with QJZG led to varying degrees of recovery in both inflammatory and apoptotic processes. Real-time quantitative PCR (RT-qPCR) and Western blot analyses further revealed that QJZG influences SS by modulating the expression of IL-1β, TNF-α, IL-6, and apoptotic-related cytokines. The results suggest that the therapeutic effects of QJZG in treating SS may be mediated through the TNF signaling and apoptosis pathways. This is likely achieved by downregulating pro-inflammatory factors such as IL-1β, TNF-α, and IL-6, which in turn alleviates inflammation-induced damage to the salivary glands.
Similar content being viewed by others

Background
Sjögren’s syndrome (SS) is a chronic systemic autoimmune disorder characterized by the production of inflammatory factors and the extensive infiltration of lymphocytes into exocrine glands, leading to persistent inflammation and dysfunction of exocrine glands, including the lacrimal, salivary, and minor lip glands1,2,3. The most prominent symptoms are dry eyes and dry mouth, resulting from dysfunction of the lacrimal and salivary glands. In addition to these hallmark features, patients with SS frequently experience fatigue, joint pain, swelling, and in some cases, joint deformities as well as other organ systems4. Severe complications, such as mucosal-associated lymphoid tissue lymphoma and non-Hodgkin’s lymphoma, are also commonly observed in SS patients5. The incidence of SS is relatively high, with more than 90% of patients being women. One study reported an incidence of 60.82 cases per 100,000 individuals in the general population, with the highest incidence observed at 56 years of age6. Additionally, the disease is persistent and often recurrent, significantly impacting the quality of life of those affected7.
Currently, due to the unclear pathogenesis and individual differences, there is no specific treatment for SS, and clinical management primarily targets the affected organs. Patients with mild symptoms often find relief through gland secretion stimulation or increased hydration8. For those with more extensive organ involvement, treatment may include nonsteroidal anti-inflammatory drugs to alleviate joint pain, hydroxychloroquine, glucocorticoids, and immunosuppressive agents for systemic management. However, these therapies are associated with significant drawbacks, including adverse reactions and high costs9,10. Consequently, the search for novel anti-SS therapies remains a key focus in rheumatology.
Traditional Chinese Medicine (TCM) takes a holistic approach, combining both disease diagnosis and syndrome differentiation. This approach offers unique advantages in improving clinical symptoms, slowing disease progression, and enhancing the quality of life for patients with SS. Qihuang Jianpi ZiShen Granule (QJZG) is a TCM formula developed based on the extensive clinical experience of Professor Huang, a renowned rheumatologist at the First Affiliated Hospital of Anhui University of Chinese Medicine. This formula is designed to modulate immune function, alleviate renal injury, and provide adjunctive therapeutic effects.11. It is derived from two classical TCM formulations: Liuwei Dihuang decoction and Sijunzi decoction12. Clinically, QJZG has been utilized to treat various rheumatic diseases, including systemic lupus erythematosus13, rheumatoid arthritis14, and ankylosing spondylitis15, demonstrating significant therapeutic effects. Furthermore, the formula has generally demonstrated a favorable safety profile in clinical practice, with only occasional gastrointestinal reactions and mild liver function abnormalities being reported13,16. In recent years, clinical studies have shown that QJZG has promising therapeutic potential in alleviating symptoms such as dry eyes, dry mouth, dry skin, and fatigue, while also improving the overall quality of life for patients with SS16,17.
Interferon-γ (IFN-γ), a type II interferon, plays a crucial role in modulating the activity of specific innate immune cells. In the salivary glands of patients with SS, it is produced in large quantities by immune cells. IFN-γ exerts a pro-inflammatory effect on the salivary glands by stimulating the production of cytokines and chemokines18. Previous studies have indicated a correlation between IFN-γ levels and the degree of inflammatory infiltration in SS19, further exacerbating its pathogenesis20,21,22. Based on this, we have established an IFN-γ-induced human salivary gland (HSG) cell model to investigate the effects of QJZG on SS inflammation and apoptosis. The total glycosides of paeony (TGP), a compound extracted from the root of the peony plant, have been shown to alleviate SS symptoms by reducing inflammatory cytokines and inhibiting the inflammasome pathway23,24,25. Given these therapeutic effects, TGP was selected as a positive control in this study to compare with the effects of QJZG.
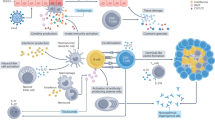
This study aims to investigate the mechanisms underlying the therapeutic effects of QJZG in treating SS through network pharmacology, bioinformatics, molecular docking, transcriptomic sequencing, and in vitro cell experiments, thereby laying the integration of TCM into the modern clinical treatment of SS. The study framework is illustrated in Fig. 1.

Study Framework. This study first employed network pharmacology and bioinformatics to identify the core ingredients, hub targets, and signaling pathways associated with QJZG in the treatment of SS. Subsequently, molecular docking, transcriptomic sequencing, and in vitro cell experiments were conducted to validate the findings from the network pharmacology analysis.
Materials and methods
Network Pharmacology
Screening for differentially expressed genes (DEGs) of SS
To identify DEGs of SS, we searched the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo) using the term “Sjögren’s syndrome” and obtained the GSE66795 dataset, annotated with the GPL10558 platform. This dataset includes whole blood samples from 131 fully phenotyped SS patients and 29 healthy controls. DEGs were identified by comparing SS samples with control samples using the “limma” package 3.62.2 in the R environment (v.4.4.1, https://www.r-project.org). Genes with significant expression differences were selected based on the following criteria: |log2FC (fold change)| > 1 and an adjusted p-value (q-value) < 0.05.
Identification of key co-expression modules of SS by weighted gene co-expression network analysis (WGCNA)
We performed WGCNA on the top 25% of the most variable genes from the GSE66795 dataset. Data preprocessing was initially conducted using the “goodSamplesGenes” function from the “WGCNA” R package 1.73 to filter out outlier genes and samples. The optimal soft threshold power for constructing the gene co-expression adjacency matrix was determined using the pickSoftThreshold function. This adjacency matrix was then transformed into a Topological Overlap Matrix (TOM), followed by hierarchical clustering with dynamic tree cutting to identify co-expression modules, with a minimum module size of 50 genes. Finally, we selected key gene modules that were significantly correlated with clinical traits of SS, using a correlation p-value threshold of < 0.05.
Identification of SS-related targets
The targets associated with SS were identified by searching the keyword “Sjögren’s syndrome” across several databases, including GeneCards (https://www.genecards.org), DisGeNET (https://disgenet.org), NCBI (https://www.ncbi.nlm.nih.gov), TTD (https://db.idrblab.net/ttd), and DrugBank (https://go.drugbank.com). Genes identified from DEGs, WGCNA, and the aforementioned databases were then merged and deduplicated to generate the final set of SS-related targets.
Screening of active ingredients of QJZG and related targets
QJZG is composed of eight Chinese herbs: Astragalus mongholicus, Atractylodes macrocephala, Dioscorea polystachya, Prepared Rehmannia glutinosa, Poria cocos, Cuscuta chinensis, Rubus chingii, and Rosa laevigata. The active ingredients of these herbs were initially screened using the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP, https://www.tcmsp-e.com). The screening criteria were set to include ingredients with a drug-likeness (DL) score ≥ 0.18 and oral bioavailability (OB) ≥ 30%. The related targets of the identified active ingredients were subsequently retrieved from the TCMSP platform. The target names were then standardized using the UniProt Knowledgebase (https://www.uniprot.org), and duplicates were removed, resulting in a refined list of target genes associated with the active ingredients of QJZG.
Acquisition of intersecting targets
The final set of SS-related targets was matched with the targets associated with the active ingredients of QJZG, using the Venny 2.1 tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html). A Venn diagram was generated to visualize the overlap between these target sets. The intersecting targets were considered potential therapeutic targets of QJZG for the treatment of SS.
“Herb-Ingredients-Targets” network and extraction of core ingredients
The active ingredients of QJZG and their intersecting targets were imported into Cytoscape 3.8.0 software (https://cytoscape.org) to construct the “Herb-Ingredients-Targets” network. Topological analysis of the ingredient nodes was performed using the CytoNCA plugin. Key ingredients were identified based on three centrality parameters: Betweenness Centrality (BC), Closeness Centrality (CC), and Degree Centrality (DC). A higher value of these parameters indicates that a node is closer to the center of the network26. Ingredients that exceeded the average values for these parameters were selected, and this process was repeated twice to identify the core ingredients27.
Protein-protein interaction (PPI) network and prediction of hub targets
The intersecting targets were uploaded to the STRING platform (https://cn.string-db.org) using the “Multiple Proteins” option. The organism was set to Homo sapiens, with a minimum interaction score of ≥ 0.85, and disconnected nodes were hidden to construct the PPI network. The resulting network’s TSV file was then imported into Cytoscape 3.8.0, where the CytoNCA plugin was used to analyze the network nodes based on six key parameters: BC, CC, DC, Eigenvector Centrality (EC), Local Average Connectivity (LAC), and Network Centrality (NC). Nodes that exceeded the average values for these parameters were selected, and this process was repeated twice to identify the hub targets.
Enrichment analysis of gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG)
GO functional and KEGG pathway enrichment28,29,30 analyses of the intersecting targets were performed using R packages (org.Hs.eg.db 3.20.0, enrichplot 1.26.6, clusterProfiler 4.14.6, and others). GO and KEGG terms with p-values and q-values < 0.05 were considered statistically significant. The results were ranked by q-value in ascending order. The top 10 GO functional terms and top 20 KEGG pathways were visualized, and pathway maps for key KEGG pathways were generated.
Immune cell infiltration analysis
We conducted an immune infiltration analysis to assess the abundance and activity of immune cells in SS, utilizing the CIBERSORT deconvolution algorithm to evaluate the relative proportions of 22 immune cell types in each sample from the GSE66795 dataset. Differences between SS patients and healthy controls were compared. Additionally, a Pearson’s correlation analysis was performed to examine the relationship between the expression of hub target genes and immune cell content, with statistical significance set at p-values < 0.05.
Molecular docking
The 2D structures of the core ingredients were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov). These structures were converted to 3D format using ChemBio3D Ultra 14.0 software (https://www.chemdraw.com.cn), followed by optimization based on minimizing the free energy, and saved in “Mol2” format, representing the small molecule ligands. The corresponding 3D structures of the hub targets were downloaded from the Protein Data Bank (PDB) (https://www.rcsb.org). Using PyMOL 3.1 software (https://www.pymol.org), water molecules, and bound ligands were removed, and the structures were saved in “pdb” format, representing the protein receptors. AutoDock Tools 1.5.6 software (https://autodock.scripps.edu) was used to identify the rotatable bonds in the small molecule ligands, add hydrogens to the target proteins, and compute partial charges. Both the small molecule ligands and protein receptors were then saved in “pdbqt” format. The docking grid parameters were set with a spacing of 1.0 Å, and the box size was adjusted to cover all active sites and the entire receptor. Molecular docking simulations were performed using AutoDock Vina 1.1.2 software (https://autodock.scripps.edu), and binding energies were calculated. A lower binding energy indicates a more stable docked complex31. A portion of the molecular docking results was visualized using PyMOL software.
Experimental verification
Reagents
Gibco RPMI 1640 Medium (basic) (batch No: C11875500BT) was purchased from KeJing (Guangzhou, China). Fetal bovine serum (FBS) (batch No: C0235) was obtained from Beyotime (Beijing, China). Recombinant human IFN-γ (batch No: HY-P7025) was sourced from MedChemExpress (Shanghai, China). The Cell Counting Kit-8 (CCK-8) (batch No: KGA9306), goat anti-rabbit IgG (batch No: KGC6207), and goat anti-mouse IgG (batch No: KGC6206) antibodies were acquired from KeyGEN (Jiangsu, China). RNA extraction reagent (batch No: G3013), chloroform substitute (batch No: G3014), SweScript All-in-One RT SuperMix for qPCR (batch No: G3337), 2×Universal Blue SYBR Green qPCR Master Mix (batch No: G3326), and Annexin V-IF488/PI Cell Apoptosis Detection Kit (batch No: G1513) were purchased from Servicebio (Wuhan, China). Gene primers were synthesized by GeneralBiol (Chuzhou, China). Antibodies targeting TNF-α (batch No: 6945 S), IL-6 (batch No: 12153 S), and IL-1β (batch No: 12703 S) were acquired from Cell Signaling Technology (Danvers, MA, USA). Polyvinylidene fluoride (PVDF) membranes (batch No: IPVH00010) were purchased from Millipore (Darmstadt, Germany).
Drug Preparation
The QJZG formulation was provided by the Preparation Center of the First Affiliated Hospital of Anhui University of Chinese Medicine (batch No: 20240219). TGP capsules were sourced from Ningbo Lihua Pharmaceutical Co., Ltd. (batch No: H20055058).
QJZG was dissolved in RPMI 1640 Medium to prepare a stock solution at a concentration of 100 mg/mL. IFN-γ was diluted to 30 µg/mL using an appropriate diluent. TGP was dissolved in DMSO to create a stock solution at 100 mg/mL. As required by the experiment, the QJZG stock solution was further diluted to 4 mg/mL, IFN-γ was diluted to 10 ng/mL, and TGP was diluted to 5 µg/mL using the complete medium. The final concentration of DMSO was kept below 0.1% to minimize potential cytotoxicity.
Cell culture, IFN-γ-induced SS model, and treatment
HSG cells (batch No: JNO-H0638) were purchased from Jennio (Guangzhou, China). The cells were cultured in RPMI 1640 Medium supplemented with 10% FBS and 1% penicillin-streptomycin. The cultures were maintained in a humidified incubator with 95% air and 5% CO₂ at 37 °C.
To establish the SS model, HSG cells were treated with 10 ng/mL IFN-γ, as described in previous studies32,33,34, followed by treatment with the corresponding drugs. HSG cells in the logarithmic growth phase were seeded into 6-well plates at a density of 2 × 10⁵ cells per well. The cells were then divided into the following treatment groups: blank control group, model group (10 ng/mL IFN-γ), QJZG group (10 ng/mL IFN-γ + 4 mg/mL QJZG), and positive control group (10 ng/mL IFN-γ + 5 µg/mL TGP).
CCK-8 assay
Before drug treatment, the CCK-8 assay was used to determine the optimal concentration of QJZG and evaluate its effect on cell viability. HSG cells in the logarithmic growth phase were seeded into 96-well plates at a density of 4 × 10³ cells per well, with a blank control well included. Each experimental group was set up with six replicate wells. After cell adhesion, 100 µL of complete medium containing different concentrations of QJZG (0, 0.5, 1, 2, 4, and 8 mg/mL) was added to the wells, while the control group received an equal volume of complete medium. After 24 and 48 h of treatment, 10 µL of CCK-8 solution was added to each well, and the cells were incubated for one more hour. The optical density at 450 nm was measured.
Transcriptome sequencing
After 10 h of treatment, cells from the blank control, model, and QJZG groups were collected, with three technical replicates per group. The cells were lysed using TRIzol reagent, left standing at room temperature for 5 min, and then immediately stored in liquid nitrogen. The samples were subsequently sent to Biomarker Technologies Co., Ltd. for RNA extraction, library construction, and transcriptome sequencing. Upon obtaining the transcriptome data, DEGs between the model and control groups, as well as between the model and QJZG groups, were identified using the “limma” R package. The criteria for identifying DEGs included a |log2FC| > 1 and a q-value < 0.05. The DEGs identified in both comparisons were then intersected, and the overlapping genes were subjected to subsequent enrichment analysis.
Cell apoptosis
HSG cells in the logarithmic growth phase were seeded at 2 × 10⁵ cells/well in a 12-well plate and grouped as described in the “Cell culture” Section. After 24 h, cells were washed, collected, and resuspended in a pre-cooled Binding Buffer (1–5 × 10⁶ cells/mL). A 100 µL aliquot was mixed with 5 µL Annexin V-IF488 and 5 µL PI, vortexed gently, and incubated in the dark at room temperature for 8–10 min. Afterward, 400 µL of pre-cooled Binding Buffer was added, and the mixture was gently mixed. A 6 µL aliquot was placed on a glass slide for fluorescence microscopy. Annexin V-IF488 stained early apoptotic cells green, while PI stained late apoptotic cells red.
Western blot
After 24 h of treatment, cells were lysed on ice using a mixture of radioimmunoprecipitation assay (RIPA) buffer and protease inhibitor. Protein concentration was determined using a BCA protein assay kit. The protein extracts were mixed with 5× loading buffer and boiled at 95 °C for 10 min. Electrophoresis was performed on a 10% SDS-PAGE gel, followed by transfer to a PVDF membrane. The membrane was blocked with 5% skim milk in TBST for 2 h at room temperature. After three washes with TBST, the membrane was incubated overnight at 4 °C with primary antibodies against TNF-α, IL-6, and IL-1β (all 1:1000). Subsequently, the membrane was incubated with secondary antibodies (1:1000) for 1 h at room temperature. Protein signals were detected using an ECL chemiluminescence reagent. The gray values of the protein bands were quantified using ImageJ software to calculate relative expression levels.
Quantitative real-time PCR (qRT-PCR)
After treating the cells according to the protocol in the “Cell culture” Section for 10 h, the cells were collected. Total RNA was extracted using the Trizol reagent, and 1 µg of RNA was reverse transcribed into complementary DNA (cDNA). QRT-PCR was performed to amplify the cDNA of the target genes. The primer sequences are provided in Table 1. The reaction mixture (20 µL) contained 10 µL SYBR Green Mix, 1 µL of each forward and reverse primer, 2 µL of cDNA template, and 6 µL of nuclease-free water. The amplification conditions were as follows: pre-denaturation at 95 °C for 30 s, denaturation at 95 °C for 15 s, and annealing at 60 °C for 30 s, for a total of 40 cycles. β-actin was used as the internal reference gene. Gene expression levels were calculated using the 2−ΔΔCt method.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.5. Each experiment was independently repeated at least three times. One-way ANOVA was used to analyze differences among groups, and P < 0.05 is considered to indicate a statistically significant difference.
Result
Results of network Pharmacology
Acquisition of SS-related targets
We initially performed differential expression analysis on the GSE66795 dataset, comparing SS patients with healthy controls, which revealed 29 DEGs (Fig. 2A). Subsequently, WGCNA was conducted using data from the same dataset. Six were selected as the optimal soft threshold (R2 = 0.85) to ensure that the interactions among genes conform maximally to a scale-free distribution (Fig. 2B). Modules associated with clinical characteristics of SS were then identified, with a total of nine modules constructed based on the gene dendrogram and the dynamic tree-cutting algorithm (Fig. 2C). The correlation between these modules and the samples (SS patients or healthy controls) was assessed, and the MEblack module, which showed the highest correlation, was identified as the key module (Fig. 2D). As a result, the 346 genes within the key module were considered to be highly associated with SS (cor = 0.88, p-value = 2.9e-113) (Fig. 2E). Additionally, we screened 1,269 potential targets associated with SS from GeneCards, DisGeNET, NCBI, TTD, and DrugBank. By integrating the targets identified through these three approaches, we obtained a final set of 1,570 potential SS-related targets (Fig. 2F).

Identification of Anti-SS Targets of QJZG. (A) Volcano plot of DEGs. (B) Determination of the optimal soft threshold power. (C) Dendrogram of clustered modules. (D) Heatmap showing the correlation between module eigengenes and SS. (E) Gene significance for the black module. (F) Venn diagram illustrating targets associated with SS. (G) Venn diagram depicting the overlap between QJZG targets and SS-related targets.
Acquisition of active ingredients and anti-SS targets of QJZG
Using the TCMSP platform, a total of 70 active ingredients were identified in QJZG, derived from the following herbs: 20 from Astragalus mongholicus, 7 from Atractylodes macrocephala, 16 from Dioscorea polystachya, 2 from Prepared Rehmannia glutinosa, 15 from Poria cocos, 11 from Cuscuta chinensis, 7 from Rubus chingii, and 6 from Rosa laevigata, with 10 ingredients shared across multiple herbs (Supplementary Table S1). Subsequently, 253 related targets for these active ingredients were retrieved from the TCMSP platform for further analysis. The intersection between SS-related targets and those of the active ingredients in QJZG was visualized using a Venn diagram (Fig. 2G), revealing 99 intersecting targets (Supplementary Table S2). These intersecting targets are considered potential therapeutic candidates for QJZG in the treatment of SS.
“Herb-Ingredients-Targets” network analysis results
The “Herb-Ingredients-Targets” network comprises 134 nodes and 271 edges. As shown in Figs. 3 and 35 active ingredients interact either directly or indirectly with 99 intersecting targets, while an additional 35 active ingredients lack corresponding targets within this set. After two rounds of topological analysis, based on the criteria outlined in the “extraction of core ingredients” Section, five core ingredients were identified, with topological parameters (BC, CC, DC) exceeding the respective mean values of 543.35, 0.2121, and 10.00. These core ingredients are quercetin, β-sitosterol, 7-O-methylisomucronulatol, formononetin, and kaempferol. The detailed topological parameters for these ingredients are provided in Supplementary Table S3. These core ingredients were subsequently selected for molecular docking analysis.

“Herb-Ingredients-Targets” Network. Diamond nodes represent intersecting targets, with the size of each node reflecting the number of active ingredients associated with that target. Square nodes represent the active ingredients of QJZG, each labeled with its corresponding TCMSP ID. The color of the pie chart within each square node indicates the source herb of the ingredient, with color codes assigned as follows: 1: Atractylodes macrocephala, 2: Poria cocos, 3: Rubus chingii, 4: Astragalus mongholicus, 5: Rosa laevigata, 6: Dioscorea polystachya, 7: Prepared Rehmannia glutinosa, and 8: Cuscuta chinensis. If a pie chart displays multiple colors, this indicates that the ingredient is derived from more than one herb.
PPI network analysis results
To identify the hub targets of QJZG in the treatment of SS, we constructed a PPI network using the STRING platform. The network comprised 86 nodes and 404 edges. Following the selection criteria described in the “prediction of hub targets” Section, we applied two rounds of filtering, resulting in the identification of seven hub targets: JUN, TP53, IL-1β, AKT1, TNF, IL-6, and BCL2. The topological parameters—BC, CC, DC, EC, LAC, and NC—for these targets were all higher than their respective averages, with values of 6.19, 0.62, 11.00, 0.18, 6.36, and 7.75, respectively. Detailed topological parameters for these targets are presented in Supplementary Table S4, and the filtering process is depicted in Fig. 4.

Screening process for hub targets in the PPI network.
GO and KEGG enrichment analysis results
The results of the GO functional enrichment analysis are shown in Fig. 5A. A total of 2,287 GO terms were enriched, including 2,117 associated with biological processes (BP), 40 with cellular components (CC), and 130 with molecular functions (MF). The enriched BP terms included “response to lipopolysaccharide”, “response to molecules of bacterial origin” and “regulation of miRNA transcription”, among others. The enriched CC terms were primarily related to “membrane raft,” “membrane microdomain” and “external side of the plasma membrane,” among others. Additionally, the enriched MF terms were primarily associated with “cytokine receptor binding”, “cytokine activity” and “G protein-coupled neurotransmitter receptor activity”, among other functions.

Results of GO and KEGG enrichment analyses. (A) The top 10 terms from GO functional enrichment analysis. (B) The top 20 pathways from KEGG pathway enrichment analysis.
The results of the KEGG pathway enrichment analysis, presented in Fig. 5B, identified 161 significantly enriched pathways. Among these, the most prominent include the “Lipid and atherosclerosis,” “Fluid shear stress and atherosclerosis,” “AGE-RAGE signaling pathway in diabetic complications,” “IL-17 signaling pathway,” “TNF signaling pathway,” and “Apoptosis,” among others. Notably, both the TNF and IL-17 signaling pathways are strongly implicated in the inflammatory response observed in SS, with apoptosis playing a critical role in its pathogenesis. Since TNF can induce the production of IL-1β and IL-635, and these three cytokines were identified as hub targets in the PPI network analysis, we focused on the TNF signaling pathway and its associated targets (IL-1β, TNF, and IL-6) for further investigation. This included an analysis of immune cell infiltration, molecular docking, and experimental validation. A diagram of the TNF signaling pathway is shown in Supplementary Figure S1.
Results of immune cell infiltration analysis
The results of the immune cell infiltration analysis are presented in Fig. 6. Among the 22 immune cell types tested, 18 were expressed in the samples. There are significant differences in the expression of memory-activated CD4+ T cells, monocytes, M0 macrophages, and activated dendritic cells between the SS group and the control group (p-value < 0.05, Wilcoxon test) (Fig. 6A). IL-1β expression was positively correlated with neutrophil levels, while negatively correlated with naive B cells, resting NK cells, resting mast cells, naive CD4+ T cells, and CD8+ T cells. TNF expression was positively correlated with both monocytes and naive B cells, but negatively correlated with activated mast cells and memory-activated CD4+ T cells (Fig. 6B-D). Additionally, IL-6 expression was positively correlated with both naive B cells and CD8+ T cells. These findings suggest that IL-1β, TNF, and IL-6 may play key roles in the pathogenesis and progression of SS by modulating immune responses.

Correlation between immune cells and inflammatory biomarkers in SS. (A) Comparison of immune cell infiltration levels between SS and control samples. (B-D) Correlation histograms depicting the relationships between hub targets (IL-1β, TNF, and IL-6) and various immune cell types.
Molecular Docking results
As shown in Supplementary Table S5, the binding energies between the core ingredients and the selected target proteins are all below − 5 kcal/mol, indicating strong affinities for the proteins. The docking results with the lowest binding energies for each target protein-core ingredient pair were visualized using PyMOL software, including quercetin-IL-1β (PDB ID: 5r8k), quercetin-IL-6 (PDB ID: 1alu), and β-sitosterol-TNF-α (PDB ID: 7kp9), as shown in Fig. 7.

Molecular Docking Results. (A) Quercetin-IL-1β. (B) Quercetin-IL-6. (C) β-Sitosterol-TNF-α. The golden dashed lines indicate hydrogen bonds.
The formation of stable complexes between the hydrophobic small molecules and the active sites of the target proteins is primarily driven by hydrogen bonding. For instance, the visualization results show that quercetin interacts with the GLU-25 residue in IL-1β via a hydrogen bond (Fig. 7A). Additionally, quercetin forms hydrogen bonds with the GLN-175 and ARG-179 residues in IL-6 (Fig. 7B). Although β-sitosterol does not form hydrogen bonds with TNF-α, the binding energy between them remains remarkably low (− 11 kcal/mol), suggesting a strong binding potential.
Experimental verification
QJZG viability assay on HSG cells
HSG cells were treated with QJZG alone or in combination with 10 ng/ml IFN-γ for 24–48 h. CCK-8 assays demonstrated that, compared to the control group, the proliferative capacity of HSG cells showed a positive correlation with the QJZG concentration, ranging from 0 to 4 mg/ml (Fig. 8). The most pronounced proliferative effect was observed at 4 mg/ml, while concentrations of 8 mg/ml resulted in drug-induced toxicity, which inhibited cell growth. Additionally, regardless of whether QJZG was administered alone or in combination with IFN-γ, a significant reduction in HSG cell viability was observed after 48 h of treatment. Based on these findings, a treatment condition of 4 mg/ml QJZG for 24 h was selected for subsequent experiments.

Cell viability of HSG cells. (A, C) HSG cells were treated with QJZG for 24 h and 48 h. (B, D) IFN-γ-stimulated HSG cells were treated to QJZG for durations of 24 h and 48 h. Statistical significance between groups is indicated by the following p-values: ***P < 0.001, **P < 0.01, *P < 0.05, nsP > 0.05.
Transcriptome validation
As shown in Fig. 9A-B, the transcriptome analysis revealed that, compared to the control group, 464 genes were upregulated and 140 genes were downregulated in the model group. Additionally, compared to the QJZG group, 128 genes were upregulated and 54 genes were downregulated in the model group. The intersection of these two datasets identified 131 DEGs, which were considered potential therapeutic targets of QJZG for SS. Consistent with the network pharmacology analysis, enrichment analysis was performed on these 131 DEGs, as shown in Fig. 9C-E. Compared to the network pharmacology findings, two biological processes (BP) terms—“response to lipopolysaccharide” and “response to molecules of bacterial origin”, two molecular functions (MF) terms—“cytokine receptor binding” and “cytokine activity”, and three KEGG pathways—“TNF signaling pathway,” “Influenza A”, and “Hepatitis C”, were validated through our transcriptome analysis, further supporting the network pharmacology results.

Transcriptome Validation Results. (A) Volcano plot showing DEGs between the model and control groups. (B) Volcano plot showing DEGs between the model and QJZG groups. (C) Venn diagram illustrating the intersection analysis for potential therapeutic targets of QJZG for SS. (D) GO enrichment analysis of 131 DEGs identified in the transcriptome analysis. (E) KEGG enrichment analysis of 131 DEGs identified in the transcriptome analysis.
QJZG reduces the apoptosis induced by IFN-γ in HSG cells
Network pharmacology analysis suggested that apoptosis is one of the key pathways through which QJZG exerts its effects in SS. The expression levels of the apoptotic genes Bcl-2 and Bax were measured using RT-qPCR, with the experimental groups defined as described in the “Cell culture” Section. The results indicated that, compared to the control group, the IFN-γ treatment led to a significant downregulation of Bcl-2 (p-value < 0.001) and upregulation of Bax (p-value < 0.001). This effect was reversed by treatment with either QJZG or TGP (p-value < 0.001) and no significant difference was observed between the QJZG and TGP groups (Fig. 10A-B). Annexin V/PI apoptosis staining further revealed that all four experimental groups exhibited varying levels of early apoptosis, with the IFN-γ group showing a significantly higher proportion of late-stage apoptosis compared to the other groups (Fig. 10C). These findings suggest that QJZG may mitigate SS through modulation of the apoptotic pathway.

QJZG reduces IFN-γ-induced apoptosis in HSG cells. (A-B) mRNA expression levels of Bcl-2 and Bax in IFN-γ-induced HSG cells, measured by RT-qPCR (n = 3). (C) Fluorescence of HSG cell apoptosis detected by Annexin V/PI assay following treatment with IFN-γ alone or in combination with QJZG/TGP, where green fluorescence marks early apoptotic cells and red fluorescence marks late apoptotic cells. Statistical significance between groups is indicated by the following p-values: ***P < 0.001, **P < 0.01, *P < 0.05, nsP > 0.05.
QJZG reduces the protein expression of inflammatory factors induced by IFN-γ in HSG cells
Inflammation plays a critical role in the pathogenesis of SS. Western blot analysis demonstrated that stimulation of HSG cells with IFN-γ led to significantly upregulated the expression of TNF-α (p-value < 0.001), IL-6 (p-value < 0.005), and IL-1β (p-value < 0.001). Treatment with QJZG and TGP effectively alleviated these increases, reducing the upregulated levels of these inflammatory markers. Compared to the model group, the QJZG-treated group showed a significant reduction in the expression levels of TNF-α, IL-6, and IL-1β (p-value < 0.05). However, no significant difference in the expression levels of these inflammatory factors was observed between the QJZG and TGP groups (Fig. 11A-D).

QJZG reduces IFN-γ-induced inflammation in HSG cells. (A) Western blot analysis of TNF-α, IL-6, and IL-1β expression in SS-HSG cells treated with IFN-γ alone or in combination with QJZG/TGP. (B-D) Quantification of TNF-α, IL-6, and IL-1β protein expression (n = 3). (E-G) mRNA expression levels of TNF-α, IL-6, and IL-1β in IFN-γ-induced SS-HSG cells, measured by RT-qPCR. Statistical significance between groups is indicated by the following p-values: ***P < 0.001, **P < 0.01, *P < 0.05, nsP > 0.05.
Effect of QJZG on mRNA expression of inflammatory factors in HSG cells
The mRNA expression levels of TNF-α, IL-6, and IL-1β in HSG cells were assessed by RT-qPCR. Compared to the control group, the model group exhibited a significant upregulation in the expression of TNF-α, IL-6, and IL-1β (p-value < 0.05). However, in the QJZG and TGP groups, the expression levels of these inflammatory markers were significantly reduced compared to the model group (p-value < 0.05). No significant differences were observed between the QJZG and TGP groups. (Fig. 11E-G).
Discussion
The pathogenesis of SS is not fully understood, but it is characterized by chronic inflammation, a hallmark of autoimmune diseases. This inflammation is perpetuated by prolonged lymphocyte infiltration, which triggers the production of a variety of inflammatory mediators. Upon antigen stimulation, salivary gland epithelial cells secrete pro-inflammatory factors that further promote immune cell infiltration. These epithelial cells also serve as antigen-presenting cells, which initiate T lymphocyte proliferation and activation. This cascade of events ultimately leads to glandular injury and disrupts the glandular microenvironment, contributing to the disease’s progression36,37. Currently, there is no specific treatment for SS in clinical practice. For patients with severe SS or those with coexisting conditions, treatment often includes a combination of glucocorticoids, hydroxychloroquine, disease-modifying antirheumatic drugs, immunosuppressants, and biologic agents. These treatments aim to alleviate disease progression and manage the associated symptoms38.
QJZG is composed of eight Chinese herbs, each contributing to the formula based on TCM theory17. Network pharmacology results further support this formulation. In the ‘Herb-Ingredients-Targets’ network, the total degree values of the nodes corresponding to the active ingredients of each herb indicate that Astragalus mongholicus and Cuscuta chinensis occupy the top two positions (Supplementary Table S6), underscoring their pivotal roles in the formulation. Modern pharmacological studies have also revealed that Astragalus polysaccharide, an active ingredient derived from Astragalus mongholicus, can enhance cardiac function in rat models of SS39. Additionally, Cuscuta chinensis has shown notable anti-inflammatory properties and the potential to alleviate oxidative stress40. QJZG has demonstrated promising efficacy in treating rheumatological and immunological diseases. Clinical studies have shown that QJZG is effective in managing SS, but its precise role and underlying mechanisms in SS remain unclear16,17. Our findings suggest that QJZG inhibits the production of inflammatory cytokines in IFN-γ-induced SS-HSG cells and may help alleviate the inflammatory environment in salivary gland cells.
TGP is a compound extracted from the dried root of Paeonia lactiflora. Modern pharmacological studies have shown that TGP can relieve SS by reducing inflammatory cytokines, inhibiting the activation of the NLRP3 inflammasome in the submandibular gland, regulating the Th1/Th2 immune balance, and improving the structure of intestinal microecology. TGP also has anti-inflammatory, analgesic, anti-stress, and immunomodulatory effects, which can alleviate musculoskeletal involvement and dryness symptoms in patients24,25,41. Our experiments showed that the efficacy of QJZG in relieving inflammation was comparable to that of TGP, suggesting that QJZG has potential for clinical application in the treatment of SS.
Based on the “Herb-Ingredients-Targets” network, the core ingredients of QJZG were selected using three centrality parameters: BC, CC, and DC. BC reflects the bridging role of a node in the network, identifying key nodes that control the flow of information. CC indicates the efficiency of a node’s propagation, with a higher closeness centrality typically signifying that the node can more effectively influence other nodes. DC represents the number of connections a node has, serving as a measure of the node’s direct influence. The higher the values of these three parameters, the more important the corresponding ingredients are within the network26. Through analysis, the core ingredients were identified as quercetin, β-sitosterol, 7-O-methylisomucronulatol, formononetin, and kaempferol.
Quercetin and kaempferol are bioactive compounds that can be extracted from the traditional Chinese medicinal plants Rubus chingii, Astragalus mongholicus, Rosa laevigata, and Cuscuta chinensis42. β-Sitosterol is a sterol compound that is present in Rubus chingii, Rosa laevigata, and Cuscuta chinensis. 7-O-methylisomucronulatol is a specific compound isolated from Astragalus mongholicus. Formononetin is an isoflavonoid that is also derived from Astragalus mongholicus43. Quercetin may protect against salivary gland damage induced by SS by reducing cell apoptosis and inflammation. β-Sitosterol has been shown to enhance the expression of CHRM3, stimulate salivary secretion, and potentially improve the prognosis of SS. Although 7-O-methylisomucronulatol, a unique active compound in Astragalus mongholicus, has been less explored in current research, it is involved in the “Herb-Ingredients-Targets” network, interacting with targets like CHRM3, a key factor in the pathogenesis of SS44. Formononetin exhibits anti-inflammatory effects in autoimmune diseases by inhibiting the IFN signaling pathway and modulating IL-6, IL-3, and other inflammatory factors45,46, suggesting its potential to modulate inflammatory responses in such conditions. In rheumatic diseases, kaempferol may reduce inflammation by inhibiting pyroptosis and suppressing excessive immune responses, potentially through the regulation of T-cell proliferation47. Collectively, these findings underscore the therapeutic potential of QJZG’s core ingredients in treating SS.
The results of the PPI network analysis highlight several hub targets of QJZG in the treatment of SS, including JUN, TP53, IL-1β, AKT1, TNF, IL-6, and BCL2. Previous research has established that TNF-α, IL-6, IL-1β, and Bcl-2 are closely linked to the onset and progression of SS48,49. Among these, TNF-α plays a pivotal role in the pathogenesis of SS by promoting the production of inflammatory cytokines and inducing apoptosis in salivary gland cells50. IL-6, primarily involved in inflammation and the regulation of mature B cells may mitigate SS by inhibiting abnormal B cell activation, thus reducing the synthesis and secretion of IgG51. The apoptotic regulators Bcl-2 and Bax modulate epithelial cell apoptosis in SS and are associated with the severity of inflammation49,52. Additionally, IL-17 expression is markedly elevated in SS patients, and the Akt/mTOR signaling pathway is implicated in the regulation of IL-17 activity53. TP53, which plays a central role in DNA repair, cell cycle regulation, and the pathogenesis of systemic autoimmune diseases, is expressed in various human and murine tissues54.
KEGG pathway enrichment analysis revealed several signaling pathways, including the TNF and apoptosis pathways, that are integral to the pathophysiology of SS. These pathways play pivotal roles in the disease’s development. The TNF signaling pathway, a well-characterized inflammation-related cascade, regulates numerous biological processes such as cell apoptosis, differentiation, proliferation, inflammation, and immune system modulation55. The downstream cytokines TNF, IL-6, and IL-1β not only function synergistically but also influence one another’s secretion35,56,57,58. This cytokine cascade initiates a strong inflammatory response, recruiting additional inflammatory cells to the site of injury and further amplifying immune activation59. In the present study, we concentrated on the inflammatory response involving TNF-α, IL-6, and IL-1β for preliminary validation. The results from immune cell infiltration analysis and molecular docking corroborate the reliability of the predictions derived from network pharmacology. Future research will build upon this foundation to conduct more comprehensive and detailed validation of QJZG’s predicted targets and signaling pathways in the treatment of SS.
The CCK-8 and apoptosis assays provided evidence that QJZG significantly increased the survival rate of HSG cells subjected to IFN-γ-induced stress, while simultaneously reducing cellular apoptosis. Transcriptome analysis further confirmed that QJZG exerts its therapeutic effects on SS by modulating the TNF signaling pathway. Western blotting and qRT-PCR analyses revealed that QJZG effectively suppressed the overexpression of pro-inflammatory cytokines, including TNF-α, IL-6, and IL-1β, in IFN-γ-stimulated HSG cells. These findings suggest that QJZG may serve as a promising therapeutic strategy for SS, potentially through the modulation of key factors within the TNF signaling pathway. This mechanism aligns with predictions made by network pharmacology analysis, further supporting the potential of QJZG as a treatment for SS.
Conclusions
In summary, we investigated the mechanism through which QJZG alleviates inflammation in SS using network pharmacology and validated these findings through preliminary cell experiments. The results suggest that the therapeutic effects of QJZG may be mediated via the TNF signaling pathway, potentially by downregulating pro-inflammatory factors such as TNF-α, IL-6, and IL-1β, thereby reducing inflammation-induced damage to the salivary glands. This study provides evidence supporting the use of Chinese medicine in treating SS. However, further research is required to fully elucidate the underlying mechanisms of QJZG in SS. Future studies will focus on identifying upstream targets and using bioinformatics to explore how QJZG modulates SS through the TNF pathway or other related pathways.
Data availability
The dataset used in this study is available in an online repository. The repository name and accession number can be found in the article or supplementary materials. For further inquiries, please contact the corresponding authors.
References
Rosso, P. et al. Involvement of substance P (SP) and its related NK1 receptor in primary Sjögren’s syndrome (pSS) pathogenesis. Cells 12 (10), 1347. https://doi.org/10.3390/cells12101347 (2023).
Nishihata, S. Y. et al. The toll-like receptor 7-mediated ro52 antigen-presenting pathway in the salivary gland epithelial cells of Sjögren’s syndrome. J. Clin. Med. 12 (13), 4423. https://doi.org/10.3390/jcm12134423 (2023).
Zhao, T., Zhang, R., Li, Z., Qin, D. & Wang, X. Novel and potential future therapeutic options in Sjögren’s syndrome. Heliyon 10 (19), e38803. https://doi.org/10.1016/j.heliyon.2024.e38803 (2024).
St Clair, E. W. et al. CD40 ligand antagonist Dazodalibep in Sjögren’s disease: A randomized, double-blinded, placebo-controlled, phase 2 trial. Nat. Med. 30 (6), 1583–1592. https://doi.org/10.1038/s41591-024-03009-3 (2024).
Sun, Y. et al. Targeting alarmin release reverses Sjogren’s syndrome phenotype by revitalizing Ca2 + signalling. Clin. Transl Med. 13 (4), e1228. https://doi.org/10.1002/ctm2.1228 (2023).
Qin, B. et al. Epidemiology of primary Sjögren’s syndrome: A systematic review and meta-analysis. Ann. Rheum. Dis. 74 (11), 1983–1989. https://doi.org/10.1136/annrheumdis-2014-205375 (2015).
Baldini, C., Fulvio, G., La Rocca, G. & Ferro, F. Update on the pathophysiology and treatment of primary Sjögren syndrome. Nat. Rev. Rheumatol. 20 (8), 473–491. https://doi.org/10.1038/s41584-024-01135-3 (2024).
Yang, Y. Q. et al. Iguratimod suppresses plasma cell differentiation and ameliorates experimental Sjögren’s syndrome in mice by promoting TEC kinase degradation. Acta Pharmacol. Sin. 45 (9), 1926–1936. https://doi.org/10.1038/s41401-024-01288-7 (2024).
Both, T., Dalm, V. A. S. H., van Hagen, P. M. & van Daele, P. L. A. Reviewing primary Sjögren’s syndrome: beyond the dryness - from pathophysiology to diagnosis and treatment. Int. J. Med. Sci. 14 (3), 191–200. https://doi.org/10.7150/ijms.17718 (2017).
Li, M. et al. Role of JAK-STAT signaling pathway in pathogenesis and treatment of primary Sjögren’s syndrome. Chin. Med. J. 136 (19), 2297–2306. https://doi.org/10.1097/CM9.0000000000002539 (2023).
Shang, S., Li, M., Jiang, H. & Huang, C. The protective effects of Qihuang Jianpi Zishen Decoction on Mrl/lpr mice and its mechanism. Pak J. Pharm. Sci. 35 (6), 1627–1635 (2022).
Ge, Y., Huang, C., Cheng, Y. & Xu, C. Clinical observation of Qihuang Jianpi Zishen granules as adjuvant treatment for spleen-kidney deficiency type Ii lupus nephritis. Shanxi J. Tradit Chin. Med. 38 (4), 22–25. https://doi.org/10.20002/j.issn.1000-7156.2022.04.008 (2022).
Li, Y. et al. Clincial efficacy of Qihuang Jianpi Zishen granules in treating cardiac involvement in systemic lupus erythematosus. Chin. J. Exp. Tradit Med. Form. 29 (22), 104–111. https://doi.org/10.13422/j.cnki.syfjx.20240891 (2023).
Cheng, Y., Huang, C., Qian, A., Zhu, Y. & Pang, L. Observation of the clinical efficacy of Qihuang Jianpi Zishen granule in the treatment of rheumatoid arthritis. Lishizhen Med. Mater. Med. Res. 35 (6), 1411–1414. https://doi.org/10.3969/j.issn.1008-0805.2024.06.30 (2024).
Tang, Z. et al. Clinical efficacy of Qihuang Jianpi Zishen granules on ankylosing spondylitis with spleen and kidney deficiency type and its effects on serum inflammatory factors. Chin. J. Tradit Chin. Med. Form. Phar. 37 (12), 7535–7539 (2022).
Tang, Z., Huang, C., Cheng, Y. & Li, S. Clinical effect of Qihuang Jianpi Zishen granule on Sjögren’s syndrome with deficiency of both Qi and Yin. J. Anhui Univer Chin. Med. 40 (4), 41–45. https://doi.org/10.3969/j.issn.2095-7246.2021.04.011 (2021).
Zhu, Y., Huang, C., Qian, A. & Cheng, Y. Clinical observation of Qi Huang Jian Pi Zi Shen granules in the treatment of patients with spleen-kidney deficiency type Sjögren’s syndrome. Lishizhen Med. Mater. Med. Res. 35 (13), 3005–3008. https://doi.org/10.3969/j.issn.1008-0805.2024.13.15 (2024).
Verstappen, G. M., Pringle, S., Bootsma, H. & Kroese, F. G. M. Epithelial-immune cell interplay in primary Sjögren syndrome salivary gland pathogenesis. Nat. Rev. Rheumatol. 17 (6), 333–348. https://doi.org/10.1038/s41584-021-00605-2 (2021).
Hall, J. C. et al. Molecular subsetting of interferon pathways in Sjögren’s syndrome. Arthritis Rheumatol. 67 (9), 2437–2446. https://doi.org/10.1002/art.39204 (2015).
Schoenborn, J. R. & Wilson, C. B. Regulation of interferon-γ during innate and adaptive immune responses. Adv. Immunol. 96, 41–101. https://doi.org/10.1016/S0065-2776(07)96002-2 (2007).
Ríos-Ríos, W. D. J., Sosa-Luis, S. A. & Torres-Aguilar, H. T cells subsets in the immunopathology and treatment of Sjogren’s syndrome. Biomolecules 10 (11), 1539. https://doi.org/10.3390/biom10111539 (2020).
Schroder, K., Hertzog, P. J., Ravasi, T. & Hume, D. A. Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75 (2), 163–189. https://doi.org/10.1189/jlb.0603252 (2004).
Xiao, L., Xiao, W. & Zhan, F. Targets of total glucosides of Paeony in the treatment of Sjogren syndrome: A network Pharmacology study. J. Chin. Med. Assoc. 86 (4), 375–380. https://doi.org/10.1097/JCMA.0000000000000884 (2023).
Cui, Y. Y. et al. Efficacy and safety of total glucosides of Paeony in treating primary Sjögren’s syndrome: A propensity-matched study. Eur. Rev. Med. Pharmacol. Sci. 28 (10), 3523–3531. https://doi.org/10.26355/eurrev_202405_36287 (2024).
Jiang, T. et al. Total glucosides of Paeony alleviates experimental Sjögren’s syndrome through inhibiting NLRP3 inflammasome activation of submandibular gland cells. Clin. Exp. Rheumatol. 41 (12), 2502–2510. https://doi.org/10.55563/clinexprheumatol/7kbuok (2023).
Tang, Y., Li, M., Wang, J., Pan, Y. & Wu, F. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 127, 67–72. https://doi.org/10.1016/j.biosystems.2014.11.005 (2015).
Barabási, A. L., Gulbahce, N. & Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 12 (1), 56–68. https://doi.org/10.1038/nrg2918 (2011).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1). https://doi.org/10.1093/nar/gkae909 (2025). D672-D677.
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Wang, W. et al. Network pharmacology, molecular docking, and in vitro experimental verification of the mechanism of Guanxining in treating diabetic atherosclerosis. J. Ethnopharmacol. 324, 117792. https://doi.org/10.1016/j.jep.2024.117792 (2024).
Zhang, Q., Yang, X. R. & Deng, Y. Iguratimod alleviates experimental Sjögren’s syndrome by inhibiting NLRP3 inflammasome activation. Cell. Biochem. Biophys. 82 (3), 2275–2283. https://doi.org/10.1007/s12013-024-01337-2 (2024).
Bulosan, M. et al. Inflammatory caspases are critical for enhanced cell death in the target tissue of Sjögren’s syndrome prior to disease onset. Immunol. Cell. Biol. 87 (1), 81–90. https://doi.org/10.1038/icb.2008.70 (2009).
Niu, R. J. Study on the suppressive of Ginsenoside Rh1 on the inflammation and apoptosis of human salivary gland cells induced by interferon-γ. Jilin University, Jilin, China. (2022). https://link.cnki.net/doi/10.27162/d.cnki.gjlin.2022.005677
Turner, N. A. et al. Mechanism of TNFalpha-induced IL-1alpha, IL-1beta and IL-6 expression in human cardiac fibroblasts: effects of Statins and Thiazolidinediones. Cardiovasc. Res. 76 (1), 81–90. https://doi.org/10.1016/j.cardiores.2007.06.003 (2007).
Xu, J. et al. Lactate-induced MtDNA accumulation activates cGAS-STING signaling and the inflammatory response in Sjögren’s syndrome. Int. J. Med. Sci. 20 (10), 1256–1271. https://doi.org/10.7150/ijms.83801 (2023).
Tian, Y., Yang, H., Liu, N., Li, Y. & Chen, J. Advances in pathogenesis of sjögren’s syndrome. J. Immunol. Res. 2021 (5928232). https://doi.org/10.1155/2021/5928232 (2021).
Thorlacius, G. E., Björk, A. & Wahren-Herlenius, M. Genetics and epigenetics of primary Sjögren syndrome: implications for future therapies. Nat. Rev. Rheumatol. 19 (5), 288–306. https://doi.org/10.1038/s41584-023-00932-6 (2023).
Wang, F. et al. Astragalus polysaccharides improved the cardiac function in Sjögren’s syndrome model rats based on Keap 1-Nrf2/ARE signaling pathway: a mechanism exploration. Chin. J. Inter Tradit West. Med. 34 (5), 566–574 (2014).
Liang, Y. et al. H(2)O(2)-challenged KGN human granulosa cell line. BMC Complement. Med. Ther. 24 (1), 189. https://doi.org/10.1186/s12906-024-04491-5 (2024). Wine- and stir-frying processing of Cuscutae Semen enhance its ability to alleviate oxidative stress and apoptosis via the Keap 1-Nrf2/HO-1 and PI3K/AKT pathways.
Lu, W. W. et al. The effect of total glucoside of Paeony on gut microbiota in NOD mice with Sjögren’s syndrome based on high-throughput sequencing of 16SrRNA gene. Chin. Med. 15 (1), 61. https://doi.org/10.1186/s13020-020-00342-w (2020).
Chang, L. et al. Quercetin ameliorates salivary gland apoptosis and inflammation in primary Sjögren’s syndrome through regulation of the leptin/OB-R signaling. Drug Dev. Res. 83 (6), 1351–1361. https://doi.org/10.1002/ddr.21964 (2022).
Wu, F. et al. Exploring the target and mechanism of radix paeoniae Alba on Sjogren’s syndrome. Comb. Chem. High. Throughput Screen. 26 (6), 1224–1232. https://doi.org/10.2174/1386207325666220823144054 (2023).
Appel, S. et al. Potential association of muscarinic receptor 3 gene variants with primary Sjogren’s syndrome. Ann. Rheum. Dis. 70 (7), 1327–1329. https://doi.org/10.1136/ard.2010.138966 (2011).
Xu, H. T. et al. Formononetin attenuates psoriasiform inflammation by regulating interferon signaling pathway. Phytomedicine 128, 155412. https://doi.org/10.1016/j.phymed.2024.155412 (2024).
Sugimoto, M. et al. Formononetin attenuates H2O2-induced cell death through decreasing ROS level by PI3K/akt-Nrf2-activated antioxidant gene expression and suppressing MAPK-regulated apoptosis in neuronal SH-SY5Y cells. Neurotoxicology 85, 186–200. https://doi.org/10.1016/j.neuro.2021.05.014 (2021).
He, X. et al. Study of Kaempferol in the treatment of rheumatoid arthritis through modulation of the NLRP3/CASP1/GSDMD axis and T-cell activation: based on network pharmacology, single-cell analysis, and experimental validation. Int. Immunopharmacol. 143 (Pt 1), 113357. https://doi.org/10.1016/j.intimp.2024.113357 (2024).
Chen, C., Liang, Y., Zhang, Z., Zhang, Z. & Yang, Z. Relationships between increased Circulating YKL-40, IL-6 and TNF-α levels and phenotypes and disease activity of primary Sjögren’s syndrome. Int. Immunopharmacol. 88, 106878. https://doi.org/10.1016/j.intimp.2020.106878 (2020).
Benchabane, S. et al. Association between increased bcl-2, Fas and FasL levels and inflammation extent in labial salivary glands during primary Sjögren’s syndrome. Endocr. Metab. Immune Disord Drug Targets. 22 (3), 328–338. https://doi.org/10.2174/1871530321666210809155147 (2022).
Kang, E. H., Lee, Y. J., Hyon, J. Y., Yun, P. Y. & Song, Y. W. Salivary cytokine profiles in primary Sjögren’s syndrome differ from those in non-sjögren Sicca in terms of TNF-α levels and th-1/th-2 ratios. Clin. Exp. Rheumatol. 29 (6), 970–976 (2011).
Xu, X. et al. Decoding the mechanism of CheReCunJin formula in treating sjögren’s syndrome based on network pharmacology and molecular docking. Evid. Based Complement. Alternat. Med. 1193846. (2022). https://doi.org/10.1155/2022/1193846 (2022).
Manganelli, P. & Fietta, P. Apoptosis and Sjögren syndrome. Semin Arthritis Rheum. 33 (1), 49–65. https://doi.org/10.1053/sarh.2003.50019 (2003).
Wang, J. et al. MiR-let-7d-3p regulates IL-17 expression through targeting AKT1/mTOR signaling in CD4 + T cells. Vitro Cell. Dev. Biol. Anim. 56 (1), 67–74. https://doi.org/10.1007/s11626-019-00409-5 (2020).
Cui, J. et al. Novel immune-related genetic expression for primary Sjögren’s syndrome. Front. Med. 8, 719958. https://doi.org/10.3389/fmed.2021.719958 (2022).
Varfolomeev, E. & Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 101, 26–32. https://doi.org/10.1016/j.cyto.2016.08.035 (2018).
Mori, T. et al. IL-1β and TNFα-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int. Immunol. 23 (11), 701–712. https://doi.org/10.1093/intimm/dxr077 (2011).
Otis, J. S. et al. Pro-inflammatory mediation of myoblast proliferation. PloS One. 9 (3), e92363. https://doi.org/10.1371/journal.pone.0092363 (2014).
Luo, G., Hershko, D. D., Robb, B. W., Wray, C. J. & Hasselgren, P. O. IL-1beta stimulates IL-6 production in cultured skeletal muscle cells through activation of MAP kinase signaling pathway and NF-kappa B. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284 (5), R1249–1254. https://doi.org/10.1152/ajpregu.00490.2002 (2003).
Ji, L. et al. Identification of bioactive compounds and potential mechanisms of scutellariae radix-coptidis rhizoma in the treatment of atherosclerosis by integrating network Pharmacology and experimental validation. Biomed. Pharmacother. 165, 115210. https://doi.org/10.1016/j.biopha.2023.115210 (2023).
Funding
This study was supported by the Clinical Medical Research Transformation Project of Anhui Province (202304295107020095, 202304295107020088), the Bozhou Key Research and Development Project (bzzc2022015), and the Natural Science Research Project of Anhui Educational Committee (2022 AH051494).
Author information
Authors and Affiliations
Contributions
HW: Writing – original draft & Writing – review & editing, Project administration, Visualization, Validation, Software, Supervision, Resources, Methodology, Formal analysis, Data curation. ZM: Writing – original draft & Writing – review & editing, Visualization, Validation, Software, Resources, Methodology, Formal analysis, Data curation. NW: Writing – review & editing, Validation, Methodology, Formal analysis, Data curation. XW: Writing – review & editing, Project administration, Validation, Software, Supervision, Resources, Methodology. XY: review & editing, Validation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Ma, Z., Wu, N. et al. Integrated network pharmacology, transcriptomics, and experimental validation to explore the mechanism of Qihuang Jianpi Zishen granules against Sjögren’s syndrome. Sci Rep 15, 21176 (2025). https://doi.org/10.1038/s41598-025-03653-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03653-w
