
Abstract
Bladder cancer is the fourth most common malignant tumor in men, with limited therapeutic biomarkers and heterogeneous responses to immunotherapy. Disulfide bond-driven cell death has emerged as a critical regulator of tumor progression and immune microenvironment remodeling. By integrating data from TCGA and GEO cohorts, we developed a Disulfide-Related Prognostic Signature (DRPS) using ten machine learning algorithms. Single-cell RNA sequencing (scRNA-seq) elucidated the cell subtype-specific expression patterns of disulfide bond regulatory genes, while immune microenvironment and drug sensitivity analyses validated its clinical translational potential. qRT-PCR experiments confirmed differential expression patterns of core genes in bladder cancer cell lines. The DRPS model, optimized by the StepCox[backward] algorithm, demonstrated robust prognostic accuracy across four validation cohorts (mean C-index = 0.658). High-risk patients exhibited an enhanced immunosuppressive microenvironment characterized by infiltrated activated cancer-associated fibroblasts, upregulated APC co-inhibition pathways, and elevated immune checkpoint expression. Notably, high DRPS scores were associated with primary resistance to immunotherapy but showed increased sensitivity to anti-tumor agents such as Elephantine and Leflunomide. This study establishes a novel DRPS that serves as a predictive indicator for bladder cancer prognosis and pan-cancer immunotherapy efficacy.
Similar content being viewed by others


Introduction
Bladder cancer (BLCA) stands as one of the most prevalent malignancies of the urinary system globally, characterized by significant clinical heterogeneity that spans a broad spectrum from non-muscle-invasive (NMIBC) to muscle-invasive (MIBC) lesions1. Despite advancements in multimodal therapies, including surgery, chemotherapy, radiotherapy, and immunotherapy, which have improved survival outcomes for some patients, those with advanced disease continue to face high recurrence rates, drug resistance, and poor prognosis2. Current clinical indicators, such as tumor stage and grade, offer limited predictive power for individualized prognosis, and there is a lack of effective biomarkers to guide treatment selection. Recent molecular subtyping studies have highlighted the critical role of tumor microenvironment heterogeneity and immune evasion mechanisms in BLCA progression. However, translating these molecular insights into actionable clinical targets remains a significant challenge3,4.
Disulfide bonds, crucial for protein structural and functional stability, play a pivotal role in cellular survival, oxidative stress, and metabolic regulation. The recently proposed “disulfide death” mechanism reveals that abnormal disulfide bond accumulation can induce programmed cell death by disrupting the cytoskeletal network, offering a novel direction for cancer therapy5. In BLCA, the overexpression of disulfide bond regulatory genes such as SLC7A11 is closely associated with metabolic reprogramming, chemotherapy resistance, and the formation of an immunosuppressive microenvironment6,7,8. For instance, SLC7A11 overexpression enhances tumor cell sensitivity to glucose deprivation, and glucose transporter inhibitors (e.g., BAY-876) selectively induce cell death in BLCA cells with high SLC7A11 expression9,10. Additionally, long non-coding RNAs (e.g., MIR4435-2HG, ARHGAP5-AS1) modulate disulfide bond-related pathways, influencing tumor progression and suggesting that disulfide bond homeostasis imbalance represents a potential therapeutic target in BLCA11,12,13.
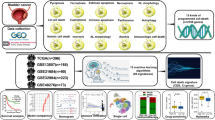
This study aims to integrate single-cell transcriptomic sequencing with multiple machine learning algorithms to systematically dissect the heterogeneous expression patterns of disulfide bond-related genes in BLCA and construct a prognostic prediction model. By leveraging single-cell resolution to uncover cell subtype-specific expression patterns of disulfide bond regulatory genes within the tumor microenvironment, combined with machine learning to identify key driver genes, this approach overcomes the limitations of traditional bulk sequencing, enabling the precise identification of molecular markers closely associated with clinical outcomes. Finally, quantitative real-time PCR (qRT-PCR) experiments were employed to validate the expression of key genes (Fig. 1). The findings will deepen our understanding of the role of disulfide bond-related genes in BLCA, provide a theoretical foundation for developing targeted therapies based on metabolic reprogramming, and offer novel tools for individualized prognosis assessment and immunotherapy response prediction, ultimately optimizing clinical decision-making.

Workflow of our study.
Methods and materials
Data acquisition
RNA-seq data for BLCA were extracted from The Cancer Genome Atlas (TCGA) (n = 412). Pathologic subtypes included 409 cases of uroepithelial carcinoma (99.3%) and 3 cases of squamous cell carcinoma (0.7%). According to the American Joint Committee on Cancer (AJCC) TNM staging system, cases were categorized as NMIBC (stage Ta/Tis/T1, n = 4) and MIBC (stage T2-T4, n = 392). After quality control, 396 cases with complete prognostic information were retained for analysis. To validate our findings, we also retrieved data from the GEO database (https://www.ncbi.nlm.nih.gov/geo/), including GSE13507 (n = 165), GSE31684 (n = 90), and GSE32894 (n = 223). Detailed information on each dataset is provided in Supplementary Table 2. Inclusion criteria for TCGA and GEO data include: (1) pathologic diagnosis of bladder cancer as migratory cell carcinoma, adenocarcinoma, and squamous cell carcinoma; (2) complete prognostic information. Exclusion criteria included: (1) postoperative follow-up time of less than one month or death within one month after surgery; (2) presence of other malignancies. Differential gene expression analysis was performed on the integrated dataset using the R package “limma” to identify differentially expressed genes (DEGs) meeting the criteria of |Log2FC| > 1.5 and p-value < 0.05. Disulfide bond-related genes were manually curated from the Genecards (https://www.genecards.org/) (Supplementary Table 1). Venn diagrams were used to identify disulfide bond-related DEGs.
Integrative machine learning algorithms constructed an optimal DRPS
Univariate Cox analysis was applied to identify potential predictive biomarkers (p-value < 0.05) among the DEGs in BLCA. Based on these potential prognostic biomarkers, an integrative machine learning analysis was conducted to screen for the Disulfide-Related Prognostic Signature (DRPS). Ten machine learning algorithms were employed, including Random Survival Forest (RSF), Elastic Net (Enet), Lasso, Ridge, StepCox, CoxBoost, Cox Partial Least Squares Regression (plsRcox), Supervised Principal Component (SuperPC), Generalized Boosted Regression Model (GBM), and Survival Support Vector Machine (survival-SVM). The DRPS prediction process was as follows: (1) In the TCGA dataset, 101 algorithm combinations were fitted to the prediction model based on a leave-one-out cross-validation framework; (2) All models were tested in three GEO cohorts; (3) For each model, Harrell’s concordance index (C-index) was calculated across all TCGA and GEO datasets, and the model with the highest average C-index was selected as the optimal one.
The optimal cutoff point was determined using the “surv_cutpoint” function in the R package “survminer.” BLCA patients were then stratified into high- and low-risk groups based on their risk scores. Kaplan-Meier survival curves were plotted, and ROC and C-index curves were generated using the “survivalROC” R package. Univariate and multivariate Cox analyses were performed to identify prognostic risk factors in BLCA patients. A dynamic nomogram for predicting BLCA patient survival was constructed using the “nomograms” R package. The predictive performance of the nomogram was evaluated using calibration curves, ROC curves, and the concordance index (C-index).
Immune infiltration analysis and enrichment analysis for DRPS
The R package “immunedeconv” was used to assess immune cell abundance in BLCA patients. “Immunedeconv” integrates seven state-of-the-art algorithms (CIBERSORT, MCP-counter, QUANTISEQ, XCELL, CIBERSORT-ABS, TIMER, and EPIC) to evaluate immune cell infiltration. The relative expression of immune checkpoints and genes in different BLCA case groups was visualized using the R packages “ggplot” or “ggpubr.” Gene Set Enrichment Analysis (GSEA) was performed using the “clusterProfiler” package to uncover potential biological functions and mechanisms associated with high- and low-risk groups.
Immunotherapy response prediction and drug sensitivity analysis
Multiple metrics, such as tumor immune dysfunction and exclusion (TIDE) scores and immune escape scores, were used to evaluate the ability of DRPS to predict immunotherapy response in BLCA patients. Drug sensitivity analysis was performed using the “oncoPredict” R package based on data from the Genomics of Drug Sensitivity in Cancer (GDSC) database to determine the IC50 values of each drug in BLCA cases. Spearman’s correlation analysis was applied to examine the relationship between drug IC50 values and risk scores to identify potential therapeutic agents.
Single-cell analysis
Single-cell RNA sequencing (scRNA-seq) data from the GSE130001 dataset were processed using the “Seurat” R package. Cells were filtered based on gene counts and mitochondrial gene percentage, removing those with too few genes or excessively high mitochondrial gene content. The quality control process consists of excluding cells with fewer than 50 detected genes (min.features = 50, adjusted for potential RNA degradation in cancer specimens as per 10x Genomics technical guidelines14) or fewer than 3 cells expressing genes (min.cells = 3) to remove low quality initial cells. Cells with more than 5% mitochondrial gene expression (percent.mt < 5) were excluded using the subset function. After stringent quality control, 21,342 cells were retained for downstream analysis. Data normalization was performed using the “NormalizeData” function. The “FindVariableFeatures” function was used to identify the top 1500 highly variable genes. Principal Component Analysis (PCA) was conducted on the normalized data using the “RunPCA” function. Batch correction across different samples was performed using the “Harmony” function, and dimensionality reduction and cluster identification were achieved using Uniform Manifold Approximation and Projection (UMAP). The “SingleR” package was used to annotate different cell clusters, and the expression levels of DRPS in different cell groups were analyzed and visualized. Cell clusters were annotated using the Single and HumanPrimaryCellAtlasData reference databases, and marker identification required a minimum gene detection rate of 25% (min.pct = 0.25).
Cell lines and cell culture
The normal bladder cell line SV-HUC-1 and the BLCA cell line T24 were purchased from the Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China), with cell line authenticity confirmed by short tandem repeat (STR) profiling and mycoplasma contamination testing (performed quarterly using the MycoAlert PLUS Mycoplasma Detection Kit, Lonza). SV-HUC-1 cells were cultured in Ham’s F-12 K medium (HyClone, China) supplemented with 10% fetal bovine serum (Gibco, Australia), 1% penicillin/streptomycin (Beyotime, China), and 0.5% Plasmocin Prophylactic (InvivoGen) to prevent mycoplasma contamination. T24 cells were maintained in RPMI 1640 medium (HyClone, China) containing 10% fetal bovine serum, 1% penicillin/streptomycin, and 0.5% Plasmocin Prophylactic. All cell lines were propagated under standard conditions (37 °C, 5% CO₂) and routinely monitored for morphological consistency and growth rate stability.
Verification of key gene expression
Total RNA was extracted from cells using TRIzol (Takara Bio, Dalian, China) and reverse-transcribed into cDNA using oligo(dT) primers. RT-qPCR was performed using SYBR Premix Ex Taq (Takara Bio) on an ABI 7900HT detection system (Thermo Fisher Scientific Inc.). Relative expression levels were calculated using the 2-ΔΔCT method. Primer sequences are listed in Supplementary Table 3.
Statistical analyses
Differences between continuous variables were evaluated using the Wilcoxon rank-sum test or t-test. Pearson’s or Spearman’s correlation analysis was used to assess the relationship between two continuous variables. Differences in Kaplan-Meier survival curves were tested using the two-sided log-rank test. Statistical analysis and graphical visualization were performed using R (version 4.4.2) and GraphPad Prism 10.1 software. A p-value < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001. ns: not significant.
Results
Identification of prognostic genes associated with disulfide in BLCA
A total of 407 DEGs were identified from TCGA-BLCA, filtered by |Log2FC| ≥ 1.5 and p-value < 0.05 (Supplementary Fig. 1A). Subsequently, an intersection with the disulfide bond-related gene set yielded 59 disulfide-related DEGs (Supplementary Fig. 1B). Further univariate Cox analysis identified 22 prognostic genes.
Synthesized multiple machine learning algorithms developed a prognostic model
The 22 potential prognostic biomarkers were subjected to an integrative machine learning program incorporating 10 algorithms. This process generated 89 prognostic models, with the C-index of each model in the training and testing cohorts shown in Fig. 2A. The model developed by StepCox[forward] was selected as the optimal prognostic signature, achieving the highest average C-index of 0.658. Eight core genes were ultimately selected as disulfide-related signature biomarkers. The risk score was calculated as follows: Risk Score = 0.224 × CALR exp + 0.320 × HLA-G exp + 0.012 × HSPA5 exp + 0.049 × THBS1 exp + 0.157 × TFRC exp + 0.155 × LTBP1 exp + 0.166 × IGF1 exp + 0.088 × NPR3 exp. Using the optimal cutoff value, BLCA cases were stratified into high- and low-risk groups. As shown in Figs. 2B-E, high-risk BLCA patients consistently exhibited worse overall survival across multiple datasets. The AUCs for 1-, 3-, and 5-year survival were 0.721, 0.710, and 0.717 in the TCGA cohort; 0.743, 0.689, and 0.672 in the GSE13507 cohort; 0.754, 0.701, and 0.637 in the GSE32894 cohort; and 0.661, 0.641, and 0.673 in the GSE31684 cohort.

Development of a prognostic DRPS by integrative machine learning procedure. (A) The C-index of prognostic models of TCGA and GEO cohorts. The survival curve of different DRPS score groups and their corresponding ROC curve in TCGA (B), GSE13507 (C), GSE32984 (D), and GSE31684 (E) cohorts.
Clinically relevant performance characterization of prognostic signatures
The C-index and clinical characteristics of the DRPS were evaluated to assess their performance in predicting clinical outcomes in BLCA cases. As shown in Fig. 3A-B, univariate and multivariate Cox regression analyses revealed that risk score was an independent risk factor for clinical outcome in BLCA cases, which outweighed the importance of traditional clinical factors including age and pathological stage (HR = 2.335, 95% CI 1.884–2.895, p < 0.001; HR = 2.060, 95% CI 1.660–2.556, p < 0.001). In the TCGA, GSE13507, GSE32894, and GSE31684 datasets, the AUC and C-index for predicting clinical outcomes were higher than those for age, sex, tumor grade, and clinical stage (Figs. 3C–D). A nomogram was constructed to predict the clinical fate of BLCA patients based on risk score and stage (Fig. 3E). Calibration plots demonstrated good agreement between nomogram predictions and actual observations for 1-, 3-, and 5-year survival in the TCGA cohort (C-index = 0.696, 95% CI = 0.656–0.735, Fig. 3F). These data indicate that DRPS can accurately and consistently predict the clinical outcomes of BLCA cases.

Evaluation of the performance of DRPS. (A–B) Univariate and multivariate Cox regression suggested DRPS as a risk factor for the prognosis of bladder cancer patients. (C–D) The ROC and C-index compare the performance of DRPS, age, gender, tumor grade, and clinical stage in predicting the prognosis of bladder cancer patients in TCGA cohorts. (E) Nomogram developed based on DRPS, age, gender, tumor grade, and clinical stage. (F) Calibration evaluated the role of nomogram in predicting the clinical outcome of bladder cancer patients in TCGA cohorts.
Analysis of the relationship between DRPS and tumor immune microenvironment
Using multiple immune analysis algorithms, we investigated the association between disulfide-related genes and immune cell infiltration. Our findings revealed positive correlations between disulfide-related genes and most immune cells, particularly cancer-associated fibroblasts, M1 and M2 macrophages, and activated myeloid dendritic cells (Figs. 4A-B). Additionally, the high-risk group exhibited higher levels of APC co-inhibition, APC co-stimulation, T cell co-inhibition, T cell co-stimulation, macrophages, neutrophils, and Treg cells compared to the low-risk group (Fig. 4C). The tumor immune landscape was classified into wound healing (C1), IFN-g dominant (C2), inflammatory (C3), and lymphocyte-depleted (C4) subtypes. High-risk BLCA patients had a higher proportion of C1 and C2 subtypes (Fig. 3D, p = 0.001). Using the ESTIMATE algorithm, we found that the high-risk group had higher ESTIMATE and immune scores than the low-risk group (Fig. 5A).

The tumor immune-microenvironment landscape of bladder cancer with different DRPS scores. (A) The relationship between DRPS and immune infiltration in BLCA is based on seven state-of-the-art algorithms. (B) DRPS score was positively correlated with the abundance of Cancer-associated fibroblast, Macrophage M1, M2 and Myeloid dendritic cell activated. (C) ssGSEA analysis revealed immune-related functions of different DRPS rating groups. (D) The immune landscape in different DRPS score groups.

DRPS score acted as an indicator for predicting the benefits of immunotherapy in bladder cancer. (A) TME scores between different risk groups. (B) The level of immune checkpoints in bladder cancer with different DRPS scores. (C–E) The TIDE score, and TMB score in bladder cancer with different CDS scores. (F–G) Waterfall maps of high DRPS score groups (F) and low DRPS score groups (G). (H–I) GSEA enrichment analysis in the high-risk group (H) and the low-risk group (I).
Drug treatment response prediction and drug sensitivity analysis
The correlation between immune checkpoint expression and risk groups suggested a high likelihood of benefiting from immune checkpoint blockade (ICB) therapy. The high-risk group exhibited higher expression levels of immune checkpoints, including CD28, CD48, and CD80 (Fig. 5B, all p < 0.05). High TIDE scores indicated a greater likelihood of immune escape and poorer response to immunotherapy. As shown in Fig. 5C, high-risk BLCA patients had higher TIDE scores (p < 0.05). As shown in Fig. 5D–E, there was no significant difference in tumor mutational burden (TMB) scores between the high and low risk groups. Whole-exome sequencing analysis of 195 tumor samples revealed that 93.3% (181/194) carried at least one driver gene mutation, with TP53, TTN, and KMT2D being the most frequently mutated. Waterfall plots demonstrated high TMB, with a median TMB of 94 mutations/Mb (Figs. 5F–G). High TMB populations may benefit from immune checkpoint inhibitors. Considering the critical role of targeted and immunotherapy in BLCA treatment, we investigated IC50 values. High-risk BLCA patients exhibited lower IC50 values (p < 0.05), indicating greater sensitivity to anti-tumor drugs (Fig. 6). Of these, the clinical first-line agents cisplatin and gemcitabine were more responsive in the high-risk group.

The IC50 value of drugs in bladder cancer patients with high DRPS score.
The underlying biological function of risk groups
Functional enrichment analysis was performed to explore the reasons for differences in clinical outcomes, immune infiltration, and treatment responses between risk groups. High-risk BLCA patients were primarily enriched in focal adhesion, ECM-receptor interaction, and systemic lupus erythematosus pathways (Fig. 5H). In contrast, the low-risk group showed enrichment in pathways related to pentose and glucuronate interconversions, ribosome, and starch and sucrose metabolism (Fig. 5I). This analysis indicated that the high-risk group constructed an immunosuppressive microenvironment through extracellular matrix remodeling and chronic inflammatory stimulation, which was characterized by activation of the adhesion plaque pathway to promote tumor metastasis, aberrant interactions of ECM receptors to form a physical barrier, and persistent inflammatory responses triggered by aberrant expression of autoimmune-related genes. The low-risk group, on the other hand, maintains metabolic homeostasis through pentose metabolic reprogramming and ribosome biosynthesis, preserving immune recognition.
Single-cell analysis for core biomarkers in BLCA
Single-cell analysis was performed using the GSE130001 dataset to characterize signature genes comprehensively. Data were clustered using the Seurat R package, and UMAP visualization identified three distinct cell types: endothelial cells, epithelial cells, and fibroblasts (Fig. 7A). Dot plots illustrated the enrichment of core genes across these cell types (Fig. 7B). CALR and HSPA5 were significantly expressed in all three cell types (Fig. 7C).

Single-cell RNA sequencing in GSE130001. (A) Cluster analysis identified 3 distinct cell clusters, which were visualized and annotated using t-distribution random neighbor embedding (t-SNE). (B) Bubble plot displaying the expression patterns of biomarkers across cell types. (C) The t-SNE plot illustrates the expression trends of biomarkers across cell types.
Validation of core gene expression in BLCA
The mRNA expression levels of the eight core genes were further validated by qRT-PCR (Figs. 8A–H). Compared to controls, CALR, HSPA5, HLA-G, and TFRC were significantly upregulated in the BLCA group, while LTBP1 and IGF1 were downregulated. The remaining genes showed differences in mRNA expression but were not statistically significant.

(A–H) mRNA expression levels of core genes were detected in different cell lines.
Discussion
BLCA is a highly heterogeneous malignancy with complex clinical features, and its prognosis varies significantly due to differences in tumor stage, molecular subtypes, and microenvironment characteristics. Although existing clinical indicators such as TNM staging provide a foundational basis for prognosis assessment, they remain insufficient to precisely explain the heterogeneity in treatment responses and survival outcomes among patients, highlighting the urgent need to explore novel molecular biomarkers. Recently, disulfide death triggered by disulfide bond homeostasis imbalance has been shown to play a critical role in tumor progression and immune microenvironment remodeling. This mechanism, involving abnormal crosslinking of cytoskeletal proteins leading to cell death, may represent a key factor influencing the malignant biological behavior of BLCA15. Studies have demonstrated that disulfide bond-related genes not only participate in tumor cell proliferation and invasion but also modulate immune checkpoint expression and immune cell infiltration, thereby shaping the immunosuppressive tumor microenvironment16. Therefore, systematically dissecting the molecular characteristics of disulfide bond-related genes in BLCA and their interactions with the immune microenvironment will provide new theoretical insights for optimizing prognosis assessment and developing personalized treatment strategies.
In this study, we constructed an integrative machine learning program incorporating ten algorithms to develop a DRPS for bladder cancer. The DRPS demonstrated robust and stable performance in predicting overall survival, with 1-, 3-, and 5-year AUC values of 0.721, 0.710, and 0.717, respectively, in the TCGA cohort. Furthermore, the DRPS risk score served as an independent prognostic factor for overall survival in the TCGA, GSE13507, GSE31684, and GSE32894 cohorts. Further analysis revealed that BLCA patients with lower DRPS risk scores exhibited higher immune-activated cell infiltration, higher C1 and C2 immune phenotypes, higher TMB scores, and lower TIDE and immune escape scores, suggesting that these patients may derive greater benefit from immunotherapy.
Based on existing evidence, the eight core genes in the DRPS may contribute to bladder cancer pathogenesis through multidimensional mechanisms. CALR, an endoplasmic reticulum chaperone, is associated with BLCA prognosis. Low CALR expression may mediate immune escape via CD47, while chemotherapy-induced CALR exposure can activate dendritic cell-mediated anti-tumor immunity17,18. HLA-G, a non-classical MHC-I molecule, though its role in BLCA remains unclear, is known to inhibit NK/T cell function, contributing to immune escape in other cancers19. HSPA5, upregulated in BLCA, promotes an immunosuppressive microenvironment by regulating PD-L1 expression and endoplasmic reticulum stress and is associated with ferroptosis resistance20,21. THBS1 may bidirectionally regulate angiogenesis and the immune microenvironment through the TGF-β signaling pathway, though its specific role in BLCA requires further validation22,23. TFRC, a hub gene in iron metabolism, promotes epithelial-mesenchymal transition via the circTFRC/miR-107 axis, and its overexpression is associated with a hypoxic microenvironment and reduced CD8 + T cell infiltration in BLCA22. LTBP1 regulates TGF-β activation and participates in tumor stroma remodeling, potentially influencing BLCA fibrosis24. The IGF1/IGF-1R axis drives BLCA cell proliferation by activating the MAPK/PI3K pathway, and its high expression correlates with advanced clinical stages and recurrence risk25. NPR3 may modulate oxidative stress responses, maintain intracellular redox balance, and regulate cell growth, differentiation, and apoptosis26. Together, these genes form a network involving immunogenic cell death regulation, metabolic reprogramming, growth factor signaling, and microenvironment remodeling, collectively shaping the immunosuppressive and malignant progression characteristics of BLCA.
Functional enrichment analysis was performed to elucidate why BLCA patients in different DRPS risk groups exhibit significant differences in clinical outcomes, immune infiltration, and immunotherapy responses. The results showed that the high-risk group was significantly enriched in ECM-receptor interaction and focal adhesion pathways. Aberrant activation of ECM receptors can promote epithelial-mesenchymal transition, enhancing tumor cell motility and invasiveness27. Dysregulation of focal adhesion-related genes (e.g., VEGF) may support metastasis by promoting angiogenesis and anti-apoptotic effects28. Thus, the enrichment of high-risk DRPS is closely associated with the invasive and metastatic potential of BLCA. Notably, the positive correlation between DRPS score and cisplatin sensitivity suggests that this model may optimize conventional chemotherapeutic drug selection. In contrast, the remarkable sensitivity of experimental drugs such as Elephantine reveals novel therapeutic directions targeting the homeostatic regulatory network of disulfide bonds. Further validation of these findings by organoid drug sensitivity testing is needed in the future.
This study employed machine learning algorithms and single-cell analysis to develop a DRPS for bladder cancer. Additional in vivo and in vitro validation studies will provide a more comprehensive understanding, aiming to improve the performance of DRPS in predicting clinical outcomes in BLCA. However, our study has certain limitations. All analyses were conducted at the RNA level and do not represent protein-level results. Validation using an internal clinical cohort would strengthen our findings. Notably, the TCGA-BLCA cohort predominantly comprised MIBC cases (n = 392) with only 4 NMIBC samples, reflecting the inherent difficulty of obtaining transcriptomic data from early-stage bladder cancer patients due to tissue availability limitations. This severe imbalance in subtype representation may introduce bias in evaluating the model’s ability to stratify prognosis between NMIBC and MIBC subtypes. Future studies with larger NMIBC cohorts are warranted to validate the generalizability of DRPS across disease stages. Furthermore, the underlying mechanisms and pathways of DRPS in BLCA require further exploration.
Conclusion
This study integrated multi-omics data and machine learning algorithms to construct a novel DRPS, which serves as a predictive indicator for bladder cancer prognosis and immunotherapy efficacy. Single-cell resolution analysis revealed that the core genes CALR and HSPA5 exacerbate tumor microenvironment heterogeneity by regulating the interaction networks among endothelial cells, epithelial cells, and fibroblasts. The immunosuppressive phenotype mediated by these genes is characterized by increased infiltration of cancer-associated fibroblasts, activation of APC co-inhibition pathways, and abnormal overexpression of immune checkpoints such as CD28 and CD80. The study found that high-risk patients exhibited significantly elevated TIDE scores and immune escape features, suggesting potential primary resistance to PD-1/PD-L1 inhibitors. In contrast, drug sensitivity analysis indicated that anti-tumor agents such as Elephantin and Leflunomide may offer therapeutic advantages for this subgroup. Experimental validation confirmed the differential expression patterns of core genes in bladder cancer cell lines.
Data availability
The analyzed datasets generated during the study were sourced from the TCGA database (https://portal.gdc.cancer.gov) and the GEO database (https://www.ncbi.nlm.nih.gov/geo).
References
Dyrskjøt, L. et al. Bladder cancer. Nat. Rev. Dis. Primers. 9(1), 58 (2023).
Lopez-Beltran, A. et al. Advances in diagnosis and treatment of bladder cancer. Bmj 384, e076743 (2024).
Warrick, J. I. et al. Intratumoral heterogeneity of bladder cancer by molecular subtypes and histologic variants. Eur. Urol. 75(1), 18–22 (2019).
Tran, L. et al. Advances in bladder cancer biology and therapy. Nat. Rev. Cancer. 21(2), 104–121 (2021).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell. Biol. 25(3), 404–414 (2023).
Jiang, Y. & Sun, M. SLC7A11: The achilles heel of tumor? Front. Immunol. 15, 1438807 (2024).
Wang, J. et al. Inhibition of endoplasmic reticulum stress cooperates with SLC7A11 to promote disulfidptosis and suppress tumor growth upon glucose limitation. Adv. Sci. (Weinh) 12(7), e2408789 (2025).
Zhong, L. et al. Development and validation of a disulfidptosis and disulfide metabolism-related risk index for predicting prognosis in lung adenocarcinoma. Cancer Cell. Int. 24(1), 2 (2024).
Wang, J. et al. Tumor suppressor BAP1 suppresses disulfidptosis through the regulation of SLC7A11 and NADPH levels. Oncogenesis 13(1), 31 (2024).
Kang, Z. et al. Disulfidptosis-related subtype and prognostic signature in prostate cancer. Biol. Direct 19(1), 97 (2024).
Li, B. et al. Analysis of long non-coding RNAs associated with disulfidptosis for prognostic signature and immunotherapy response in uterine corpus endometrial carcinoma. Sci. Rep. 13(1), 22220 (2023).
Yao, H. et al. Establishment of disulfidptosis-related LncRNA signature as biomarkers in colon adenocarcinoma. Cancer Cell. Int. 24 (1), 183 (2024).
Jia, X. et al. Constructed risk prognosis model associated with Disulfidptosis LncRNAs in HCC. Int. J. Mol. Sci. 24(24) (2023).
Danielski, K. Guidance on processing the 10x genomics single cell gene expression assay. Methods Mol. Biol. 2584, 1–28 (2023).
Zhou, Z. et al. Construction and verification of a prognostic model for bladder cancer based on disulfidptosis-related angiogenesis genes. PeerJ 13, e18911 (2025).
Guo, S. et al. Disulfidptosis related immune genes drive prognostic model development and tumor microenvironment characterization in bladder urothelial carcinoma. Sci. Rep. 15 (1), 8130 (2025).
Fucikova, J. et al. Calreticulin and cancer. Cell. Res. 31(1), 5–16 (2021).
Oresta, B. et al. Mitochondrial metabolic reprogramming controls the induction of Immunogenic cell death and efficacy of chemotherapy in bladder cancer. Sci. Transl Med. 13(575) (2021).
Loustau, M. et al. HLA-G Neo-Expression on tumors. Front. Immunol. 11, 1685 (2020).
Li, T. et al. New progresses on cell surface protein HSPA5/BiP/GRP78 in cancers and COVID-19. Front. Immunol. 14, 1166680 (2023).
Qin, J. et al. Expression of transferrin Receptor/TFRC protein in bladder cancer cell T24 and its role in inducing iron death in bladder cancer. Int. J. Biol. Macromol. 274 (Pt 1), 133323 (2024).
Tang, R. et al. TFRC, associated with hypoxia and immune, is a prognostic factor and potential therapeutic target for bladder cancer. Eur. J. Med. Res. 29(1), 112 (2024).
Zhang, X. et al. Upregulation of THBS1 is related to immunity and chemotherapy resistance in gastric cancer. Int. J. Gen. Med. 14, 4945–4957 (2021).
Cai, R. et al. LTBP1 promotes esophageal squamous cell carcinoma progression through epithelial-mesenchymal transition and cancer-associated fibroblasts transformation. J. Transl Med. 18(1), 139 (2020).
Liu, L. & Li, X. A review of IGF1 signaling and IGF1-related long noncoding RNAs in chemoresistance of Cancer. Curr. Cancer Drug Targets 20(5), 325–334 (2020).
Li, S. et al. NPR3, transcriptionally regulated by POU2F1, inhibits osteosarcoma cell growth through blocking the PI3K/AKT pathway. Cell. Signal. 86, 110074 (2021).
Machado Brandão-Costa, R. et al. Extracellular matrix derived from high metastatic human breast cancer triggers epithelial-mesenchymal transition in epithelial breast cancer cells through Αvβ3 integrin. Int. J. Mol. Sci., 21(8) (2020).
Shi, Z. et al. The role of VEGF in Cancer angiogenesis and tumorigenesis: Insights for anti-VEGF therapy. Cytokine 189, 156908 (2025).
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
Contributions
H.H.: Writing—original draft preparation, investigation. S.H.Y.: Conceptualization, and methodology. G.L.L., W.Y.F.: Conceptualization, study design, and supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, H., Shao, H., Wang, Y. et al. Disulfide bond-related gene signature development for bladder cancer prognosis prediction and immune microenvironment characterization. Sci Rep 15, 18826 (2025). https://doi.org/10.1038/s41598-025-03974-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-03974-w
