
Abstract
The association between oxidative stress and osteoporosis (OP) has been substantiated by numerous studies; however, the precise underlying mechanism remains elusive. Hence, we employed bioinformatics methodologies to investigate this phenomenon. OP-related datasets (GSE56815 and GSE7158) were utilized in this study. Key module genes linked to oxidative stress-related genes (OS-RGs) were acquired through weighted gene coexpression network analysis (WGCNA). By crossing key module genes and differentially expressed genes (DEGs) from differential expression analysis, candidate genes were obtained. Subsequently, diagnostic genes were obtained through receiver operating characteristic (ROC) curve analysis and expression evaluation. Furthermore, a nomogram model was developed utilizing these genes to assess the collective predictive capacity of the diagnostic genes for OP comprehensively. Additionally, gene set variation analysis (GSVA), immune analysis, and construction of a molecular regulatory network were implemented to further understand the mechanism of the diagnostic genes in OP. We also preliminarily verified the effect of NAPG on osteogenic differentiation through experiments such as ALP, ARS and Western Blot. A total of 101 candidate genes were identified by crossing 395 DEGs and 1,730 key module genes. Importantly, NAPG, NCOA1, and TRIM44 were identified as diagnostic genes associated with oxidative stress in OP. The nomogram model showed the potential predictive ability of OP. Moreover, the GSVA results demonstrated that low expression of NAPG, NCOA1, and TRIM44 was enriched in the oestrogen response early signalling pathway. Moreover, these diagnostic genes were strongly correlated with multiple immune-related genes in the two datasets. Additionally, we identified several important factors that have regulatory relationships with diagnostic genes, such as MEF2A, STAT3, YY1, CREB1, hsa-mir-132-3p, and hsa-mir-148a-3p. Finally, it was verified at the protein and cellular levels that NAPG inhibits osteogenic differentiation and may play a crucial role in osteoporosis. NAPG, NCOA1, and TRIM44 were found to be associated with the diagnosis of OP, suggesting novel opportunities for diagnosing and treating OP.
Similar content being viewed by others

Introduction
Osteoporosis (OP) is the most prevalent skeletal disease and is characterized by low bone mineral density (BMD) and structural deterioration of bone tissues, increasing susceptibility to fractures1. In particular, approximately 80% of fractures are related to OP2. Approximately half of women and one third of men are affected by OP at some point in their lives3. Since bone loss occurs with age, the incidence of osteoporosis also increases with age4. Moreover, oestrogen deficiency can lead to increased bone remodelling, which leads to a loss of bone density5. With the ageing of the population, the prevalence of OP is gradually increasing, which places a considerable economic and health burden on society4. Currently, bone densitometry and dual X-rays are common diagnostic methods for OP6. Recently, the identification of biomarkers, such as RANKL and ESR1, has become an effective method for early diagnostic strategies for OP7.
Oxidative stress (OS) is an imbalance between oxidants and antioxidants, leading to the disruption of REDOX signalling and the control of molecular damage8. OS is reportedly associated with a variety of diseases, such as neurodegenerative disease9, cardiovascular diseases10, and cancer11. Additionally, OS is a major driver of the pathophysiological processes of cartilage and bone and plays a decisive role in determining the cell fate of OP12. OS increases reactive oxygen species (ROS), promotes the occurrence of osteoclasts, and reduces the occurrence of osteoblasts13. The superoxide produced by osteoclasts directly leads to bone degradation; in contrast, osteoblasts produce antioxidants, such as GPX, to prevent the generation of ROS14. In brief, OS has a positive effect on osteoblast activity and mineralization15. However, few studies have addressed whether OS has a causative role in triggering OP, and studying the potential pathogenesis of OS-related genes (OS-RGs) may help to identify potential causative factors and REDOX-related therapeutic targets for OP.
In this study, OP-related microarray datasets from the Gene Expression Omnibus (GEO) database were used. The diagnostic genes for OP were subsequently identified through weighted gene coexpression network analysis (WGCNA), differential expression analysis, and protein‒protein interaction (PPI) network construction. The functions and molecular regulatory relationships of the diagnostic genes were subsequently explored to elucidate the mechanism of OP. Overall, our study aims to explore OS in OP by a set of bioinformatics analyses and provide a reference for further studies of OP.
Materials and methods
Data extraction
The GEO database (http://www.ncbi.nlm.nih.gov/geo/) was utilized in this study to access OP-related datasets, namely, GSE56815 (platform: GPL96) and GSE7158 (platform: GPL570). The GSE56815 dataset comprises 40 peripheral blood monocytes (PBMs) samples with low bone mineral density (BMD) and 40 PBMs samples with high BMD, whereas the GSE7158 dataset consists of 12 PBMs samples with low BMD and 14 PBMs samples with high BMD. 1,046 genes related to oxidative stress were extracted from GeneCards Database (https://www.genecards.org/) using “oxidative stress” as the keyword and relevance score > 1 as filtering criteria16.
WGCNA
With the OS-RGs as the background gene set, the single-sample gene set enrichment analysis (ssGSEA) algorithm of the GSVA package (v 1.52.3) was used to calculate the ssGSEA score for each sample in the GSE56815 dataset, and the differences in these scores between the high-BMD samples and low-BMD samples were compared via the Wilcoxon test (p < 0.05). Furthermore, receiver operating characteristic (ROC) curve analysis was implemented with the pROC package (v 1.18.5) to explore whether this score had diagnostic performance in distinguishing the two groups.
Key module genes linked to OS-RGs were acquired via the WGCNA package (v 1.72-5). First, clustering analysis was adopted for all samples in the GSE56815 dataset. Through the clustering of the samples, it was determined whether there were outliers that needed to be filtered out to ensure the accuracy of the subsequent analysis. To ensure that the intergene interactions maximally conformed to the scale-free distribution, a soft threshold was determined for the data. The optimal soft threshold (β) was determined when the scale-free topology model fit (R2) exceeded the threshold value of 0.85 and the mean value of the neighbourhood function also gradually approached 0. On the basis of the determined optimal soft threshold (β), the minimum number of genes per gene module was set to 30 in accordance with the criteria of hybrid dynamic tree cutting, thus clustering genes into different modules. The ssGSEA score of OS-RGs was used as a phenotypic trait, followed by calculating the correlation coefficient between the module and this score by Pearson correlation analysis. Modules with significant maximum positive and negative correlations were selected and defined as key modules [|correlation coefficient (cor)| > 0.3 & p < 0.05]. The genes in these key modules were defined as the key module genes.
Differential expression analysis
With the application of the limma package (v 3.60.4), differential expression analysis was implemented between high and low BMD groups in the GSE56815 dataset with the aim of identifying differentially expressed genes (DEGs) (adj.p < 0.05)17. To understand the distribution of DEGs as a whole, the ggplot2 package (v 3.4.1) was used to create a volcano plot. The ComplexHeatmap package (v 2.15.1) was subsequently used to draw a heatmap of the expression of the top 15 upregulated and downregulated genes sorted by their adj.p values.
Functional enrichment analysis and PPI network construction
The intersection of DEGs and key module genes was determined via the Venn diagram package (v 1.7.1) to identify genes linked to both OP and oxidative stress, which were recorded as candidate genes. The ClueGo plug-in was employed to interpret the biological functions of candidate genes, and the result was visualized by Cytoscape software18,19,20. Moreover, these candidate genes were entered into the STRING database (http://www/string-db.org/), followed by the construction of a PPI network with the objective of probing their interactions at the protein level.
Identification of diagnostic genes
The GSE56815 and GSE7158 datasets were utilized to conduct receiver operating characteristic (ROC) curve analysis, and the ability of the candidate genes to discriminate between high-BMD and low-BMD samples was assessed by calculating their area under the ROC curve (AUC) values. Importantly, candidate genes demonstrating robust discriminatory capacity with AUC values exceeding 0.7 in both datasets were designated diagnostic genes. Simultaneously, the visualization of diagnostic gene expression was depicted using box plots in both the GSE56815 and GSE7158 datasets. In addition, Spearman analysis was conducted using the psych package (v 2.2.9) to investigate the correlations among diagnostic genes.
Establishment and evaluation of the nomogram
The diagnostic genes identified by the aforementioned analysis were utilized to construct a nomogram model employing the rms package (v 6.8-1) in the GSE56185 dataset, aiming to comprehensively assess the collective predictive capacity of these diagnostic genes for OP. Moreover, calibration curves and decision curve analysis (DCA) were adopted to evaluate the accuracy of the predictive capability of the nomogram model. The DCA was implemented with the rmda package (v 1.6). Additionally, ROC curve analysis was implemented in both the GSE56815 and GSE7158 datasets to evaluate the predictive performance of the nomogram model.
Gene set variation analysis (GSVA)
In the GSE56815 dataset, all samples were stratified into high-expression and low-expression groups on the basis of the median expression of diagnostic genes. A comparative analysis was subsequently conducted to evaluate the differences in the GSVA scores of samples from both groups in the HALLMARK pathway derived from the Molecular Signatures Database (MSigDB) (https://www.gsea-msigdb.org/gsea/msigdb).
Immune analysis and construction of a molecular regulatory network
To further investigate the relationship between diagnostic genes and the immune response in OP, Spearman analysis was separately conducted on the GSE56815 and GSE7158 datasets to explore the associations between diagnostic genes and 3 immunoinhibitors, 17 immunostimulators, and 12 immune checkpoints. In addition, the miRNet database (https://www.mirnet.ca/miRNet/home.xhtml) was used to predict transcription factors (TFs) and miRNAs that target diagnostic genes.
Correlation analysis between diagnostic genes and clinical features
To further investigate the discriminative ability of diagnostic genes between OP and control samples, a total of 15 OP and 10 control samples were collected from Qilu Hospital with approval from the Medical Ethics Committee of Qilu Hospital, Shandong University. The total RNA of PBMs in those samples was extracted using TRIzol reagent (Solarbio, Beijing). The RNA from each sample was subsequently reverse transcribed into cDNA for real-time PCR. The primers used in the present study are listed in Table 1. The relative expression of diagnostic genes was calculated via the 2−△△ct method. The discriminatory power of the diagnostic genes was assessed by constructing an ROC curve. Additionally, Spearman correlation analysis was conducted using clinical feature information from 14 OP patients to explore the relationships between diagnostic genes and clinical characteristics. These clinical features included hip bone mineral content (BMC), hip bone mineral density (BMD), hip T score, lumbar spine BMC, lumbar spine BMD, lumbar spine T score, and calcium (Supplementary Table 1).
Western blot analysis of NAPG in bone tissues
Bone tissue samples (5 pairs of osteoporosis and control groups) were homogenized in RIPA lysis buffer containing protease inhibitors (Solarbio, China). Protein concentrations were quantified using the BCA assay (Beyotime, China). Equal amounts of protein (30 µg) were separated by SDS-PAGE and transferred to PVDF membranes. After blocking with 5% skim milk, membranes were incubated overnight at 4 °C with anti-NAPG primary antibody (1:1000, Abcam, USA) and anti-GAPDH (1:5000, Proteintech, China). HRP-conjugated secondary antibodies (1:5000, Proteintech, China) were applied for 1 h at room temperature. Grayscale analysis was carried out using ImageJ software. Group differences were analyzed using one-way ANOVA and Tukey’s multiple comparison test, with a P-value of less than 0.05 denoting statistical significance.
Cell culture and osteogenic differentiation
MC3T3-E1 pre-osteoblasts (Cobioer, China) were cultured in α-MEM (Gibco, USA) supplemented with 10% FBS and 1% penicillin/streptomycin. For osteogenic differentiation, cells were treated with 50 µg/mL ascorbic acid and 10 mM β-glycerophosphate (EpiZyme, China) for 0, 3, 7, and 14 days respectively. Medium was refreshed every 3 days.
NAPG overexpression and knockdown
NAPG overexpression plasmids (oe-NAPG) and siRNA targeting NAPG (si-NAPG) were synthesized by Ribobio (China). MC3T3-E1 cells were transfected using Lipofectamine 3000 (Thermo Fisher Scientific, USA). Scrambled siRNA and empty vector served as controls (oe-NC、si-NC). Transfection efficiency was validated by qPCR and Western blot.
Functional assays in MC3T3-E1 cells
Alizarin Red Staining (ARS) : The transfected MC3T3-E1 cells were continuously cultured for 14 days, and the osteogenic induction medium was replaced every 3 days. After the culture, the medium was discarded, the cells were gently washed twice with PBS, and fixed with 4% paraformaldehyde for 30 min. Then, they were stained with 0.1% alizarin red staining solution (Beyotime, China) for 30 min. During the staining process, the culture plate was gently shaken to ensure uniform staining. After staining, the cells were washed with PBS until the washing solution was colorless, and the formation of mineralized nodules was observed and photographed under a microscope.
Alkaline Phosphatase Assay (ALP): The transfected MC3T3-E1 cells were cultured for 7 days, and then the cells were collected. According to the instructions of the ALP activity detection kit (Beyotime, China), the cells were lysed with a cell lysis buffer, and the supernatant was collected by centrifugation. The supernatant was reacted with the ALP substrate. The total area of blue nodules in ALP and red nodules in ARS were detected by an Olympus IX73 inverted microscope (Olympus, Tokyo, Japan).
Western Blot: In the osteoblast differentiation experiment, MC3T3-E1 cells were collected on days 0, 3, 7, and 14 of differentiation, respectively. The operation method of Western Blot is the same as that in 2.10. The primary antibodies involved are: ALP (1:1000, Abcam, USA), COL1A1 (1:1000, Cell Signaling Technology, Germany), OPN (1:800, Proteintech, China), β-catenin (1:1000, Cell Signaling Technology, Germany), RUNX2 (1:800, Abcam, USA) and GAPDH (1:5000, Proteintech, China).
Ethics statement
The Medical Ethics Committee of Qilu Hospital, Shandong University, granted approval for this study. A total of 40 female patients aged between 20 and 75 years volunteered to participate in the study. Prior to their involvement, all the subjects provided informed consent. The experimental methods employed in this study were conducted in accordance with the relevant regulations and guidelines.
Statistical analysis
The data were analysed via R software (v 4.1.0). The Wilcoxon test was used to assess the differences between different groups. A p value less than 0.05 was considered statistically significant.
Results
Identification of the key module genes linked to OS-RGs
The Wilcoxon test demonstrated that the ssGSEA score of OS-RGs was significantly higher in the high-BMD group than in the low-BMD group, suggesting that oxidative stress does have an effect on OP (Fig. 1A). Moreover, the ROC curve demonstrated that there was a certain ability to distinguish the two groups (Fig. 1B). The clustering analysis results indicated that there were no outliers in the GSE56815 dataset (Fig. 1C). The sample grouping, ssGSEA score of OS-RGs, and sample clustering tree were subsequently combined for visualization (Fig. 1D). Immediately thereafter, the optimal soft threshold (β) was chosen to be 6 on the basis of the criteria that R2 exceeded 0.85 and that the mean value of the adjacency function gradually approached zero (Fig. 1E). After that, 12 gene modules were obtained (Fig. 1F). Furthermore, red and blue were selected as key modules, because red (cor = -0.6, p = 4 × 10− 8) and blue (cor = 0.63, p = 4 × 10− 9) exhibited significant maximum negative and positive correlations with ssGSEA scores, respectively. Among these two key modules, 1,730 key module genes were obtained.

Identification of the key module genes linked to oxidative stress-related genes (OS-RGs). (A) The difference in the ssGSEA score of OS-RGs between the high-BMD group and the low-BMD group in the GSE56815 dataset. (B) Receiver operating characteristic (ROC) curve analysis of the ssGSEA score of OS-RGs in the GSE56815 dataset. (C) Clustering of data samples. (D) Scale-free soft threshold distribution. (E) Clustering dendrogram of modules from WGCNA. (F) Heatmap of the relationships between gene modules and traits.
Exploration of the functions of candidate genes
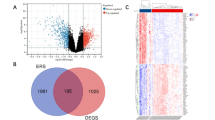
With the application of the limma package, 395 DEGs were selected from the GSE56815 dataset. Among them, 276 genes were notably upregulated in the low-BMD group, whereas 119 genes were notably downregulated in the low-BMD group (Fig. 2A-B). A total of 101 candidate genes were subsequently identified by crossing 395 DEGs and 1,730 key module genes (Fig. 2C). Furthermore, enrichment analysis revealed that a total of 101 candidate genes were significantly enriched in various signalling pathways, including the neurotrophin signalling pathway, the cellular response to ketone pathway, the IL-17 signalling pathway, and the regulation of phosphatase activity (Fig. 2D). Additionally, the PPI results revealed significant interactions between VCP, NFKB2, PSMD3, PSMA1, and other candidate genes (Fig. 2E). These findings suggest the presence of intricate interactions among candidate genes, potentially influencing disease progression by modulating specific signalling pathways.

Exploration of the functions of candidate genes. (A) Volcano plot of differentially expressed genes (DEGs) in the GSE56815 dataset. The blue dots indicate significantly downregulated genes, and the red dots indicate significantly upregulated genes. (B) Heatmap of the top 15 upregulated and downregulated DEGs in the GSE56815 dataset. (C) Venn diagram of 101 candidate genes. (D) Enrichment analysis of 101 candidate genes. (E) Protein‒protein interaction (PPI) analysis of 101 candidate genes.
NAPG, NCOA1, and TRIM44 were identified as diagnostic genes for OP
ROC curve analysis revealed that the AUC values of NCOA1, TRIM44, ZIC1, RMB5, and NAPG in the GSE56815 (Fig. 3A) and GSE7158 (Fig. 3B) datasets exceeded 0.7, indicating their superior ability to discriminate between the low-BMD and high-BMD groups. Moreover, box plots revealed that the expressions of NAPG, NCOA1, and TRIM44 in the GSE56815 (Fig. 3C) and GSE7158 (Fig. 3D) datasets were higher in the low-BMD group. Consequently, these genes (NAPG, NCOA1, and TRIM44) were identified as diagnostic genes associated with oxidative stress in OP and were utilized for subsequent analyses. Furthermore, a strong or moderate positive correlation between TRIM44 and NCOA1 was observed in GSE56815 (cor = 0.7, p < 0.05) and GSE7158 datasets (cor = 0.44, p < 0.05) (Fig. 3E-F).

Identification of diagnostic genes for OP. (A-B) ROC curve analysis of candidate genes in both the GSE56815 and GSE7158 datasets. (C-D) Box plot showing the consistent expression trends of the three diagnostic genes in both the GSE56815 and GSE7158 datasets. (E-F) Correlations between diagnostic genes in the GSE56815 and GSE7158 datasets.
The diagnostic genes exhibited excellent predictive ability for OP
The predictive ability of the diagnostic genes as a whole for OP was evaluated. A nomogram model was created on the basis of NAPG, NCOA1, and TRIM44. Within this model, a higher total point demonstrated an increased probability of OP (Fig. 4A). The calibration curve demonstrated a close resemblance between the slope of the nomogram model and the ideal curve, further emphasizing its predictive accuracy (Fig. 4B). Moreover, the nomogram model exhibited greater net benefit than a single diagnostic gene in DCA, highlighting its superior performance (Fig. 4C). In addition, ROC curve analysis further confirmed the excellent ability of the nomogram model to distinguish high-BMD and low-BMD samples in both the GSE56815 (Fig. 4D) and GSE7158 datasets (Fig. 4E).

Establishment and verification of the nomogram model. (A) The nomogram model for predicting the incidence of OP by NAPG, NCOA1, and TRIM44. (B) Calibration curves of the nomogram model. (C) Decision curve analysis of the nomogram model. (D-E) ROC curve analysis for the nomogram model in both the GSE56815 and GSE7158 datasets.
Specific signalling mechanisms of diagnostic genes
GSVA was implemented to probe the specific signalling mechanisms of NAPG, NCOA1, and TRIM44 in OP. In the GSE56185 dataset, high expression of NAPG was enriched in oxidative phosphorylation, MYC targets V1, peroxisome, protein secretion, fatty acid metabolism and other signalling pathways, whereas low expression of NAPG was enriched in the Wnt/beta-catenin signalling pathway, p53 pathway, myogenesis and oestrogen response early among other signalling pathways (Fig. 5A). Additionally, low expression of NCOA1 was enriched in the oestrogen response early and late signalling pathways (Fig. 5B). High expression of TRIM44 was enriched in peroxisomes, MYC targets V1, and the fatty acid metabolism signalling pathway, whereas low expression of TRIM44 was enriched in the hedgehog signalling pathway, myogenesis, oestrogen response early and late signalling pathways, and other pathways (Fig. 5C). These three diagnostic genes exhibited shared enrichment in signalling pathways, including both early and late oestrogen response pathways. These findings suggest that these diagnostic genes might modulate disease progression by either activating or inhibiting the functionality of these signalling pathways.

Specific signalling mechanisms of diagnostic genes. (A–C) Gene set variation analysis for NAPG, NCOA1, and TRIM44.
Link between diagnostic genes and OP immunity
We performed correlation analysis between the expression of various immune genes and the three diagnostic genes in both the GSE56815 and GSE7158 datasets. Our findings revealed that in the GSE56185 dataset, NAPG was significantly negatively correlated with all three immunoinhibitors, with CSF1R showing the strongest negative association (r = -0.43) (Fig. 6A). Among the 17 immunostimulators examined, TNFRSF25 (r = -0.36) and TNFRSF8 (r = -0.48) were strongly negatively correlated with three diagnostic genes, particularly TRIM44 (Fig. 6B). Additionally, TNFSF18 (r = 0.37) and TNFSF9 (r = 0.36) were strongly positively correlated with NCOA1. Furthermore, among the 12 immune checkpoints investigated, CCL24 demonstrated the strongest positive correlation with TRIM44 (r = -0.47) (Fig. 6C). Moreover, in the GSE7158 dataset, only NCOA1 exhibited a significant negative correlation with the immunoinhibitor CSF1R (r = -0.49) (Fig. 6D). Among the 17 immunostimulators, NAPG was strongly positively correlated with TNFSF4 (r = 0.53), whereas both NCOA1 (r = -0.56) and TRIM44 (r = -0.39) were negatively correlated with CD28. The most robust positive correlation was observed between NCOA1 and IL6 (r = 0.58), whereas TRIM44 demonstrated the strongest negative correlation with LTA (r = -0.6) (Fig. 6E). Among the 12 immune checkpoints, only NAPG and NCOA1 were strongly correlated with some of them. Specifically, NAPG presented the strongest positive correlation with CXCL5 (r = 0.53), whereas NCOA1 presented the strongest positive correlation with CCL20 (r = 0.56) (Fig. 6F). These findings revealed a strong correlation between diagnostic genes and OP immunity, thereby offering a novel reference for OP treatment.

Link between diagnostic genes and OP immunity. (A) Correlations between diagnostic genes and immunoinhibitors in the GSE56185 dataset. (B) Correlations between diagnostic genes and immunostimulators in the GSE56185 dataset. (C) Correlations between diagnostic genes and immune checkpoints in the GSE56185 dataset. (D) Correlations between diagnostic genes and immunoinhibitors in the GSE7158 dataset. (E) Correlations between diagnostic genes and immunostimulators in the GSE7158 dataset. (F) Correlations between diagnostic genes and immune checkpoints in the GSE7158 dataset.
Relationships between regulatory factors and diagnostic genes
By utilizing the miRNet database, we investigated the associations between diagnostic genes and TFs, as well as miRNAs. Among the predicted TFs, MEF2A was found to concurrently regulate NAPG, NCOA1, and TRIM44, while STAT3, YY1, and CREB1 were observed to simultaneously regulate NAPG and NCOA1 (Fig. 7A). With respect to miRNAs, we included only those that exhibited regulatory effects on a minimum of two genes, such as hsa-mir-132-3p, hsa-mir-148a-3p, and hsa-miR-135b-3p (Fig. 7B).

Relationships between regulatory factors and diagnostic genes. (A) TF‒mRNA regulatory network. (B) miRNA‒mRNA regulatory network.
Diagnostic genes strongly negatively correlated with multiple clinical features
We first assessed the expression of NAPG, NCOA1, and TRIM44 in our own clinical samples. We found that their expression was significantly increased in OP (Fig. 8A and C), which was consistent with the sequencing results. The ROC curve analysis of the clinical samples revealed that the AUC values for NAPG, NCOA1, and TRIM44 exceeded 0.8 (Fig. 8D), indicating their diagnostic potential in distinguishing between the OP and control groups. Furthermore, correlation analysis revealed a strong negative association between NAPG and Hip BMC (Fig. 8E). Therefore, to further verify the mechanism of NAPG in osteoporosis, we conducted a series of experiments. At the protein level, Western Blot was used to detect the expression of NAPG in bone tissues of the osteoporosis group and the control group, with a sample size of 5 pairs. The results showed that the protein expression level of NAPG in the bone tissues of the osteoporosis group was significantly higher than that in the control group. Grayscale analysis was performed using ImageJ software (Supplementary Fig. 1A). This result further confirmed the association between NAPG and osteoporosis at the protein level, suggesting that NAPG is highly expressed in osteoporotic bone tissues. In addition, we used the MC3T3-E1 cell line and detected the expression changes of NAPG during osteoblast differentiation by Western Blot. The results showed that with the progression of osteoblast differentiation (cell samples were collected on days 0, 3, 7, and 14 of induced differentiation), the expression of NAPG gradually decreased (Supplementary Fig. 1B). This indicates that the expression change of NAPG is closely related to the osteoblast differentiation process, revealing from the cell level that NAPG inhibits osteoblast differentiation. We also investigated the effects of overexpressing and interfering with NAPG on osteoblast differentiation. The results of ARS demonstrated that the number of mineralized nodules formed in the MC3T3-E1 cell group over-expressing NAPG (oe-NAPG) was significantly less than that in the control group (oe-NC), while the number of mineralized nodules in the cell group with NAPG expression interfered (si-NAPG) was significantly more than that in the control group (si-NC) (Supplementary Fig. 1C). This visually indicates that NAPG inhibits the mineralization ability of MC3T3-E1 cells, strongly supporting the hypothesis that NAPG inhibits osteoblast differentiation. The results of ALP assay showed that the ALP activity in the cell group over-expressing NAPG (oe-NAPG) was significantly lower than that in the control group (oe-NC), and the ALP activity in the cell group with NAPG expression interfered (si-NAPG) was significantly higher than that in the control group (si-NC) (Supplementary Fig. 1D). Since ALP activity is one of the important markers of osteoblast differentiation, this result further confirmed the inhibitory effect of NAPG on osteoblast differentiation. Furthermore, we detected the protein expression levels of osteoblast markers ALP, COL1A1, OPN, β-catenin, and RUNX2 by Western Blot. The results showed that after over-expressing NAPG, the protein expression levels of these osteoblast markers were significantly decreased, while after interfering with NAPG expression, their expression levels were significantly increased (Supplementary Fig. 1E). In conclusion, we provided more direct and powerful evidence at the protein level and in cell function experiments for the association between NAPG and osteoporosis, further supporting the conclusion that NAPG plays an important role in the development of osteoporosis.

Correlation analysis between diagnostic genes and clinical features. (A) ROC curve analysis of clinical samples. (B) Correlations between diagnostic genes and clinical features.
Discussion
OP is a major risk factor for fracture, and as with most degenerative and age-related diseases, OS has been identified as a key driver of the underlying pathogenesis of OP12. OS induces osteoblast apoptosis and varying degrees of specific factors, such as receptor activator kappa B ligand (RANKL), sclerostin, and fibroblast growth factor 23, thereby impacting bone remodelling and high bone resorption15. Here, our study screened three novel diagnostic genes related to OS for OP, namely, NAPG, NCOA1, and TRIM44 by bioinformatic strategies and validated their performance in OP diagnosis via expression detection and ROC curves using clinical samples. In particular, a nomogram model constructed with diagnostic genes has potential to predict OP. In conclusion, these three diagnostic genes are pivotal for clinical practice, especially early diagnosis before the occurrence of a fracture.
In this study, three diagnostic genes, NAPG, NCOA1, and TRIM44, were identified via the PPI algorithm, ROC curves, and expression levels. Specifically, these three diagnostic genes, which were coenriched in the oestrogen response (ER) early signalling pathway, were upregulated in the OP group. Many studies have demonstrated that an oestrogen-related decrease in ERalpha may lead to a decrease in bone mass in OP patients21,22,23. The AUC values of the nomogram were 0.840 for GSE56815 and 0.857 for GSE7158. Furthermore, MEF2A was found to concurrently regulate NAPG, NCOA1, and TRIM44.
NAPG encodes the N-ethyl-maleimide-sensitive fusion protein (NSF) attachment protein gamma, which mediates platelet exocytosis and controls membrane fusion events24,25. Xu et al. reported that the NAPG mutation (c.784 A > G) may increase the risk of hereditary haemorrhagic telangiectasia (HHT) in China26. Here, our study confirmed the high expression of NAPG in the PBMC samples of OP patients, but there are no published studies supporting this finding. In the GSVA of the NAPG results, E2F targets were activated in the high-expression subgroup. Specifically, Dong et al. reported that E2F transcription factor 2 (E2F2) was highly expressed in traumatic fracture (TF) model mice27. Overall, the specific mechanism by which NAPG affects OP remains to be further explored.
Nuclear receptor coactivator 1 (NCOA1) is a member of the p160/steroid receptor coactivator (SRC) family (also known as SRC-1) and plays important roles in the regulation of gene expression mediated by various steroid receptors28,29. Indeed, NCOA1 has been reported to play an important role in a variety of diseases, such as oesophageal carcinoma29, obesity30, and glioblastoma31. Two relevant studies in network pharmacology proved that NCOA1 is an effective target for treating OP in Gushukang (GSK) and GuBen-ZengGu granules (GBZGG)32,33. Mödder et al. also confirmed trabecular osteopenia in both male and female SRC-1 knockout (KO) mice34. Interestingly, NCOA1 was overexpressed in the OP group; therefore, the role of NCOA1 in OP patients needs further expression validation.
Tripartite motif containing 44 (TRIM44) is a member of the tripartite motif (TRIM) family and functions as a deubiquitinating enzyme35,36. TRIM44 is a prognostic marker that is upregulated in many carcinomas and is associated with poor prognosis37,38,39. In particular, the expression of TRIM44 is upregulated in multiple myeloma, which promotes SQSTM1 oligomerization under OS40,41. Moreover, high expression of TRIM44 leads to decreased responsiveness to proteasome inhibitor treatments37,41. In our study, TRIM44 was upregulated in OP patients, as mentioned earlier, and we speculate that it may cause dysregulation of OS in OP patients. Furthermore, Wang et al. selected TRIM44 as a hub gene for the diagnosis of OP on the basis of microarray data42. In summary, these three diagnostic genes provide new perspectives for the early diagnosis of OP. Maasalu et al.43 used whole-blood transcriptome and identified CACNA1G, ALG13, SBK1, GGT7, MBNL3, and RIOK3 as potential biomarkers for OP. They evaluated their function and diagnostic performance by ROC, correlation analysis and IPA analysis. In our study, we used the transcriptome profile of PBMs, which may act as precursors of osteoclasts and be used as a cell model to study bone-related disease44. Moreover, our study mainly focused on the oxidative stress-related genes in diagnosing OP. In addition, there were another two papers aimed at identifying oxidative stress-related genes in OP. One study by Zhao et al.45 used RNA expression profile in human mesenchymal stem cells between four non-OP controls and five primary OP patients, and identified PPP1R15A, CYB5R3, BCL2L1, ABCD1, MAPKAPK2, HSP90AB1, CSF1, rela, P4HB, AKT1, HSP90B1, and CTNNB1 as key genes involved in the progression of OP. However, the sample size they used is small, and the diagnostic capability is not validated in OP. Another study by Liu et al.46 used RNA expression profile in PBMs of 20 postmenopausal OP and 20 controls, and identified DDIT3 and FOXO3 as postmenopausal OP biomarkers. They mainly focused on postmenopausal OP, however, clinical validation is lacking in their study. In our study, we used the transcriptome profile in PBMs of 40 OP and 40 controls, which had larger sample size, making the results more robust and reliable. In addition, we validated the diagnostic performance of NAPG, NCOA1 and TRIM44 in real clinical samples, demonstrating their reliability. Therefore, different sources of samples and analysis strategies may yield different results, both of which need further experimental validations.
After screening these three diagnostic genes, we further explored the relationships between these genes and immune-related genes, including immunoinhibitors, immunostimulators, and immune checkpoints. Our results revealed that these diagnostic genes were strongly correlated with multiple immune-related genes. To our knowledge, immunity is involved in various bone metabolic processes, especially OP47. Zhang et al. reported that understanding the connection between immune cells and bone remodelling processes can help in evaluating the pathogenesis of OP and that identifying immune checkpoints is an excellent direction for immunotherapies in OP patients48. Additionally, Song et al. screened five hub genes related to immunity via machine learning and qRT‒PCR49.
MEF2A is a common TF that regulates these three diagnostic genes and is worthy of further exploration. MEF2A can protect primary neurons from cell damage induced by OS50. Tang et al. reported that changes in MEF2A expression can affect the expression of chondrogenic marker genes, such as Runx2 and Sox951.
In addition, we conducted verification through further experiments. At the protein level, we found that the protein expression of NAPG in the bone tissues of the osteoporosis group was significantly higher than that in the control group, which further confirmed the association between NAPG and osteoporosis. In cell experiments, as osteoblasts differentiated, the expression of NAPG gradually decreased. Moreover, overexpression of NAPG inhibited osteoblast differentiation, while interference with NAPG promoted it. The results of Alizarin Red staining and alkaline phosphatase activity assays demonstrated that NAPG inhibited the mineralization ability and alkaline phosphatase activity of MC3T3 - E1 cells. Additionally, the Western Blot results showed that overexpression of NAPG decreased the protein expression levels of osteoblast markers such as ALP, COL1A1, OPN, β-catenin, and RUNX2, while interfering with NAPG expression increased the expression levels of these markers. These results provided more direct and powerful evidence at the protein level and in cell function experiments for the association between NAPG and osteoporosis, further supporting the conclusion that NAPG plays an important role in the development of osteoporosis.
In this study, we identified three diagnostic genes—NAPG, NCOA1, and TRIM44—that show potential for the early diagnosis of osteoporosis (OP). However, several limitations should be acknowledged. First, this study lacks more comprehensive cell and animal experiments for verification. Second, the relatively small sample sizes of the GSE56815 and GSE7158 datasets may limit the generalizability of our conclusions. Although we validated the expression levels of the key genes in real clinical samples, the current sample size remains insufficient for robust validation of the nomogram. Furthermore, although a positive correlation between TRIM44 and NCOA1 was observed in two datasets, further experimental studies are required to confirm a functional relationship between these genes. To strengthen our findings, future studies should include larger clinical cohorts to validate the nomogram, as well as animal models of OP to explore the oxidative stress-related biological functions of these candidate genes in vivo.
Data availability
Data availabilityAll research data in this article can be obtained by contacting the corresponding author.
Change history
06 November 2025
The original online version of this Article was revised: In the original version of this Article Zhenchuan Liu, Xu Yang and Zexin Wang were omitted as equally contributing authors. The statement has now been added and reads: “Zhenchuan Liu, Xu Yang and Zexin Wang contributed equally to this work.”
References
Brown, J. P. Long-Term Treatment of Postmenopausal Osteoporosis.Endocrinology and metabolism (Korea), 36(3), 544–552. (2021).
Bessette, L. et al. The care gap in diagnosis and treatment of women with a fragility fracture. Osteoporos. International: J. Established as Result Cooperation between Eur. Foundation Osteoporos. Natl. Osteoporos. Foundation USA. 19 (1), 79–86 (2008).
Wright, N. C. et al. The recent prevalence of osteoporosis and low bone mass in the united States based on bone mineral density at the femoral neck or lumbar spine. J. Bone Mineral. Research: Official J. Am. Soc. Bone Mineral. Res. 29 (11), 2520–2526 (2014).
Wu, D. et al. T-Cell mediated inflammation in postmenopausal osteoporosis. Front. Immunol. 12, 687551 (2021).
Ji, M. X. & Yu, Q. Primary osteoporosis in postmenopausal women. Chronic Dis. Translational Med. 1 (1), 9–13 (2015).
Aibar-Almazán, A. et al. Current status of the diagnosis and management of osteoporosis. Int. J. Mol. Sci. 23 (16), 9465 (2022).
Estrada, K. et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 44 (5), 491–501 (2012).
Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 4, 180–183 (2015).
Teleanu, D. M. et al. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int. J. Mol. Sci. 23 (11), 5938 (2022).
Senoner, T. & Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients, 11(9), 2090. (2019).
Jelic, M. D., Mandic, A. D., Maricic, S. M. & Srdjenovic, B. U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 17 (1), 22–28 (2021).
Riegger, J., Schoppa, A., Ruths, L., Haffner-Luntzer, M. & Ignatius, A. Oxidative stress as a key modulator of cell fate decision in osteoarthritis and osteoporosis: a narrative review. Cell. Mol. Biol. Lett. 28 (1), 76 (2023).
Iantomasi, T. et al. Oxidative stress and inflammation in osteoporosis: molecular mechanisms involved and the relationship with MicroRNAs. Int. J. Mol. Sci. 24 (4), 3772 (2023).
Yuan, Y., Yang, J., Zhuge, A., Li, L. & Ni, S. Gut microbiota modulates osteoclast glutathione synthesis and mitochondrial biogenesis in mice subjected to ovariectomy. Cell Prolif., 55(3), e13194. (2022).
Marcucci, G. et al. Oxidative stress and natural antioxidants in osteoporosis: Novel preventive and therapeutic approaches. Antioxid. (Basel Switzerland). 12 (2), 373 (2023).
Stelzer, G. et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protocols Bioinf. 54, 1301–13033 (2016).
Lill, M. et al. Peripheral blood RNA gene expression profiling in patients with bacterial meningitis. Front. NeuroSci. 7, 33 (2013).
Bindea, G. et al. ClueGO: A cytoscape plug-in to Decipher functionally grouped gene ontology and pathway annotation networks. Bioinf. (Oxford England). 25 (8), 1091–1093 (2009).
Wei, Z. & Wang, J. Exploration of the core pathway of inflammatory bowel disease complicated with metabolic fatty liver and two-sample Mendelian randomization study of the causal relationships behind the disease. Front. Immunol. 15, 1375654 (2024).
Aldakheel, F. M. et al. Comprehensive computational analysis of differentially expressed MiRNAs and their influence on transcriptomic signatures in prostate cancer. Sci. Rep. 15 (1), 3646 (2025).
Armstrong, V. J. et al. Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires Estrogen receptor alpha. J. Biol. Chem. 282 (28), 20715–20727 (2007).
Jessop, H. L. et al. Osteoblast-like cells from Estrogen receptor alpha knockout mice have deficient responses to mechanical strain. J. Bone Mineral. Research: Official J. Am. Soc. Bone Mineral. Res. 19 (6), 938–946 (2004).
Sun, X. et al. Octyl Itaconate inhibits osteoclastogenesis by suppressing Hrd1 and activating Nrf2 signaling. FASEB Journal: Official Publication Federation Am. Soc. Experimental Biology. 33 (11), 12929–12940 (2019).
Lemons, P. P., Chen, D., Bernstein, A. M., Bennett, M. K. & Whiteheart, S. W. Regulated secretion in platelets: Identification of elements of the platelet exocytosis machinery. Blood 90 (4), 1490–1500 (1997).
Pappu, P. et al. Next generation sequencing analysis of lung cancer datasets: A functional genomics perspective. Indian J. Cancer. 53 (1), 1–7 (2016).
Xu, Y. et al. NAPG mutation in family members with hereditary hemorrhagic telangiectasia in China. BMC Pulm. Med. 21 (1), 197 (2021).
Dong, R. et al. Modulation of the microRNA-6089/E2F transcription factor2 axis by querceting: implications for osteoblast viability, proliferation, migration, and osteogenic differentiation in fracture healing. J. Physiol. Pharmacology: Official J. Pol. Physiological Soc. 75 (2), 173–183 (2024).
Chen, X., Liu, Z. & Xu, J. The cooperative function of nuclear receptor coactivator 1 (NCOA1) and NCOA3 in placental development and embryo survival. Mol. Endocrinol. (Baltimore Md). 24 (10), 1917–1934 (2010).
Wang, L. et al. The oncogenic roles of nuclear receptor coactivator 1 in human esophageal carcinoma. Cancer Med. 7 (10), 5205–5216 (2018).
Cacciottolo, T. M. et al. Obesity due to steroid receptor Coactivator-1 deficiency is associated with endocrine and metabolic abnormalities. J. Clin. Endocrinol. Metab. 107 (6), e2532–e2544 (2022).
Gong, M. et al. Steroid receptor coactivator-1 enhances the stemness of glioblastoma by activating long noncoding RNA XIST/miR-152/KLF4 pathway. Cancer Sci. 112 (2), 604–618 (2021).
Wang, S., Zhu, S., Li, X. & Yang, Z. Study on the mechanism of action of the traditional Chinese medical prescription Gushukang in treating osteoporosis based on network Pharmacology and experimental verification. Curr. Comput.-Aided Drug Design. https://doi.org/10.2174/0115734099282620240521102006 (2024). Advance online publication.
Wang, K. et al. Network Pharmacological analysis and animal experimental study on osteoporosis treatment with GuBen-ZengGu granules. Evidence-based Complement. Altern. Medicine: eCAM. 2023, 9317557 (2023).
Mödder, U. I. et al. The skeletal response to Estrogen is impaired in female but not in male steroid receptor coactivator (SRC)-1 knock out mice. Bone 42 (2), 414–421 (2008).
Liu, T. Y. et al. Role of the tripartite motif (TRIM) family in female genital neoplasms. Pathology-Research Pract. 250, 154811 (2023).
Allen, M. D. & Bycroft, M. The solution structure of the ZnF UBP domain of USP33/VDU1. Protein Science: Publication Protein Soc. 16 (9), 2072–2075 (2007).
Wang, Y., Lyu, L., Vu, T. & McCarty, N. TRIM44 enhances autophagy via SQSTM1 oligomerization in response to oxidative stress. Sci. Rep. 14 (1), 18974 (2024).
Kawaguchi, T. et al. Overexpression of TRIM44 is related to invasive potential and malignant outcomes in esophageal squamous cell carcinoma. Tumour Biology: J. Int. Soc. Oncodevelopmental Biology Med. 39 (6), 1010428317700409 (2017).
Liu, S., Yin, H., Ji, H., Zhu, J. & Ma, R. Overexpression of TRIM44 is an independent marker for predicting poor prognosis in epithelial ovarian cancer. Experimental Therapeutic Med. 16 (4), 3034–3040 (2018).
Lyu, L., Chen, Z. & McCarty, N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy 18 (4), 783–798 (2022).
Vu, T., Wang, Y., Fowler, A., Simieou, A. & McCarty, N. TRIM44, a novel prognostic marker, supports the survival of Proteasome-Resistant multiple myeloma cells. Cells 13 (17), 1431 (2024).
Wang, Z. et al. Mining potential drug targets for osteoporosis based on CeRNA network. Orthop. Surg. 15 (5), 1333–1347 (2023).
Maasalu, K. et al. Featured article: Transcriptional landscape analysis identifies differently expressed genes involved in follicle-stimulating hormone induced postmenopausal osteoporosis. Experimental biology and medicine. (Maywood N J). 242 (2), 203–213 (2017).
Zhou, Y. et al. A novel approach for correction of crosstalk effects in pathway analysis and its application in osteoporosis research. Sci. Rep. 8 (1), 668 (2018).
Zhao, Y. et al. Revealing oxidative stress-related genes in osteoporosis and advanced structural biological study for novel natural material discovery regarding MAPKAPK2. Front. Endocrinol. 13, 1052721 (2022).
Liu, D. et al. Identification of biomarkers associated with oxidative stress-related genes in postmenopausal osteoporosis. Cellular and molecular biology. France) 69 (6), 186–192 (2023). (Noisy-le-Grand.
Saxena, Y., Routh, S. & Mukhopadhaya, A. Immunoporosis: role of innate immune cells in osteoporosis. Front. Immunol. 12, 687037 (2021).
Zhang, W. et al. Immunoporosis: role of immune system in the pathophysiology of different types of osteoporosis. Front. Endocrinol. 13, 965258 (2022).
Hao, S. et al. Identification of key immune genes of osteoporosis based on bioinformatics and machine learning. Front. Endocrinol. 14, 1118886 (2023).
Zhang, L. et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 10 (6), 1015–1035 (2014).
Chen, C. et al. Mef2a is a positive regulator of Col10a1 gene expression during chondrocyte maturation. Am. J. Translational Res. 15 (6), 4020–4032 (2023).
Acknowledgements
This work was supported in part by the National Key Research and Development Program of China (2020YFC2009004) and the National Natural Science Foundation of China (81900804、82402758).
Author information
Authors and Affiliations
Contributions
Author contributionsZ. L.: Writing—review & editing, writing—original draft, methodology, formal analysis, and validation. X. Y.: Validation, methodology, and conceptualization. Z. W.:Validation and methodology. Q. L.: Writing—review & editing and conceptualization. H. G.: Writing—review & editing and funding acquisition. Q. M.: Writing—review & editing. Y. Z.: Writing—review & editing, supervision, and project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Z., Yang, X., Wang, Z. et al. Identification and verification of oxidative stress-related genes in the diagnosis of osteoporosis. Sci Rep 15, 23222 (2025). https://doi.org/10.1038/s41598-025-06118-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-06118-2
