
Abstract
Spinal cord injury (SCI) is a severe neurological disorder, with glucocorticoids like methylprednisolone commonly used for treatment. However, their efficacy and risks remain controversial. Programmed cell death (PCD) mechanisms have been increasingly implicated in SCI pathology. This study aimed to identify differentially expressed genes (DEGs) related to glucocorticoid receptors and PCD and to construct a diagnostic model to guide glucocorticoid use in SCI treatment. SCI datasets (GSE5296, GSE47681, GSE151371, and GSE45550) were analyzed using protein-protein interaction networks, consensus clustering, GSVA for PCD pathway enrichment, and WGCNA. A total of 113 diagnostic models were developed through 12 machine learning algorithms, with the optimal model, “Lasso + Stepglm[both],” featuring six genes: Abca1, Cdh1, Glipr1, Glt8d2, Il10ra, and Pde5a. Validation through qRT-PCR confirmed the differential expression of four genes (Abca1, Glipr1, Il10ra, and Cdh1), which demonstrated strong predictive performance. Pathway enrichment of GRRDEGs was analyzed using GO, KEGG, and Bayesian network methods, and immune cell infiltration was assessed via CIBERSORT. In this study, we identified GR- and PCD-related DEGs in SCI and constructed a diagnostic model that may improve understanding of SCI molecular mechanisms and inform future investigations of glucocorticoid use.
Similar content being viewed by others


Introduction
Spinal cord injury (SCI) is a critical neurological condition that frequently results in motor and sensory impairments and, in severe cases, paralysis, profoundly affecting quality of life and societal productivity1. Despite advances in clinical interventions such as surgical decompression, pharmacotherapy, and rehabilitation, achieving meaningful neural functional recovery remains a substantial challenge2. To date, no universally effective therapeutic strategy has been developed to reverse or fully repair SCI-induced functional deficits, highlighting the urgent need to explore novel treatment approaches.
Glucocorticoids, particularly methylprednisolone (MP), are among the most commonly used pharmacological agents for SCI treatment. Administered intravenously within 8 h post-injury, MP is believed to exert therapeutic effects by improving microcirculation, controlling inflammatory responses, inhibiting neuronal necrosis, and enhancing neuronal activity3. These effects can contribute to the recovery of neurological function in SCI patients. However, the clinical application of MP remains controversial due to variability in patient response and potential adverse effects, such as gastrointestinal bleeding, sepsis, pulmonary infections, steroid-induced myopathy, and delayed wound healing4. As a result, MP therapy is not universally recommended as a standard treatment for SCI, and many clinicians advocate its selective use based on individualized assessment5.
Recent studies have highlighted the significant role of programmed cell death (PCD) mechanisms-such as necrosis, apoptosis, and autophagic cell death-in SCI pathology. Necrosis is often associated with exacerbation of inflammation, further aggravating spinal tissue damage6, while apoptosis involves activation of signaling pathways that lead to the programmed death of neurons and glial cells7. Emerging evidence also points to the role of autophagic cell death in neuronal injury8. These complex and interconnected cell death mechanisms present a challenge for understanding the pathological processes underlying SCI. While glucocorticoids have shown therapeutic potential, their precise mechanisms of action remain unclear, particularly concerning their impact on PCD pathways.
In recent years, machine learning technology has shown significant potential in SCI classification and mechanism analysis. Many studies have constructed diagnostic models by integrating multimodal data. For instance, a study utilized9 the random forest algorithm to conduct a joint analysis of transcriptome data and immune infiltration characteristics, dividing the SCI mouse model into inflammatory and neurodegenerative subgroups, revealing the dynamic changes in the M1/M2 macrophage ratio. However, it did not delve deeply into the interaction between glucocorticoid receptors and the programmed cell death pathway. Another study10 screened out the hub genes related to the pathological process of SCI by combining the support vector machine with the WGCNA co-expression module. However, its model construction relied on single-modal data and lacked cross-species validation. In addition, some studies11 have identified molecular subtypes closely related to nerve injury by integrating pyroptosy-related genes and immune infiltration analysis, but the verification of the biological functions of key genes is insufficient.
In response to these challenges, this study innovatively combines the following strategies: First, multi-omics data integration: Jointly analyze cross-species GEO datasets (mice, rats, and humans), remove batch effects through sva packages, and use WGCNA to identify co-expression modules strongly correlated with SCI phenotypes, overcoming the limitations of a single data type. Second, machine learning algorithm optimization: A combination of 12 algorithms was adopted to construct 113 models and conduct ten-fold cross-validation. Eventually, the optimal model was selected, significantly enhancing the classification robustness. Thirdly, The expression differences of key genes were verified by qRT-PCR, and the regulatory relationship between them and the immune microenvironment was analyzed by CIBERSORT to ensure the biological reliability of the results.
Building on this framework, we systematically applied bioinformatic analyses-including protein-protein interaction networks, consensus clustering, gene set variation analysis (GSVA), and WGCNA—to identify glucocorticoid receptor- and PCD-related differentially expressed genes (GRRDEGs) in SCI. To further validate these findings, a mouse model of spinal cord injury was established, enabling functional assessment of candidate biomarkers in vivo.
Materials and methods
Data acquisition
The SCI datasets GSE5296, GSE47681, GSE151371, and GSE45550 were downloaded from the Gene Expression Omnibus (GEO) database12 (https://www.ncbi.nlm.nih.gov/geo/) via the R package GEOquery13. Detailed information is provided in Table 1.
Glucocorticoid receptor-related genes (GRRGs) were retrieved from the GeneCards database14 (https://www.genecards.org/) using “glucocorticoid receptor” as the search term, filtered by “protein coding” and a relevance score > 2, resulting in 352 GRRGs. These genes were converted to their mouse counterparts using the R package homologene, yielding 335 mouse GRRGs. An additional 492 mouse GRRGs were sourced from the UniProt database15 (https://www.uniprot.org/). After merging the datasets and removing duplicates, a final set of 761 GRRGs was compiled, as summarized in Table S1.
Key regulatory genes for 13 PCD pathways were collected from a published study16, resulting in a total of 884 PCD-related genes. The final list of genes is available in Table S2.
To eliminate batch effects, the GSE5296 and GSE47681 datasets were combined using the R package sva17. The resulting integrated dataset consisted of 79 SCI samples and 51 control samples. Standardization was performed with the R package limma18, followed by probe annotation and normalization. Principal component analysis (PCA)19 was conducted to validate the effectiveness of batch effect removal, as shown in Fig. S1.
Glucocorticoid receptor (GR)-related differentially expressed genes (GRRDEGs) in SCI
Samples from the integrated GEO dataset were categorized into SCI and control groups. Differential expression analysis was conducted using the R package limma, with thresholds set at |logFC| > 0.5 and p-value < 0.05 for identifying differentially expressed genes (DEGs). The Benjamini‒Hochberg (BH) method was used for p value correction. Volcano plots depicting differential expression results were generated via the R package ggplot2.
Glucocorticoid receptor-related differentially expressed genes (GRRDEGs) were identified by intersecting the DEGs with GRRGs. A Venn diagram was created to illustrate the overlap, and the expression of GRRDEGs was visualized through heatmaps generated using the R package pheatmap.
Functional analysis of GRRDEGs in SCI
A genomic map featuring 24 chromosomes was created using the RCircos package. Functional roles of GRRDEGs were evaluated through Kyoto Encyclopedia of Genes and Genomes (KEGG)20,21,22 and Gene Ontology (GO) analyses, focusing on biological processes (BPs), cellular components (CCs), and molecular functions (MFs). Key genes were further analyzed using the Metascape platform. A protein-protein interaction (PPI) network for the key genes was constructed via the STRING database23 (https://string-db.org/) with an interaction score > 0.400. The network was visualized in Cytoscape (version 3.9.0), and potential hub genes were identified using five CytoHubba algorithms24,25: maximal clique centrality (MCC), degree, stress, edge percolated component (EPC), and closeness26. Based on their scores, the top 20 GRRDEGs were selected, and a Venn diagram was generated to highlight overlapping genes for further analysis.
Immune cell infiltration analysis
CIBERSORT27, a method based on linear support vector regression, was applied to deconvolute the transcriptome expression matrix and estimate the composition and abundance of immune cells in mixed cell populations. Using the CIBERSORT algorithm with a mouse-specific gene signature matrix, immune cell infiltration scores were calculated. The proportions of immune cells were visualized in a bar plot. The differences in the proportions of infiltrating immune cells between the SCI and control samples were visualized via the R package ggplot2, and only those results with p values < 0.05 were retained. Immune cells with significant differences in proportions were selected for subsequent analysis. Spearman’s correlation analysis was performed to assess the relationships between immune cells proportions, and a heatmap of these correlations was created via the R package pheatmap. The correlations between the expression levels of hub genes and the proportions of infiltrating immune cells were similarly calculated and visualized.
Unsupervised clustering of SCI samples
Unsupervised clustering of the 79 mouse samples was performed using the expression profiles of GRRDEGs and the R package ConsensusClusterPlus28, applying the partitioning around medoids (PAM) algorithm. The optimal number of clusters was determined based on a consistency score (> 0.9), the cumulative distribution function (CDF) curve, and consensus matrix assessment, with a maximum of six subtypes considered.
GSVA of 13 cell death pathways
GSVA29 of the expression matrix was conducted via the R package GSVA. With this analysis, enrichment scores were calculated for each gene set, reflecting the activity of different cell death pathways across samples. Default parameters, such as Gaussian kernel density estimation, were used. A GSVA score matrix for all samples was obtained, with rows representing the 13 cell death pathways and columns representing the samples.
WGCNA
The WGCNA algorithm30 was employed to identify potential key genes across the results. Suitable thresholds were determined to construct weighted adjacency and topological overlap matrices (TOMs). Gene modules were analysed on the basis of their correlation with target phenotypes. Module membership (MM) represented the correlation between genes and module eigengenes, whereas gene significance (GS) reflected the correlation between genes and target phenotypes. The WGCNA results were intersected to identify key genes within coexpression modules for subsequent validation and analysis.
Bayesian pathway enrichment analysis
To infer the pathway interaction network, the CBNplot package31 was used. Sequence data from the 79 SCI mice in the combined dataset were used as a background to calculate interaction directions. The bngeneplot function was utilized to generate graphical representations, with a filtering standard set to 0.90.
ML model construction
To construct diagnostic models, 12 machine learning (ML) algorithms were employed: Lasso, Ridge, StepGLM, XGBoost, random forest (RF), elastic net (Enet), partial least squares regression for generalized linear models (plsRglm), gradient boosting machine (GBM), naive Bayes, linear discriminant analysis (LDA), glmBoost, and support vector machine (SVM). A total of 113 combinations of these algorithms were tested for variable selection and model development using the training dataset (integrated GEO dataset) with tenfold cross-validation. To mitigate overfitting risks, Lasso regularization was applied during feature selection to reduce model complexity by penalizing non-informative variables. The meta-cohort (GSE151371 and GSE45550) served as an external test set to validate model performance. The model achieving the highest average area under the receiver operating characteristic (ROC) curve (AUC) across both the training and test sets was selected as the best-performing model32.
Prediction model construction
A nomogram model was constructed with the R package rms33 using four key predictive genes to evaluate SCI occurrence. The “Total Points” represent the aggregate of the individual points assigned to each predictive variable. Decision curve analysis (DCA) and calibration plots were used to assess the prediction performance of the model. Group comparison plots based on hub gene expression levels were created to further explore the differences between SCI and control samples in the integrated GEO dataset.
Animal study
Healthy female Kunming mice, 7–8 weeks old and weighing 30–35 g, were housed under standard conditions and weres provided by Slike Jingda Laboratory Animal Co. Ltd. All animal procedures were conducted in compliance with the guidelines of the International Association for the Study of Pain (IASP) and were approved by the Ethics Committee of Gannan Medical University (Approval No. 2024 − 228). The SCI group and control group each consisted of 6 mice.
Establishment of the SCI mouse model
Anaesthesia was induced with a 1% sodium pentobarbital solution (40 mg/kg) via intraperitoneal injection. The mice were placed on a heating pad during the procedure. After the skin incision was made, soft tissue at the level of the eighth to twelfth thoracic vertebrae (T8-T10) was separated. A steel clip was inserted under the transverse process, and a laminectomy was performed at T9. The dorsal spinal cord was transected to a depth of approximately 2 mm. Haemostasis was achieved with the use of gelatine sponges. Sham-operated mice underwent the same procedures except for spinal cord transection. Tissue samples for further experiments were collected one week postsurgery from an 8–10 mm segment surrounding the injury site.The BMS locomotor scores and mechanical pain threshold of mice over 7 days are shown in Fig. S2.
RNA isolation and quantitative real-time polymerase chain reaction (qRT–PCR)
RNA was extracted using TRIzol reagent (TransGen Biotech, Beijing, China). OPN cDNA was synthesized from total RNA using the FastQuant RT Kit (TIANGEN, Shanghai, China). Quantitative real-time PCR (qRT-PCR) was carried out with SYBR Premix ExTaq (TaKaRa) following the manufacturer’s instructions. GAPDH served as the internal control, and the primer sequences are provided in Table S3. Relative gene expression levels were calculated via the 2−ΔΔCt method.
Statistical analysis
All data processing and analyses were conducted using R software (version 4.3.0). Unless otherwise specified, the statistical significance of differences between two groups for normally distributed variables was assessed using an independent Student’s t-test. For non-normally distributed variables, the Mann-Whitney U test (Wilcoxon rank-sum test) was applied. Comparisons involving three or more groups were analyzed with the Kruskal-Wallis test. Spearman correlation analysis was performed to calculate correlation coefficients between gene expression levels. Unless otherwise noted, all statistical tests were two-sided, and a p-value < 0.05 was considered statistically significant.
Software and package versions. All computations were performed in R (R Foundation for Statistical Computing, v4.3.0, https://www.r-project.org). Gene-expression matrices were retrieved with GEOquery (Gene Expression Omnibus query, v2.69., (https://bioconductor.org/packages/GEOquery) and batch effects were removed using sva (Surrogate Variable Analysis, v3.46.0, https://bioconductor.org/packages/sva). Differential expression analysis employed limma (Linear Models for Microarray & RNA-seq Data, v3.58.1, https://bioconductor.org/packages/limma); pathway activity scores were calculated with GSVA (Gene Set Variation Analysis, v1.50.0, https://bioconductor.org/packages/GSVA); and weighted gene co-expression networks were built with WGCNA (Weighted Gene Co-expression Network Analysis, v1.72-3, https://cran.r-project.org/package=WGCNA). Unsupervised consensus clustering used ConsensusClusterPlus(v1.72.0, https://bioconductor.org/packages/ConsensusClusterPlus). Machine-learning models were trained with randomForest (Breiman & Cutler Random Forests, v4.7-1.1), glmnet (Lasso / Elastic-Net GLMs, v4.1-9), xgboost (Extreme Gradient Boosting, v1.7.6), gbm (Generalized Boosted Regression Models, v2.1-8), mboost (Model-Based Boosting, v2.9-7), plsRglm (Partial Least Squares for GLM, v1.2.4), e1071 (Miscellaneous Functions of TU Wien, v1.7-16) and MASS (Modern Applied Statistics with S, v7.3-60) - all from CRAN. Receiver-operating-characteristic curves were drawn with pROC (v1.18.5), heatmaps with pheatmap (v1.0.12), circular ideograms with RCircos (v1.2.1) and general graphics with ggplot2 (Grammar of Graphics for R, v3.5.0). Bayesian pathway inference relied on CBNplot (v0.99.7, https://bioconductor.org/packages/CBNplot); nomograms were produced with rms (Regression Modeling Strategies, v6.6-1). Protein-protein interaction networks were queried from STRING (v11.5, https://string-db.org) and visualized in Cytoscape (v3.9.0, https://cytoscape.org). Immune-cell infiltration was estimated with CIBERSORTx (v1.0, http://cibersortx.stanford.edu), and functional enrichment of gene lists was performed on Metascape (v3.5, https://metascape.org).
Results
Technical roadmap
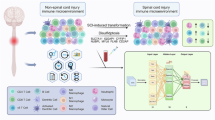
The workflow of this study comprises four steps: (1) data acquisition and batch-effect correction; (2) bioinformatic screening of GR/PCD-related differentially expressed genes; (3) construction of a machine-learning model with external validation; and (4) qRT-PCR validation in a mouse SCI model (Fig. 1).

Technology roadmap. DEGs, Differentially Expressed Genes; GRRGs, Glucocorticoid receptor-Related Genes; GRRDEGs, Glucocorticoid receptor-Related Differentially Expressed Genes; GO, Gene Ontology; GSVA, Gene Set Variation Analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; ROC, Receiver Operating Characteristic; SCI, spinal cord injury; PPI, Protein-protein Interaction; WGCNA, Weighted Gene Co-expression Network Analysis;
GRRDEGs in SCI
Differential expression analysis of the integrated GEO dataset identified 457 DEGs, including 257 upregulated and 200 downregulated genes. A volcano plot was generated to display the results (Fig. 2A). Further analysis of GRRDEGs expression differences in the integrated dataset was performed. The results were visualized using a volcano plot (Fig. 2B) and a heatmap (Fig. 2C), both generated with the R package pheatmap.

Differential gene expression analysis. (A) Volcano plot illustrating the differential expression analysis of SCI and Control samples in the combined GEO datasets. (B) Volcano plot showing the analysis of GRRDEGs in the integrated GEO dataset. (C) Heat map displaying GRRDEGs in the integrated GEO datasets.
PPI network construction and GO/KEGG pathway enrichment analysis
A PPI network of 54 GRRDEGs was constructed via the STRING database (Fig. 3A). The top 20 GRRDEGs, on the basis of the five algorithm scores, were intersected and analysed via a Venn diagram (Fig. 3B). GO and KEGG pathway enrichment analyses were conducted to investigate the BP, CC, MF, and KEGG pathways associated with the 17 GRRDEGs and their relevance to SCI. (Table S4). These genes were associated with functions such as protein kinase activator activity, interleukin-1 receptor binding, kinase regulator activity, and cytokine activity. KEGG pathway analysis indicated that these genes were significantly enriched in the PI3K-Akt, NF-κB, MAPK, IL-17, and TNF signaling pathways. The results of the GO and KEGG enrichment analyses were visualized using bar plots (Fig. 3C) and bubble charts (Fig. 3D).

PPI interaction network and GO/KEGG enrichment analysis. (A) The PPI network of GRRDEGs calculated using the STRING database. (B) Venn diagram of the top 20 GRRDEGs from five algorithms in the CytoHubba plugin. (C–D). Bar plot (C) and bubble plot (D) showing the results of GO and KEGG enrichment analysis of GRRDEGs. Pathway diagram adapted from KEGG (Kyoto Encyclopedia of Genes and Genomes)20,21,22.
Dysregulation of GRRDEGs and immune cell activation in SCI samples
The expression levels of 17 GRRDEGs were compared between control and SCI samples (Fig. 4A), revealing significant upregulation of genes such as Ccl5, Ccnd1, Cebpa, Cebpb, Egr1, Fos, Icam1, Igf1, Il1a, Il1b, IL6, Jun, Myc, Nfe2l2, Ptgs2, Tgfb1, and Tnf in SCI samples (p < 0.05). Next, the expression patterns of these genes between the spinal cord injury group and the control group, as well as their locations on the chromosomes(Fig. 4C), were visualized using a heat map(Fig. 4B). We then constructed a gene coexpression network to explore interactions between these genes (Fig. 4D), with the chord diagram highlighting the key connections, especially among Il1b, Tnf, and Jun. Correlation analysis quantified the relationships between these genes (Fig. 4E), with Ptgs2 and Cebpb showing the strongest positive correlation, whereas Il6 and Igf1 had the strongest negative correlation. These results suggest significant positive and negative correlations among the genes, supporting their synergistic or antagonistic roles in SCI.

Identification of GRRDEGs in spinal cord injury. (A) Box plot showing the expression profiles of 17 GRRDEGs. (B) Heatmap depicting the expression patterns of 17 GRRDEGs between SCI and Control samples. (C) Chromosomal locations of the 17 GRRDEGs. (D) Gene relationship circle diagram for the 17 GRRDEGs, with violet lines indicating positive correlations and green lines indicating negative correlations. (E) Relationships among the 17 GRRDEGs. Significance levels were indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
To further investigate immune system changes in SCI, immune cell infiltration analysis was conducted. The relative proportions of immune cells were visualized via a stacked bar plot (Fig. 5A). Boxplots revealed significant differences in the proportions of infiltrating immune cells between the control and SCI groups (Fig. 5B). Notably, the proportion of M2 macrophages was significantly higher in the SCI group, while M0 macrophages showed a reduced proportion. Correlation analysis of infiltrating immune cell types (Fig. 5C) identified significant relationships, such as a positive correlation between M2 macrophages and T cells CD4 memory resting, and a negative correlation between M0 macrophages and M2 macrophages. Further analysis of the relationships between immune cell proportions and GRRDEG expression (Fig. 5D) showed that Il1b expression had the strongest positive correlation with activated mast cells (p < 0.0001), while Myc expression was negatively correlated with follicular helper T cells (p < 0.0001). These findings highlight the potential immunoregulatory roles of GRRDEGs in SCI.

Combined datasets immune infiltration analysis by CIBERSORT algorithm. (A–B) The proportion of immune cells in theCombined Datasets bar graph (A) and group comparison graph (B). (C) Correlation heatmap showing immune cell infiltration abundance in the combined datasets. (D) Bubble plot illustrating the correlation between GRRDEGs and immune cell infiltration abundance in the combined datasets. * represents p value < 0.05, statistically significant; ** represents p value < 0.01, highly statistically significant; *** represents p value < 0.001 and highly statistically significant. In the proportion bar chart and group comparison chart, violet is SCI samples, and green is Control samples. green is negative correlation, violet is positive correlation, and the depth of color represents the strength of correlation.
Characterization of GR-related clusters in SCI
Clustering analysis of the 17 GRRDEGs expression profiles revealed the most stable and distinct clustering results when k = 2 (Fig. 6A). To determine the optimal k, we analysed changes in the area under the CDF curve (Fig. 6B) for k values from 26, revealing relative stability at k = 2. The consensus index distribution (Fig. 6C) further supported k = 2, as the consensus scores for each subtype were significantly greater than 0.9 at this value (Fig. 6D). PCA was used to divide the 79 SCI samples into two subtypes, Cluster 1 (C1, N = 49) and Cluster 2 (C2, N = 30), with distinct differences in GRRDEG expression levels (Fig. 6E).

Consensus clustering based on GRRDEGs expression matrices. (A) Consensus clustering matrix for k = 2. (B) Relative change in the area under the CDF delta curves. (C) Cumulative distribution function (CDF) for consensus clustering at k = 2–6. (D) Consensus clustering scores. (E) PCA plot of the two clusters, with each scatterplot point representing a sample based on the two principal components (PC1 and PC2) derived from GRRDEG expression profiles.
Immunological and functional pathway differences between clusters
To investigate the molecular features of C1 and C2, key gene expression differences were analysed, and significantly different patterns between the clusters were observed (Fig. 7A). C1 presented elevated expression levels of Tgfb1, Igf1, Cebpa, and Nfe2L2, whereas C2 presented increased expression levels of IL6, Tnf, IL1b, Lcam1, Jun, Fos, Myc, Ptgs2, Cebpb, and Egr1 (Fig. 7B). Immune cell infiltration analysis (Fig. 7C) revealed significant differences between the clusters, with a notable increase in the proportion of infiltrating M1 macrophages in C1. Correlation analysis between GRRDEGs expression and immune cell proportions (Fig. 7D) showed that Fos expression was strongly positively correlated with activated mast cells (cor = 0.814) and significantly negatively correlated with resting dendritic cells (cor = -0.65).

Gene abundances and immune characteristics of GRRDEGs. (A) and (B) display the expression patterns of 17 GRRDEGs across two distinct glucocorticoid receptor-related phenotypes using boxplots and heatmaps, respectively. (C) Boxplot showing the proportions of 22 immune-infiltrated cell types analyzed via the CIBERSORT algorithm. (D) Bubble plot illustrating the correlation between GRRDEGs and immune cell infiltration abundance. Significance levels were indicated by *p < 0.05, **p < 0.01, ***p < 0.001.
Bayesian pathway enrichment analysis of 14 GRRDEGs with differential expression between clusters revealed significant enrichment in immune-related pathways, such as the signalling by interleukins, interleukin-4 (IL-4), and interleukin-13 (IL-13) signalling pathways (Fig. 8A–C).

Bayesian network enrichment analysis. Bar plot (A) and bubble plot (B) showing the results of Bayesian network enrichment analysis of GRRDEGs. (C) Bayesian network inference of the pathway regulatory network of GRRDEGs. Each node represents a pathway; the redder and larger the node, the higher the gene expression value. The thicker the line, the stronger the regulatory effect.
PCD pathway activity and key co-expression modules
To further investigate the activation status of various cell death pathways in SCI, enrichment scores for 13 PCD-related pathways were calculated using GSVA. The results indicated increased activity in eight PCD pathways, including akaliptosis, apoptosis, disulfidptosis, ferroptosis, lysosome-dependent cell death, necroptosis, oxidative stress-induced cell death, and pyroptosis. Conversely, decreased activity was observed in three pathways: autophagy, cuproptosis, and parthatos (Fig. 9A). Seven coexpression modules were identified with the brown and turquoise modules showing a high correlation with the cluster traits (Fig. 9B).

GSVA analysis of PCD pathways and Identification of the critical gene modules. (A) GSVA analysis of 13 PCD pathways displayed as box plots comparing Control and SCI groups. (B) Module-trait correlations, with columns representing traits and rows representing module eigengenes. (C) The correlation between cluter and module eigengenes. Each column matches a trait, and each row corresponds a module eigengene. Each unit cell includes the corresponding p-value and correlation. SCI, Spinal Cord Injury.
With the use of the WGCNA algorithm, a gene coexpression network was constructed to analyse gene expression data from patients with SCI and controls, with a focus on the top 25% most highly variable genes. The soft-threshold power was set to 2, achieving a scale-free network topology with R2 = 0.9 and high average connectivity. Correlation analysis between modules and clinical traits revealed that the brown module, containing 855 genes, had the highest correlation with SCI (cor = 0.58, p = 6e-13) (Fig. S3).
For the GR-related gene clusters, the brown module, containing 996 genes, had the highest correlation with the cluster trait (cor = 0.74, p = 1e-14) (Fig. 9C).
Identification of core genes and development of diagnostic models
The module genes most correlated with SCI and clusters were intersected with the PCD-related brown and turquoise module genes, resulting in 5 and 32 genes, respectively, for a total of 37 genes (Fig. S4). We combined 12 ML algorithms under a tenfold cross-validation framework to develop the most robust diagnostic model on the basis of 37 intersecting genes. The AUC values of different combinations were visualized via a heatmap (Fig. S5), where the best model, Lasso + Stepglm[both]. The volcano plot displayed the included genes, with the six genes from the optimal model specifically annotated. The results showed that Glt8d2 and Pde5a exhibited no differential expression, while Abca1, Cdh1, Glipr1, and Il10ra were upregulated (Fig. 10A), We refer to these four genes as glucocorticoid receptor and PCD-related differentially expressed genes(GRPRDEGs). The ROC curve analysis indicated that Glipr1 had the highest diagnostic value, with an AUC of 0.906, Abca1 followed closely, with an AUC value of 0.8 (Fig. 10B). Finally, qRT-PCR results showed that, compared to the control group, the mRNA expression levels of Abca1, Glipr1, and Il10ra were significantly increased, while Cdh1 expression was decreased. In contrast, no statistically significant differences were observed for Glt8d2 and Pde5a (Fig. 10C).

Diagnostic performance of our model. (A) Volcano plot of the included genes. (B) ROC curves for the included genes. (C) qRT-qPCR validation of GRPRDEG mRNA expression. (D) Nomogram predicting the risk of SCI based on GRPRDEGs (Abca1, Glipr1, Il10ra). (E–G) ROC curves evaluating the prediction model in the combined dataset, GSE151371 dataset, and GSE45550 dataset.
A nomogram model was developed based on the GRPRDEGs (Abca1, Glipr1, and Il10ra) (Fig. 10D). Decision curve analysis (DCA) and calibration curve analysis confirmed the high predictive accuracy of the model (Fig. 6). The model’s performance was validated across both the training and external datasets, achieving AUCs of 0.920 in the combined datasets, 0.858 in GSE151371, and 0.772 in GSE45550 (Fig. 10E–G).
Discussion
SCI is a highly complex and severe neurological condition that is often accompanied by long-term functional impairments and a significant reduction in quality of life. Glucocorticoids have become a common choice for treating acute SCI because of their ability to regulate inflammatory responses, inhibit neuronal necrosis, and increase microcirculation2. However, despite the demonstrated clinical benefits of glucocorticoids, their precise molecular targets in SCI remain unclear. This ambiguity limits their potential in personalized treatment. Binds molecules associated with PCD, identifying molecular targets could lead to the optimization of therapeutic strategies, increase glucocorticoid efficacy, and reduce the risk of unnecessary complications. In future clinical applications, the expression levels of key GRRDEGs such as Abca1, Glipr1 and Il10ra, could serve as biomarkers to predict a patient’s response to glucocorticoid treatment. By measuring these biomarkers in patient blood samples, clinicians may be able to selectively administer glucocorticoids to those most likely to benefit, thereby optimizing treatment and minimizing potential side effects. Case reports and clinical trials would be valuable in further validating these biomarkers and their clinical applicability.
Notably, during rigorous model refinement, we excluded Cdh1 from the predictive signature due to discordant expression trends between RNA-seq and qRT-PCR validation. Post-revision analyses demonstrated preserved diagnostic accuracy in the training set (AUC = 0.920 vs. 0.920 pre-exclusion), while external validations showed modest yet statistically significant reductions in GSE45550 (AUC = 0.711 vs. 0.772) and GSE151371 (AUC = 0.834 vs. 0.858). Importantly, this exclusion enhanced the model’s biological plausibility by ensuring complete concordance between omics and experimental data - a critical prerequisite for clinical translation.
Our findings indicated that GRRDEGs are significantly upregulated in SCI patients compared with controls, suggesting that these genes may play a role in the pathogenesis of SCI. KEGG pathway analysis revealed significant enrichment of GRRDEGs in the PI3K-Akt, NF-κB, MAPK, IL-17, and TNF signalling pathways. A large number of studies have proved that these pathways play an important role in the occurrence and development of SCI34,35,36,37,38,39,40,41,42,43,44,45,46. We identified two distinct GR-related clusters on the basis of GRRDEGs and investigated differences in immune characteristics and BPs among these clusters. Our analysis identified distinct differences in the immune microenvironment between SCI and control samples. The proportion of infiltrating M2 macrophages was significantly increased in the SCI group, whereas the proportion of infiltrating M1 macrophages was notably reduced in C2 compared with C1. Bayesian pathway enrichment analysis of GRRDEGs between the clusters revealed significant enrichment in signalling pathways such as signalling by interleukins, interleukin-4 (IL-4), and interleukin-13 (IL-13), which are key regulators of immune inflammation. Macrophages derived from bone marrow cells play critical roles in initiating, maintaining, and resolving inflammation47. M0 macrophages polarize into two phenotypes: M1 (proinflammatory) and M2 (anti-inflammatory). M1 macrophages increase antigen presentation and secrete IL-12 and IL-2348, whereas M2 macrophages promote cell proliferation, tissue repair, angiogenesis, and phagocytosis49. Studies have shown that the transition from M1 to M2 macrophages offers clinical benefits in neurological diseases. IL-4 induces M2 polarization and reduces neuronal apoptosis, limiting secondary injury after damage50,51.
Next, we used WGCNA to identify the module genes most closely associated with SCI and clusters, and then intersected these genes with PCD-related module genes, ultimately obtaining the intersecting genes associated with all three factors. To address personal bias and inherent preferences, we combined 12 ML algorithms and evaluated their diagnostic performance, selecting the best model to eliminate potential biases. Through the integration of 113 combinations of these algorithms, the ‘Lasso + Stepglm[both]’ model was determined to be the best for gene analysis. We validated the differential expression of these six genes in SCI and control samples via qRT‒PCR. Significant differences in the expression of GRPRDEGs (Abca1, Glipr1 and Il10ra) were observed. And the GRPRDEGs were used to construct a nomogram model. DCA, calibration curve analysis, and ROC curve analysis validated the nomogram’s high reliability and predictive accuracy. The correlation results revealed that Abca1 and Glipr1 had the strongest association with PCD. Abca1 is essential for cholesterol and phospholipid metabolism, maintaining neuronal membrane integrity and synaptic transmission52, and also plays a critical role in lysosomal function; its deficiency may promote lysosome-dependent cell death53. When Abca1 expression is upregulated, it facilitates the efflux of cholesterol and phospholipids, preserves neuronal membrane stability, thereby inhibiting lipid accumulation at the injury site and reducing inflammation and apoptosis54; conversely, decreased expression may exacerbate neuroinflammation and increase the risk of cell death. Glipr1 is closely linked to apoptosis, with its overexpression potentially leading to increased cell death55 and often acting via the p53 pathway56. Upon upregulation, Glipr1 enhances apoptosis and modulates immune responses through a p53-dependent mechanism, aiding in the clearance of damaged cells57; reduced expression may impair apoptotic clearance and prolong inflammatory responses. Elevated Il10ra expression strengthens IL-10 signaling, promotes M2 macrophage polarization, and suppresses the NF-κB pathway, thereby exerting anti-inflammatory effects58, whereas insufficient expression can result in increased pro-inflammatory cytokine levels and heightened inflammation at the lesion site. Finally, reduced Cdh1 protein levels weaken cell-cell adhesion and restrict axonal regeneration59; restoration of Cdh1 expression may support barrier reconstruction and promote functional recovery.
This study has several strengths. We identified two molecular clusters associated with SCI on the basis of key GRRDEGs, thereby revealing distinct biological functions and immune statuses. Unlike previous studies, we conducted a comprehensive analysis, applying CIBERSORT, WGCNA, and 12 ML models to develop a robust model that we validated across test and external datasets. We also constructed a nomogram for clinical application and explored the relationships between hub gene expression levels and the findings provide insights into personalized treatment strategies for glucocorticoids in SCI. However, this study also has limitations. Although the model demonstrated robustness in the training set (mouse-derived data) and human blood samples (GSE151371), its performance declined in rat spinal cord samples (GSE45550), indicating potential species-specific limitations. These discrepancies may stem from heterogeneity in cross-species gene regulatory networks, such as differences in the expression regulation mechanisms of genes like Abca1 and Il10ra across species. Additionally, the tissue source variation between the training set (spinal cord tissue) and validation sets (peripheral blood and rat spinal tissue) could further challenge model generalizability. Future studies should integrate more standardized cross-species and multi-tissue data and optimize model adaptability through approaches like transfer learning or species-specific feature calibration. Furthermore, the lack of extensive clinical validation and multidimensional experimental evaluation remains a limitation that needs to be addressed in future research.
In conclusion, this study clarified the relationship between SCI and GRs, identifying two pathological subgroups with distinct immune profiles and biological processes. The “Lasso + StepGLM[both]” model was identified as the most effective for diagnosing and assessing SCI risk. These findings may improve understanding of the mechanisms underlying glucocorticoid treatment in SCI and could help to guide future work on treatment strategies for SCI patients.
Data availability
The datasets generated and/or analyzed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/.
References
Karsy, M. & Hawryluk, G. Modern medical management of spinal cord injury. Curr. Neurol. Neurosci. Rep. 19(9), 65. https://doi.org/10.1007/s11910-019-0984-1 (2019).
Sterner, R. C. & Sterner, R. M. Immune response following traumatic spinal cord injury: Pathophysiology and therapies. Front. Immunol. 13, 1084101. https://doi.org/10.3389/fimmu.2022.1084101 (2023).
Bracken, M. B. et al. Administration of methylprednisolone for 24 or 48 hours or Tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury: results of the third National acute spinal cord injury randomized controlled trial. National acute spinal cord injury study. JAMA 277(20), 1597–1604. https://doi.org/10.1001/jama.1997.03540440031028 (1997).
Zhu, B. et al. Schwann cell-derived exosomes and methylprednisolone composite patch for spinal cord injury repair. ACS Nano 17(22), 22928–22943. https://doi.org/10.1021/acsnano.3c08046 (2023).
Rabinstein, A. A. Traumatic spinal cord injury. Continuum 24(2, Spinal Cord Disorders), 551–566. https://doi.org/10.1212/CON.0000000000000581 (2018).
Shi, Z. et al. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell. Prolif.. https://doi.org/10.1111/cpr.12992 (2021).
He, W. et al. Targeted therapy of spinal cord injury: Inhibition of apoptosis is a promising therapeutic strategy. Mol. Neurobiol. 61(7), 4222–4239. https://doi.org/10.1007/s12035-023-03814-w (2024).
Li, Y. et al. Impairment of autophagy after spinal cord injury potentiates neuroinflammation and motor function deficit in mice. Theranostics 12(12), 5364–5388. https://doi.org/10.7150/thno.72713 (2022).
Cao, L. & Li, Q. Revealing potential spinal cord injury biomarkers and immune cell infiltration characteristics in mice. Front. Genet. 13, 883810. https://doi.org/10.3389/fgene.2022.883810 (2022).
Li, X., Yang, Y. & Xu, S. Screening biomarkers for spinal cord injury using weighted gene co-expression network analysis and machine learning. Neural Regen. Res. 19(12), 2723–2734. https://doi.org/10.4103/1673-5374.391306 (2024).
Zhang, P., Zhang, J. & Kou, W. Comprehensive analysis of a pyroptosis-related gene signature of clinical and biological values in spinal cord injury. Front. Neurol. 14, 1141939. https://doi.org/10.3389/fneur.2023.1141939 (2023).
DavisS, M. P. et al. GEOquery: A bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23(14), 1846–1847 (2007).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 41(Database issue), D991–D995 (2013).
Stelzer, G. et al. The genecards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinf. 54, 1301–1303 (2016).
UniProt. The universal protein knowledge base in 2021. Nucleic Acids Res. 49(D1), D480–D489. https://doi.org/10.1093/nar/gkaa1100 (2021).
Luo, Y., Liu, L. & Zhang, C. Identification and analysis of diverse cell death patterns in diabetic kidney disease using microarray-based transcriptome profiling and single-nucleus RNA sequencing. Comput. Biol. Med. 169, 107780. https://doi.org/10.1016/j.compbiomed.2023.107780 (2024).
Leek, J. T. et al. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6), 882–883 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Ben Salem, K. & Ben Abdelaziz, A. Principal component analysis (PCA). Tunis Med. 99(4), 383–389 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Szklarczyk, D. et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47(D1), D607–D613 (2019).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
Chin, C. H. et al. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 (2014).
Yang, X. et al. Study on the multitarget mechanism and key active ingredients of herba siegesbeckiae and volatile oil against rheumatoid arthritis based on network Pharmacology. Evid. Based Complement. Altern. Med. 2019, p8957245 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
Mak, T. K. et al. The cancer-associated fibroblast-related signature predicts prognosis and indicates immune microenvironment infiltration in gastric cancer. Front. Immunol. 13, 951214. https://doi.org/10.3389/fimmu.2022.951214 (2022).
Xu, M. et al. Identification and validation of immune and oxidative stress-related diagnostic markers for diabetic nephropathy by WGCNA and machine learning. Front. Immunol. 14, 1084531. https://doi.org/10.3389/fimmu.2023.1084531 (2023).
Sato, N., Tamada, Y., Yu, G. & Okuno, Y. CBNplot: Bayesian network plots for enrichment analysis. Bioinformatics 38(10), 2959–2960. https://doi.org/10.1093/bioinformatics/btac175 (2022).
Chen, B. et al. An integrated machine learning framework for developing and validating a diagnostic model of major depressive disorder based on interstitial cystitis-related genes. J. Affect. Disord. 359, 22–32. https://doi.org/10.1016/j.jad.2024.05.061 (2024).
Chen, X. et al. Neurotrophic effects of serum- and glucocorticoid-inducible kinase on adult murine mesencephalic dopamine neurons. J. Neurosci. 32(33), 11299–11308. https://doi.org/10.1523/JNEUROSCI.5910-11.2012 (2012).
Li, S. et al. EDNRB reverses methylprednisolone-mediated decrease in neural progenitor cell viability via regulating PI3K/Akt pathway and LncRNA expression. J. Mol. Neurosci. 70(3), 403–412. https://doi.org/10.1007/s12031-019-01427-4 (2020).
Cox, S. N. et al. European IgAN consortium. Multiple rare genetic variants co-segregating with Familial IgA nephropathy all act within a single immune-related network. J. Intern. Med. 281(2), 189–205. https://doi.org/10.1111/joim.12565 (2017).
Srinivasan, M. & Walker, C. Circadian clock, glucocorticoids and NF-κB signaling in neuroinflammation—Implicating glucocorticoid induced leucine zipper as a molecular link. ASN Neuro 14, 17590914221120190. https://doi.org/10.1177/17590914221120190 (2022 Jan-Dec).
Shao, J. et al. Glucocorticoid receptor inhibits the activity of NF-κB through p38 signaling pathway in spinal cord in the spared nerve injury rats. Life Sci. 208, 268–275. https://doi.org/10.1016/j.lfs.2018.07.026 (2018).
Xue, R. et al. Mesenchymal stem cell-derived exosomes promote recovery of the facial nerve injury through regulating macrophage M1 and M2 polarization by targeting the P38 MAPK/NF-κB pathway. Aging Dis. 15(2), 851–868. https://doi.org/10.14336/AD.2023.0719-1 (2024).
Zhang, J. et al. Inhibition of JNK ameliorates depressive-like behaviors and reduces the activation of pro-inflammatory cytokines and the phosphorylation of glucocorticoid receptors at Serine 246 induced by neuroinflammation. Psychoneuroendocrinology 113, 104580. https://doi.org/10.1016/j.psyneuen.2019.104580 (2020).
Yang, Y. et al. Sex-biased transgenerational transmission of betaine-induced epigenetic modifications in glucocorticoid receptor gene and its downstream BDNF/ERK pathway in rat hippocampus. Nutr. Neurosci. 25(4), 746–757. https://doi.org/10.1080/1028415X.2020.1807711 (2022).
Munoz, A. M. et al. Dexamethasone potentiates chimeric antigen receptor T cell persistence and function by enhancing IL-7Rα expression. Mol. Ther. 32(2), 527–539. https://doi.org/10.1016/j.ymthe.2023.12.017 (2024).
Wang, Y. et al. A TNF-α blocking peptide that reduces NF-κB and MAPK activity for attenuating inflammation. Bioorg. Med. Chem. 92, 117420. https://doi.org/10.1016/j.bmc.2023.117420 (2023).
McPherson, M. J. et al. An anti-TNF-glucocorticoid receptor modulator antibody-drug conjugate is efficacious against immune-mediated inflammatory diseases. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.add8936 (2024).
Fan, H. et al. Inhibiting HMGB1-RAGE axis prevents pro-inflammatory macrophages/microglia polarization and affords neuroprotection after spinal cord injury. J. Neuroinflamm. 17(1), 295. https://doi.org/10.1186/s12974-020-01973-4 (2020).
Ren, J. et al. Schwann cell-derived exosomes containing MFG-E8 modify macrophage/microglial polarization for attenuating inflammation via the SOCS3/STAT3 pathway after spinal cord injury. Cell. Death Dis. 14(1), 70. https://doi.org/10.1038/s41419-023-05607-4 (2023).
Wang, C. et al. Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J. Cell. Mol. Med. 22(2), 1148–1166. https://doi.org/10.1111/jcmm.13368 (2018).
Francos-Quijorna, I. et al. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia 64(12), 2079–2092. https://doi.org/10.1002/glia.23041 (2016).
Xin, N. et al. Neuroinduction and neuroprotection co-enhanced spinal cord injury repair based on IL-4@ZIF-8-loaded hyaluronan–collagen hydrogels with nano-aligned and viscoelastic cues. J. Mater. Chem. B 10(33), 6315–6327. https://doi.org/10.1039/d2tb01111e (2022).
Liu, Z. et al. Microarray profiling and co-expression network analysis of circulating LncRNAs and mRNAs associated with major depressive disorder. PloS One 9(3) (2014).
Van Broeckhoven, J. et al. Macrophage-based delivery of interleukin-13 improves functional and histopathological outcomes following spinal cord injury. J. Neuroinflamm. 19(1), 102. https://doi.org/10.1186/s12974-022-02458-2 (2022).
Karasinska, J. M. et al. ABCA1 influences neuroinflammation and neuronal death. Neurobiol. Dis. 54, 445–455. https://doi.org/10.1016/j.nbd.2013.01.018 (2013).
Ren, C. et al. Identification and characterization of RTVP1/GLIPR1-like genes, a novel p53 target gene cluster. Genomics 88(2), 163–172. https://doi.org/10.1016/j.ygeno.2006.03.021 (2006).
Scheuring, U. J. et al. GliPR1 knockdown by RNA interference exerts anti-glioma effects in vitro and in vivo. J. Neurooncol. 153(1), 23–32. https://doi.org/10.1007/s11060-021-03737-3 (2021).
Yan, L. et al. TPX2-p53-GLIPR1 regulatory circuitry in cell proliferation, invasion, and tumor growth of bladder cancer. J. Cell. Biochem. 119(2), 1791–1803 (2018).
Zhang, J. et al. ABCA1 deficiency-mediated glomerular cholesterol accumulation exacerbates glomerular endothelial injury and dysfunction in diabetic kidney disease. Metabolism 139, 155377. https://doi.org/10.1016/j.metabol.2022.155377 (2023).
Yale, A. R. et al. Regulation of neural stem cell differentiation and brain development by MGAT5-mediated N-glycosylation. Stem Cell. Rep. 18(6), 1340–1354 (2023).
Karanika, S. et al. GLIPR1-deltaTM synergizes with docetaxel in cell death and suppresses resistance to docetaxel in prostate cancer cells. Mol. Cancer 14, 122 (2015).
Lairson, L. L. et al. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555 (2008).
Li, K. et al. IL-10 negatively controls the primary T cell response of Tilapia by triggering the JAK1/STAT3/SOCS3 axis that suppresses NF-kappaB and MAPK/ERK signaling. J. Immunol. 210(3), 229–244. https://doi.org/10.4049/jimmunol.2200335 (2023).
Chen, Z. et al. A Cdh1-FoxM1-Apc axis controls muscle development and regeneration. Cell. Death Dis. 11(3), 180. https://doi.org/10.1038/s41419-020-2375-6 (2020).
Funding
This work was supported by the Natural Science Foundation of Jiangxi Province, China [grant number 20232ACB206050].
Author information
Authors and Affiliations
Contributions
Concept: S.C.; Design: W.L.; Definition of intellectual content: L.W.; Literature search: Y.L. and Z.C.; Experimental studies: F.L. and Z.C.; Data acquisition: F.L. and Y.L.; Data analysis: F.L.; Statistical analysis: F.L.; Manuscript preparation: L.W.; Manuscript editing: F.H.; Manuscript review: M.Z. and L.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All procedures were conducted in accordance with the guidelines of the International Association for the Study of Pain (IASP) and were approved by the Ethics Committee of Gannan Medical University (2024-228). We confirm that this study is reported in accordance with the ARRIVE guidelines and adheres to the relevant ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lu, F., Liu, Y., Chen, Z. et al. Identification and validation of glucocorticoid receptor and programmed cell death-related genes in spinal cord injury using machine learning. Sci Rep 15, 24202 (2025). https://doi.org/10.1038/s41598-025-08060-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08060-9
