
Abstract
Single-cell RNA sequencing (scRNA-seq) is a powerful technology used to investigate cellular heterogeneity. When applied to unicellular eukaryotes such as Plasmodium, scRNA-seq provides a single-cell resolution valuable to study complex infections comprised of mixed lifecycle stages and clones. Until now, the application of scRNA-seq has been mainly limited to in vitro and animal malaria models due to the challenges of working with Plasmodium natural infections in endemic settings. We validated sample preparation methods and a single-cell RNA preservation and sequencing technology in P. knowlesi for the future implementation in low-resource settings. We recovered 22,345 P. knowlesi single-cell transcriptomes containing all asexual blood stages from 6 samples to generate the most extensive P. knowlesi single-cell dataset to date. All 6 samples produced reproducible circular UMAP projections with consistent cluster localization and high gene expression correlation, regardless of the sample preparation method. Biomarker expression and life stage annotation using the Malaria Cell Atlas P. knowlesi reference dataset further confirmed these results. In conclusion, adaptable sample preparation methods combined with novel scRNA-seq preservation technology has the potential to unlock scRNA-seq for field studies which will lead to additional insights into Plasmodium biology and fundamentally transform how we study malaria natural infections.
Similar content being viewed by others


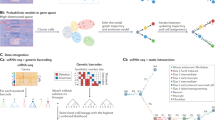
Introduction
Single-cell RNA sequencing (scRNA-seq) offers a powerful approach to unravel biological complexity of unicellular eukaryotes, as it provides unprecedented resolution into cellular heterogeneity1. This is particularly useful when investigating the expression profiles of single-celled organisms with complex life cycles and mixed life stages or mixed clones in parasite populations.
The application of scRNA-seq to Plasmodium parasites has been insightful to reconstruct cell trajectories and reveal transcriptional profiles associated with different stages in the intraerythrocytic developmental cycle across species2,3,4,5,6,7,8,9,10,11,12,13,14. Studies focusing on P. falciparum4,5,6,7,8,9 and P. berghei blood stages10,11 have begun to unravel gene expression signatures of sexual commitment and differentiation. Additional studies have described gene expression profiles of Plasmodium sporozoite development and liver stages in P. berghei2,15,16,17,18, P. falciparum19,20, and P. vivax13,14,21,22,23. This effort has primarily been led by the Malaria Cell Atlas (MCA) which has the most comprehensive resource of publicly available Plasmodium single-cell data2,3,4,10,19,24.
Despite the vast potential of scRNA-seq in malaria research, its application has been largely restricted to parasites from in vitro cultures or animal models, with only one study analyzing four Malian natural infections being published to date4. While laboratory strains are essential for studying Plasmodium biology, they are not necessarily representative of circulating parasite populations. Genome-wide transcriptomic studies have demonstrated inherent differences in gene expression between culture-adapted strains and natural infections25,26,27,28. Moreover, natural infections are often complex with individuals frequently carrying multiple clones29,30,31,32,33. Different clones within a single host can vary in their drug resistance, virulence, immune evasion or invasion strategies, providing insights into how parasites interact, compete, and evolve during an infection. The Dogga et al. scRNA-seq study, that analyzed four Malian natural infections, revealed cell clusters with significantly lower gene expression levels as compared to laboratory strains, as well as differential expression between clones within the same host in genes related to host-parasite interactions4. The capability to identify and characterize intra-host diversity using scRNA-seq will advance our understanding of the complexity of natural human infections and uncover critical aspects of gene expression in Plasmodium biology34. For these reasons, it is important to validate the application of technologies that extend single-cell transcriptomic studies to low-resource settings.
The challenges associated with applying omics techniques to study Plasmodium natural infections in humans, including the abundance of human genetic material, low parasite densities, and limitations of laboratory equipment at patient recruitment sites in low-resource endemic settings, extend beyond single-cell technologies and are also commonly shared with bulk omics approaches. A combination of leukocyte depletion and parasite enrichment methods are indispensable to remove human genetic material and increase the concentration of parasites prior to RNA preservation35,36,37,38,39,40,41,42. An additional challenge for scRNA-seq of human natural infections is the need to preserve cell integrity, which precludes the use of RNA preservation buffers which lyse cells such as Trizol (Invitrogen) or RNAProtect (Qiagen). Furthermore, methods for scRNA-seq of natural infections need to be suited for transportation between field and laboratory sites, as sample collection and processing for scRNA-seq often occur at different locations.
Traditionally, the two main single-cell technologies used for Plasmodium are droplet- and well-based technologies. While these two technologies differ in their cell isolation methodology and sequencing resolution43, they both require fresh cells and costly instruments in addition to equipment such as ultra-low temperature freezers, which are not often available in remote health centers. Recently, a new well-based technology has been developed with the advantage that it is instrument-free and provides integrated RNA preservation, making it compatible with sample collection in low-resource settings. HIVEs (Honeycomb Biotechnologies) are small devices that contain a microscopic well (pico-well) system designed to mimic a honeycomb which captures and preserves up to 60,000 single cells for the CLX version. Each pico-well contains a uniquely barcoded bead that captures mRNA using a poly-dT tail, similar to droplet sequencing. RNA preservation buffer is added to the HIVE before freezing to ensure RNA integrity and allowing the HIVEs to be stored for up to 9 months as per manufacturer’s instructions. Once frozen, HIVEs can be shipped on dry ice to a central laboratory for processing and library preparation, making it ideal for studies involving multiple collection sites or time points. During processing, HIVEs undergo bead collection, transcriptome recovery and amplification, and library preparation to create libraries that can be sequenced using standard short-read sequencing approaches.
In this study, we applied HIVE CLX single-cell sequencing technology for the first time to Plasmodium parasites and validated different parasite enrichment methods to make it deployable for the collection of cells from natural infections in low-resource endemic settings. Standardized parasite enrichment protocols combined with preservation and scRNA-seq technologies have the potential to revolutionize Plasmodium research by enabling scRNA-seq of natural infections.
Results
Processing Plasmodium natural infection samples to recover all intraerythrocytic life stages
To validate the application of HIVE technology for the analysis of Plasmodium natural infections, we tested different sample processing and loading methods, and assessed the single-cell RNA sequencing results. To create a mock natural infection sample with mixed parasite life stages, we diluted in vitro-cultured P. knowlesi A1-H.1 parasites to approximately 1% parasitemia in whole blood.
Tested methods for sample processing include two combinations of leukodepletion and parasite enrichment techniques tailored to optimally isolate all life stages present in blood circulation and eliminate uninfected human cells from natural infections, which would otherwise dominate sequencing reads. A schematic representation of the protocols of the 6 samples is provided in Fig. 1. For the PNyco method, which only requires a tube centrifuge, we used a Plasmodipur filter to remove human leukocytes, followed by a Nycodenz density gradient to enrich for parasite life stages. Here we found an enrichment to 16% parasitemia after the Nycodenz gradient by microscopy (Table S1). In the second method, MACSPS, we used a MAgnetic Cell Separation (MACS) column to enrich for trophozoite and schizont life stages in the magnetic column based on the paramagnetic properties of hemozoin, similar to the MCA method2. The first elution contains leukocytes, uninfected RBCs, and ring stages which have passed through without binding to the magnetic column. Subsequently, the first elution underwent further processing using a Plasmodipur filter to remove leukocytes, followed by a saponin lysis to remove uninfected RBCs, resulting in the isolation of the ring stage. In the second elution, the trophozoite and schizont stages bound to the magnetic column were released and recovered. The ring stages were then combined with trophozoites and schizonts recovered to obtain an enriched sample containing all life stages. For the HIVE loading, we evaluated two different methods for loading enriched parasite into the HIVEs: one utilizing a plate centrifuge to facilitate the sinking of parasites into the pico-wells, and the other allowing parasites to sink by gravity, eliminating the need for a plate centrifuge if unavailable (C and G, respectively, Fig. 1).

Experimental setup to test methods for processing Plasmodium natural infections and in vitro cultures. A mock natural infection sample was prepared by mixing P. knowlesi A1-H.1 in vitro culture with human whole blood. Two combinations of leukodepletion and parasite enrichment methods were tested (PNyco and MACSPS). In addition, we processed an in vitro-cultured P. knowlesi sample, with two different density gradients: Nycodenz (PkCultureN) and Percoll (PkCultureP). For each sample 60,000 cells were loaded into a HIVE by gravity (G) or centrifuge (C). Figure created using BioRender.com.
In addition, we also tested cultured P. knowlesi samples without mixing with whole blood. Parasite enrichment was conducted using two different density gradient centrifugation methods. Half of the cultured parasites were purified using a Percoll gradient (PkCultureP), typically employed to enrich samples for schizonts stages42, as evidenced by 66% of the recovered parasites being microscopically identified as schizonts despite schizonts only comprising 10% of the parasites in the original culture (Table S1). The other half underwent enrichment using a Nycodenz gradient (PkCultureN), commonly used for P. knowlesi35, which recovered a broader range of parasite life stages comparable to the original culture composition. Both methods led to a similar level of enrichment, (52% with Percoll vs. 45% with Nycodenz) with a similar loss of the ring stages (Table S1) and both samples were loaded using centrifugation.
Validation of single-cell RNA sequencing using HIVE technology
A total number of 22,345 P. knowlesi parasite cells were recovered across 6 samples after applying a conservative filtering approach with thresholds of 300 genes and 600 transcripts per cell and doublet removal. Doublets, or pico-wells which contained multiple cells, were identified in our dataset using Scrublet. The number of identified doublets fell within the expected range based on the number of cells loaded and recovered. All identified doublets were removed along with cluster 2 in its entirety as a majority of cells in this cluster were identified as doublets (Fig. S1). More details about the doublet detection and removal strategy applied to recover high-quality single cells are described in the methods and supplementary material (Fig. S1 and Table S2). Cell recovery per sample ranges from 2,291 to 7,532 cells depending on enrichment and loading method (Fig. 2) with a mean of 678 genes and 1,375 transcripts per cell (Fig. S2 and Table S3).

2D UMAP projections per sample by de novo clustering after doublet removal. Six clusters with comparable locations are observed in a ring formation across all sample UMAPs, with one cluster (cluster 2) being excluded from analysis during doublet removal.
We explored the single-cell transcriptome data of the 6 samples loaded in HIVEs using uniform manifold approximation and projection (UMAP), which shows the cells in a circular pattern reproducible across the UMAPs of all samples and thus, processing methods (Fig. 2). Moreover, de novo Louvain clustering on the PCA of the cell transcriptomes resulted in 7 clusters (cluster 2 was excluded during doublet removal), with consistent locations and partitioning of the clusters, despite differences in the number of cells recovered per cluster across the 6 samples (Table S4). For example, as compared to the other samples, cells in cluster 5 are less abundant in PkCultureN, PkCultureP, and in the mock natural infection sample PNycoC (percentages lower than 2%), while cells in cluster 6 are most abundant in the PkCultureP sample processed with the Percoll density gradient (31%) as compared to the other samples (percentages lower than 10%).
Next, we investigated the transcriptomic variability using pairwise correlation plots, following a pseudo-bulk approach as described in the materials and methods section. We calculated the correlation of the gene expression values between different samples after summing per gene the number of reads over all cells (Fig. 3). All samples show highly correlated gene expression profiles with Pearson’s correlation coefficients (R) greater than 0.88 (p < 0.001) in all pairwise comparisons, despite the use of the different sample types and processing methods. The strong correlation in expression profiles between the four mock natural infection samples (0.97) demonstrates that enrichment and loading methods did not affect the single-cell data generated. Correlation values of PkCultureN with mock samples ranges between 0.95 and 0.98; while correlation values of PkCultureP with mock samples is lower (0.88 and 0.90) likely reflective of the overall differences in cell abundance between clusters 5 and 6.

Pairwise correlation matrix of pseudo-bulk gene expression levels between sample processing and loading methods. Pearson correlation coefficients (R) and associated p-values are shown in red.
To examine whether HIVE sequencing technology is sensitive enough to detect gene expression at the single-cell level to study genes crucial for biological processes, we superimposed the expression levels of genes with known expression patterns to validate expression specificity (Fig. 4). Tubulin (pktubulin, PKNH_0807700), a housekeeping gene which is consistently expressed throughout the lifecycle, is identified across all clusters in the UMAP44,45. The genes merozoite surface protein 8, msp8 (PKNH_1031500), and msp1 (PKNH_0728900) were used as markers of early and late blood stage asexual parasites, respectively2. In the UMAP, the expression of msp8 is highly enriched in cluster 5 and decreases in cluster 0 in contrast to expression of msp1, which begins in cluster 0 and increases clockwise with the highest level of expression in cluster 6.

2D UMAP projections superimposed with gene expression levels (scaled and normalized counts) of marker genes show high and specific expression. First row (left to right): UMAP of all cells recovered with the de novo clustering annotation, pktubulin housekeeping gene with constant expression over the clusters, early (msp8) and late (msp1) asexual stage marker expression; Middle row (left to right): expression of genes involved in invasion (PkDBPα, PkDBPβ, DBPγ and PkNBPXa); Last row (left to right): PkNBPXb gene involved in invasion and gametocyte markers (LAP5, mget, Pks25).
Genes involved in erythrocyte invasion, Duffy Binding Protein (DBP) genes alpha, beta, and gamma (DBPα, PKNH_0623500; DBPβ, PKNH_1400800; DBPγ, PKNH_1356900) and the homologues of the P. vivax Reticulocyte Binding Protein (NBPXa, PKNH_1472300 and NBPXb, PKNH_0700200), were projected due to their high-specific expression in the late schizont stage27. These invasion genes, with the exception of DBPβ, are highly enriched in cluster 6. P. knowlesi DBPα is a known ligand of DARC in human RBCs46 and NBPXa has been identified as essential in human RBC invasion47, while the roles of DBPβ, DBPγ, and NBPXb remain unknown in human RBCs48. We then examined the expression of known gametocyte marker genes, LAP5 (PKNH_1230500), mget (PKNH_1211100), and ookinete surface protein Pks25 (PKNH_0615700)12,49. As the P. knowlesi A1-H.1 strain lost its ability to develop gametocytes when it was adapted to grow in vitro in human RBCs35, we did not identify gametocyte gene markers in the UMAP, as expected.
Additionally, we used pseudotime analysis to further explore the gene expression of the de novo clustering and predict the developmental trajectory of the cells. Based on the mapping of life-stage specific marker genes msp8 and msp1 (Fig. 4), clusters 5 and 6 were predefined as the start and end points of the pseudotime trajectory, respectively, which resulted in a single smooth trajectory without any branching (Fig. 5A), indicating a single pathway of development. The circular-shape of the UMAP captures the cyclical nature of the asexual erythrocytic developmental trajectory as it maps the progression of gene expression and provides directionality, beginning after invasion in the ring stage, through the trophozoite stage, and ending in the schizont stage.

UMAP and gene expression through the developmental trajectory. (A) UMAP of the predicted developmental trajectory using pseudotime with the start and end points predefined as clusters 5 and 6, respectively. (B) Heatmap showing the wave of gene expression (scaled and normalized counts) over the developmental trajectory by mapping the expression of the top 250 most variable genes (y-axis) by cell ordered by pseudotime and including the cluster annotation of each cell (x-axis).
The heatmap shows the top 250 genes (y-axis, Fig. 5B) with the highest variable expression over the pseudotime corresponding with the identified de novo clustering (x-axis). The continuous biological transition is reflected by the wave-like changes in gene expression, which is in agreement with previous bulk and scRNA-seq studies12,27,50, demonstrating that HIVE scRNA-seq can be used to study and map the dynamics of gene expression across the erythrocytic life cycle at the level of individual parasites.
Life stage annotation and composition by enrichment method
Cells were assigned to the ring, trophozoite, or schizont life stages based on the scmap algorithm and the P. knowlesi MCA reference dataset2. For each cell in our dataset, the scmap algorithm transfers the life stage annotation from the cell with the closest matching transcriptome in the MCA reference dataset. The annotated UMAP shows the recovery of all parasite life stages (Fig. 6A) and that the location of the life stages corresponds well with the marker gene expression results in Fig. 4. In addition, all life stages are recovered in all samples (Fig. 6B), further confirming the reproducibility of the methods used. Out of the 22,345 cells in our dataset, only 2 cells could not be assigned (NA) to a specific life stage.

Life stage annotation of HIVE scRNA-seq dataset and life stage composition (A) 2D UMAP of the HIVE scRNA-seq dataset (22,345 cells) with the life stage annotation. Cells were assigned to ring (early/late), trophozoite (early/mid/late), and schizont (early/mid/late) life stages, using scmap and the P. knowlesi reference dataset. Only 2 cells could not be assigned a life stage (NA). (B) 2D UMAPs showing the life stage annotation per sample. (C) Life stage composition recovered per sample as a percentage of the total number of cells recovered per method.
Subsequently, we compared cell and life stage recovery between sample processing and HIVE loading methods. Between the mock natural infection samples, the life stage composition remained remarkably stable between the processing (MACSPS vs. PNyco) and loading methods (centrifuge vs. gravity), although the number of cells recovered differed significantly (Fig. 6C; Table 1). Mock natural infection samples loaded by gravity had a similar recovery of cells (2,303 cells vs. 2,291, p > 0.05) and life stage composition (8% vs. 10% rings, 72% vs. 71% trophozoites, 20% schizonts, p > 0.05) regardless of processing method, MACSPS or PNyco, respectively (Table 1). Loading by gravity also showed a higher percentage recovery of ring stages as compared to loading by centrifuge (8% and 10% by gravity with MACSPS and PNyco vs. 4% and 1% when loaded by centrifuge, respectively, both p < 0.001); however, the total number of ring stage cells recovered by centrifuge is almost doubled (299 cells vs. 176 by gravity with MACSPS), which is advantageous for the analysis.
When looking at overall cell recovery, the MACSPSC method loaded by centrifuge recovered a 3-fold higher number of cells (7,532 cells vs. 2,303 by gravity) and also the most cells of each life stage: ring, trophozoite, and schizont. The same comparison could not be made for the PNyco method due to an unusually high percentage of doublets in this sample. However, our analysis shows that, after doublet removal, the remaining transcriptomes are of high quality, with consistent life stage composition and clustering patterns comparable to other samples. This suggests that such datasets can still be included in the analysis. While we cannot definitively determine the reason for the high percentage of doublets, a likely explanation is the formation of an air bubble during the HIVE loading procedure. This highlights the importance of ensuring complete filling of the HIVE, even if it requires adjusting the input volume beyond what is specified in the protocol.
In the PkCulture samples, The Nycodenz gradient (PkCultureN) recovered a significantly higher number of total cells (4,096 vs. 2,873 cells, p < 0.001) with a more even distribution over the developmental cycle than the Percoll gradient (PkCultureP). On the other hand, a Percoll gradient is highly efficient in enriching the sample for the schizont stage, with schizonts comprising 53% of the life stages recovered versus 27% with a Nycodenz gradient (p < 0.001).
Across all samples, a smaller proportion of ring stages was recovered as compared to the percentage of rings present in the starting cultures (Table 1 and Fig. S3). Before sample processing, the mock natural infection contained 38% rings, while our analysis identified 1–10% of the cells recovered as ring stages. Similarly, for the PkCulture samples, only 1 to 2% percent of cells recovered were rings, as opposed to the culture which contained 17% rings. This loss can mostly be attributed to the difficulty of separating ring-infected RBCs from non-infected RBCs during sample processing, especially when using density gradients. To investigate a potential alternative cause for the low recovery of rings, we created barcode rank plots, where cells were ordered and plotted based on the number of reads per cell (from high to low) for all cells and per life stage (Fig. 7). We found differences between the average number of reads per cell per life stage, with rings always containing the fewest reads per cell, while trophozoites had the highest number of reads in all but one sample (PkCultureP). Similar trends were also seen when mapping the number of reads per cell with cells ordered by pseudotime (Fig. S5). If we lower the filtering thresholds to 100 genes and 250 transcripts per cell, we increase the recovery of rings from 866 to 1,869 cells (Table S5), which indicates that when conservative filtering thresholds of 300 genes and 600 transcripts per cell are applied uniformly across the life stages, we are potentially filtering out rings that were successfully sequenced. When we repeated our analysis pipeline using the lower thresholds of 100 genes and 250 transcripts per cell, we found concordant UMAPs and life stage compositions, indicating that stage-flexible filtering thresholds could allow us to better refine the analysis to address targeted biological questions (Fig. S4, Table S5).

Barcode Rank Plots. For each of the six samples, the log10 value of the number of reads per cell is plotted versus the rank order (log10) when sorting them based on the number of reads per cell from high to low. This analysis is performed for all cells combined as well as for each life stage separately. The x-axis shows ranked barcodes in log10 order, while the y-axis represents reads per cell on a log10 scale.
Discussion
Our study provides the first application and validation of HIVE CLX scRNA-seq technology to unicellular organisms and demonstrates the quality of the single-cell dataset generated by showing high reproducibility between samples and benchmarking our results with the publicly available MCA dataset. HIVE technology is unique in its potential to collect natural infection samples in low-resource settings as it is instrument-free and integrates RNA preservation, which allows sample collection to be separated from library preparation and sequencing.
We tested different sample processing and enrichment methods that can be used to isolate parasites from natural infections and cultured parasites as input for scRNA-seq using the HIVE technology. The methods tested successfully recovered all asexual erythrocytic life stages present with slight differences in life stage recovery between the methods. These differences can be leveraged to study different aspects of parasite biology. While erythrocytic sexual stages were not recovered since the P. knowlesi A1-H.1 strain lost its ability to develop gametocytes, we expect the MACS and density gradient protocols to also be suitable. Efficient gametocyte enrichment protocols for MACS columns and Percoll gradients for P. falciparum and P. vivax gametocytes have previously been published51,52, and are consistent with our own observations. For the cultured samples, a Percoll density gradient enriches more prominently for the schizont life stage, while a Nycodenz gradient performs a more balanced enrichment across the trophozoite and schizont stages. Therefore, Percoll can be best used to study egress and invasion, while Nycodenz is better suited to study the developmental trajectory. Additionally, both Percoll and Nycodenz density gradient percentages can be adjusted to enrich for different species and life stages35,41. The ring stage is notoriously difficult to enrich from natural infections due to their similarity in morphology to uninfected RBC and a lack of hemozoin53. During parasite maturation inside the RBC, parasites digest hemoglobin, which lowers the density of the infected RBC (iRBC) and increases hemozoin formation (the result of heme detoxification)54,55,56. These changes in the iRBC enable the separation of trophozoite and schizont stages from uninfected RBCs using a density gradient or based on their paramagnetic hemozoin with a MACS column37,38,39. However, newly infected iRBCs (ring stage) have not yet consumed enough hemoglobin to alter the density of the RBC nor produced enough hemozoin to be captured by a MACS column.
The MACSPS method, which combines a MACS column with Plasmodipur leukodepletion and saponin RBC lysis, was designed to maximize parasite enrichment (particularly of the ring stage) and remove uninfected RBCs from natural infection samples2. When using this method and loading HIVEs by gravity, 8% of the total cells recovered were ring stages. This was highly comparable to the 10% rings stages obtained using the PNycoG method, but lower than expected after ring enrichment. Furthermore, when loading the MACSPS method by centrifuge, only 4% of the cells recovered were rings, despite the absolute number of ring cells recovered being twice as high. This discrepancy is likely due to the higher percentage of the less-dense late trophozoite and schizont stages settling into the wells with the help of the force from the centrifuge, thereby making the rings a smaller percentage of the total number of cells recovered.
Since all loading methods produce high-quality and comparable cell transcriptomes, loading by gravity initially seems ideal as it results in the highest percentage of ring recovery and may be the only option in some settings. However, the power of single-cell analysis relies on the number of cells, and thus to truly maximize cell recovery we recommend loading HIVEs using a centrifuge when available. Nevertheless, the high correlation between methods provides optimal versatility allowing for methods to be readily adapted to equipment availability. For example, a study with multiple collection sites could choose different processing and loading methods for each site and still analyze the samples together.
Additional preservation methods were not tested during our experiment due to the suitability of the integrated preservation in the HIVE CLX capture kit. RNALater (Invitrogen) which does not lyse RBCs, has previously been used in two scRNA-seq studies2,3. In cases where processing cannot begin in a timely manner, RNAlater or other preservation solutions that maintain cell integrity could be explored.
We generated, after filtering, scRNA-seq data of 22,345 P. knowlesi cells, more than five times the number of P. knowlesi cells published to date. Our de novo clustering reveals consistent clustering, with the same number of clusters in the same locations in all samples, which corresponds well with expected specific, localized biomarker expression and life stage annotation from the MCA. There is a clear shift from the egress of the schizont stage to the ring stage after invasion and continuing through the developmental trajectory, indicating biological significance of the clustering. While some overlap is seen in the life stage annotation, this is due to the use of an external reference to map life stages. Additionally, we show that there are no artifacts introduced into our dataset from the sample processing methods or preservation. The six samples included in the study were analyzed without the use of data integration methods (e.g. the anchor-based integration in Seurat57 or Harmony58) often needed to remove batch effects between samples, demonstrating the high reproducibility of our methods, even when the PkCulture samples and mock natural infection samples were loaded at different times.
The cell loading capacity of the HIVE CLX is particularly important when working with samples containing mixed populations, such as Plasmodium, where mixed iRBC and RBC populations are unavoidable despite the implementation of enrichment techniques. From the 60,000 cells loaded per HIVE, we recovered up to 7,500 cells when loading by centrifuge, representing 45% of the expected cell recovery reported by Honeycomb Biotechnologies. To capitalize on the loading capacity, Honeycomb Biotechnologies has recently made cell hashing compatible with HIVE (HIVE CALL). This new possibility opens the option of adding multiple samples to one HIVE, hereby reducing cost per sample, as long as RBC integrity is maintained during sample preparation. Future studies should also sequence at a deeper level to maximize cell recovery for natural infection samples, ensuring a minimal number of 25,000 reads per cell for each sample, compared to the average depth of 16,925 reads per cell in this study.
To ensure that only high-quality cells were analyzed, we used stringent filtering thresholds of 300 genes and 600 transcripts per cell, as per the settings suggested in the Honeycomb vignette for the analysis of HIVE scRNA-seq data with Seurat. These thresholds are conservative compared to other Plasmodium scRNA-seq studies of erythrocytic life stages, which defined thresholds at 150 genes per cell for P. knowlesi or even lower for P. falciparum2,7. While conservative thresholds ensure data quality, they may impact the number of cells recovered, especially ring stages. As shown here and in previous studies, ring stages are characterized by lower transcription levels as compared to trophozoite and schizont stages2,7,14, making them less likely to pass conservative filtering and be included in the analysis. To avoid excessive filtering, thresholds could be adapted to each life stage. Stage-flexible filtering might be particularly important for natural infection samples, which often have low parasite densities and a high proportion of rings in circulation.
HIVE scRNA-seq provides high quality sequencing data that will lead to new insights into biological processes of Plasmodium and has similar potential for other unicellular organisms. The advantages of this technology for Plasmodium include the versatility and compatibility to low-resource settings and the ability to load a large number of cells which is beneficial when parasite enrichment is limited. We highlight the potential application of HIVE technology for natural infections which could be especially valuable for species difficult to culture such as P. vivax.
Methods
Ethics statement
This study was approved by the ITM Institutional Review Board (Ref 1565/22) and the UZA Ethics Committee (ID 5491). The Institute of Tropical Medicine Antwerp (ITM) policlinic uses an opt-out consensus model for research use of blood samples collected in the clinic.
Culture of Plasmodium knowlesi
P. knowlesi A1-H.1 strain parasites were maintained between 0.5 and 10% parasitemia in culture medium consisting of RPMI-1640 (Westburg Life Sciences) supplemented with 0.32 g/L sodium bicarbonate, 5.0 g/L Albumax II (Invitrogen, cat: 11021-029), 2.0 g/L D-glucose, 25mM HEPES, 0.05 g/L hypoxanthine, 0.005 g/L gentamicin, 2mM L-glutamine with 10% (v/v) heat-inactivated horse serum (Invitrogen) at 3% hematocrit (HCT). Parasites were continuously cultured in O + blood and incubated at 37 °C under atmospheric conditions of 90% N2, 5% O2, 5% CO2. Parasitemia was determined using thin smear slides stained with 10% Giemsa stain for 10 min.
HIVE sample processing
P. knowlesi culture enrichment with Nycodenz and Percoll density gradients
A P. knowlesi infected red blood cell (iRBC) pellet of 5% parasitemia was diluted to 50% HCT. 0.75mL of diluted culture was loaded onto 5mL of either 70% Nycodenz (N) (Serumwerk Bernburg, 18003) or 60% Percoll (P) (Sigma-Aldrich, cat: GE17-0891-01) density gradient and centrifuged at 900 g for 12 min (brake 0) for Nycodenz or 2400 rpm for 10 min (brake 0) for Percoll, to separate iRBCs from non-iRBCs. The interfaces of the gradients were washed in 10mL medium without serum. The enriched iRBC pellets were resuspended in 2mL PBS-BSA buffer (PBS with 0.5% BSA).
P. knowlesi mock natural infection sample and parasite enrichment
A P. knowlesi iRBC pellet was diluted in 4mL whole blood to approximately 1% parasitemia. The infected blood sample was centrifuged at 2400 rpm for 10 min and the buffy coat was removed. The sample was split into 2 aliquots and processed through two different methods (MACSPS and PNyco) for leukocyte depletion and parasite enrichment.
MACSPS
One aliquot was processed using a MACS LD column (Miltenyi Biotec), a Plasmodipur filter (Europroxima), and saponin lysis (Sigma-Aldrich, cat: 47036). First, a MACS column, attached to a magnet, was pre-wet with PBS-BSA pre-warmed to 37 °C. The iRBC pellet was diluted to 20% HCT and loaded onto the column. The column was washed three times with 1mL of PBS-BSA which recovered elution 1 containing mostly ring stages (elution 1). The column was removed from the magnet, washed twice with 3mL PBS-BSA and pelleted to elute the hemozoin-containing trophozoite and schizont stages (elution 2). Elution 1 was passed through a pre-wet Plasmodipur filter to deplete leukocytes, pelleted, and RBCs were lysed with 2.5 V 0.15% (w/v) cold saponin (to remove non-iRBC) by pipetting 10x and pelleted for 5 min at 2400 rpm. The pellet was washed twice with PBS, combined with the elution 2 pellet, and diluted in 2mL PBS-BSA.
PNyco
The second aliquot was processed using a Plasmodipur filter and 70% Nycodenz density gradient prepared in PBS (PNyco). The aliquot was diluted to 20% HCT in PBS, passed through a pre-wet filter, and washed with 10mL PBS. The flow-through was pelleted, diluted to 50% HCT, and loaded onto a 70% Nycodenz gradient. This was centrifuged at 900 g for 12 min (brake 0), the interface was washed in PBS, and 10µL of pellet was diluted in 2mL PBS-BSA.
HIVE sample capture
Resuspended enriched iRBC pellets were further diluted in PBS-BSA to a concentration of 60,000 cells/mL using a Scepter 3.0 cell counter (Merck). A volume of 1mL of cell suspension was loaded into each HIVE to settle into the pico-wells via gravity or using a centrifuge according to modified manufacturer’s directions. The incubation time for the HIVE loading was increased from 30 min to 1 h at room temperature (gravity) and from 3 to 5 min (centrifuge). HIVEs were stored at −20 °C until library preparation to mimic using a −20 °C freezer in the field.
HIVE transcriptome recovery, library preparation, and sequencing
Transcriptome recovery and library preparation were conducted following manufacturer’s directions with provided reagents and 10 extra PCR cycles added to the whole transcriptome amplification (WTA). Transcriptome recovery involves removing the storge solution followed by cell lysis (CLX Lysis Solution and hybridization to capture poly-A transcripts (Hybridization Buffer). Beads with hybridized transcripts are recovered using the provided bead collector, CLX Bead Recovery Solution, and centrifuge to pellet the beads and transfer them to a filter plate. Library preparation continues in the filter plate using a vacuum and includes 1 st strand cDNA synthesis (CLX 1 st Strand Reaction Mix), bead clean-up (Clean-Up Reaction Mix) to remove bead oligos without cDNA, 2nd strand cDNA synthesis (2nd Strand Reaction Mix), followed by whole transcriptome amplification (WTA) where an additional 10 PCR cycles were added. After a size-selection bead clean-up, an index PCR adds the required indexes for library multiplexing and sequencing (HIVE UDI Index Plate). A final size-selection bead clean-up results in a final volume 25µL. All samples were sequenced on a NovaSeq6000 sequencer (Broad Institute, USA) according to the manufacturer’s recommendations. Sequencing data has been deposited in the NCBI sequencing read archive.
Analysis
BeeNet v1.1 was used to map sequencing reads to an updated PKNH reference (downloaded from PlasmoDB version 6259) with default parameters and an expected number of cells of 60,000. As 3’UTR annotations are absent for most P. knowlesi genes, the annotations of the 3′UTR were added in a systematic manner to enhance the accuracy of read assignments to the annotated transcript: for each protein-coding gene, the 3′UTR region was annotated by adding 2500 base pairs downstream of the stop codon. In cases where the newly designated 3′UTR overlapped with other genomic elements, the 3’UTR was truncated to avoid overlap. The resulting TCM and RCM files were read into R (version 4.2.1) and converted into a Seurat object using Seurat 4.260. The single-cell data from 6 samples were merged into one Seurat object. Cells with less than 600 transcripts per cell or less than 300 expressed genes per cell were filtered out. Doublets were removed using scrublet (version 0.2.361), Preliminary analysis (PCA analysis and clustering analysis) before excluding potential doublets showed that one cluster was significantly enriched with scrublet-predicted doublets, and as such the entire cluster was also removed from the single-cell dataset. Count data were normalized using regularized negative binomial regression (SCTransform). After principal component analysis, the Uniform Manifold Approximation and Projection (UMAP) dimensional reduction technique was applied for visualization of the single-cell transcriptomes using the first 20 principal components62. Clustering of cells was performed on the PCA output using a resolution of 0.25.
The single-cell data were aggregated into pseudo-bulk expression profiles by summing counts per gene across all cells within each sample. Pairwise correlations between samples were then calculated using the Pearson correlation coefficient via the cor function in R. Visualization of the correlation matrix was performed using the ggplot2 package providing an overview of the similarity in gene expression profiles between samples.
Annotation of the life stages as described for P. knowlesi in the Malaria Cell Atlas (MCA)2 was downloaded from https://zenodo.org/records/2843883 (pk10xIDC dataset), where the stage_pred column in the phenotypic metadata was used as life stage predictor. P. knowlesi single-cell transcriptomes obtained from the MCA database were projected on our dataset using the scmap-cell (version 1.18.063), implementation, and the corresponding annotation of the MCA cells was transferred to our dataset. Pseudotime analysis was performed using the Slingshot package (version 2.4.064), in R.
Statistical analysis was performed using R (version 4.3.3) with differences in cell recovery and life stage composition being determined using the Pearson chi-squared test.
Data availability
The dataset generated and analyzed during the current study is available in the NCBI Gene Expression Omnibus (GEO accession number GSE271911). The Malaria Cell Atlas P. knowlesi dataset (pk10xIDC dataset) used in the analysis was downloaded from https://zenodo.org/records/2843883.
References
Nötzel, C., Poran, A. & Kafsack, B. F. C. Single-Cell transcriptome profiling of protozoan and metazoan parasites. Trends Parasitol. 34, 731–734 (2018).
Howick, V. M. et al. The malaria cell atlas: single parasite transcriptomes across the complete Plasmodium life cycle. Science 365, eaaw2619 (2019).
Reid, A. J. et al. Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. eLife 7, e33105 (2018).
Dogga, S. K. et al. A single cell atlas of sexual development in Plasmodium falciparum. Science 384, eadj4088 (2024).
Mohammed, M. et al. Single-cell transcriptomics reveal transcriptional programs underlying male and female cell fate during plasmodium falciparum gametocytogenesis. Nat. Commun. 15, 7177 (2024).
Ngara, M. et al. Exploring parasite heterogeneity using single-cell RNA-seq reveals a gene signature among sexual stage Plasmodium falciparum parasites. Exp. Cell Res. 371, 130–138 (2018).
Poran, A. et al. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature 551, 95–99 (2017).
Walzer, K. A., Fradin, H., Emerson, L. Y., Corcoran, D. L. & Chi, J. T. Latent transcriptional variations of individual Plasmodium falciparum uncovered by single-cell RNA-seq and fluorescence imaging. PLoS Genet. 15, e1008506 (2019).
Brancucci, N. M. B. et al. Probing Plasmodium falciparum sexual commitment at the single-cell level. Wellcome Open. Res. 3, 70 (2018).
Russell, A. J. C. et al. Regulators of male and female sexual development are critical for the transmission of a malaria parasite. Cell Host Microbe. 31, 305–319e10 (2023).
Hentzschel, F. et al. Host cell maturation modulates parasite invasion and sexual differentiation in Plasmodium Berghei. Sci. Adv. 8, eabm7348 (2022).
Sà, J. M., Cannon, M. V., Caleon, R. L., Wellems, T. E. & Serre, D. Single-cell transcription analysis of Plasmodium Vivax blood-stage parasites identifies stage- and species-specific profiles of expression. PLoS Biol. 18, e3000711 (2020).
Hazzard, B. et al. Long read single cell RNA sequencing reveals the isoform diversity of Plasmodium Vivax transcripts. PLoS Negl. Trop. Dis. 16, e0010991 (2022).
Hazzard, B. et al. Single-cell analyses of polyclonal plasmodium Vivax infections and their consequences on parasite transmission. Nat. Commun. 15, 7625 (2024).
Afriat, A. et al. A Spatiotemporally resolved single-cell atlas of the Plasmodium liver stage. Nature 611, 563–569 (2022).
Ruberto, A. A. et al. Single-cell RNA sequencing reveals developmental heterogeneity among Plasmodium Berghei sporozoites. Sci. Rep. 11, 4127 (2021).
Bogale, H. N. et al. Transcriptional heterogeneity and tightly regulated changes in gene expression during Plasmodium berghei sporozoite development. Proc. Natl. Acad. Sci. U.S.A. 118, e2023 (2021).
Witmer, K. et al. Using scRNA-seq to identify transcriptional variation in the malaria parasite Ookinete stage. Front. Cell. Infect. Microbiol. 11, 604129 (2021).
Real, E. et al. A single-cell atlas of Plasmodium falciparum transmission through the mosquito. Nat. Commun. 12, 3196 (2021).
Mohammed, M. et al. Single-Cell transcriptomics to define Plasmodium falciparum stage transition in the mosquito midgut. Microbiol. Spectr. 11, e03671–e03622 (2023).
Mancio-Silva, L. et al. A single-cell liver atlas of Plasmodium Vivax infection. Cell Host Microbe. 30, 1048–1060e5 (2022).
Ruberto, A. A. et al. Single-cell RNA sequencing of Plasmodium Vivax sporozoites reveals stage- and species-specific transcriptomic signatures. PLoS Negl. Trop. Dis. 16, e0010633 (2022).
Ruberto, A. A. et al. Single-cell RNA profiling of Plasmodium vivax-infected hepatocytes reveals parasite- and host- specific transcriptomic signatures and therapeutic targets. Front. Cell. Infect. Microbiol. 12, 986314 (2022).
Heaton, H. et al. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods. 17, 615–620 (2020).
Duffy, S., Avery, V. M. & Routine Vitro culture of Plasmodium falciparum: experimental consequences?? Trends Parasitol. 34, 564–575 (2018).
LeRoux, M., Lakshmanan, V. & Daily, J. P. Plasmodium falciparum biology: analysis of in vitro versus in vivo growth conditions. Trends Parasitol. 25, 474–481 (2009).
Lapp, S. A. et al. Plasmodium Knowlesi gene expression differs in ex vivo compared to in vitro blood-stage cultures. Malar. J. 14, 110 (2015).
Andradi-Brown, C. et al. A novel computational pipeline for var gene expression augments the discovery of changes in the Plasmodium falciparum transcriptome during transition from in vivo to short-term in vitro culture. eLife 12, 87726 (2024).
Pacheco, M. A. et al. Multiplicity of infection and disease severity in Plasmodium Vivax. PLoS Negl. Trop. Dis. 10, e0004355 (2016).
Zhong, D. et al. Multiplicity and molecular epidemiology of Plasmodium Vivax and Plasmodium falciparum infections in East Africa. Malar. J. 17, 185 (2018).
Fola, A. A. et al. Higher complexity of infection and genetic diversity of Plasmodium Vivax than Plasmodium falciparum across all malaria transmission zones of Papua new Guinea. Am. J. Trop. Med. Hyg. 96, 630–641 (2017).
Mwesigwa, A. et al. Plasmodium falciparum genetic diversity and multiplicity of infection based on msp-1, msp-2, glurp and microsatellite genetic markers in sub-Saharan Africa: a systematic review and meta-analysis. Malar. J. 23, 97 (2024).
Friedrich, L. R. et al. Complexity of infection and genetic diversity in Cambodian Plasmodium Vivax. PLoS Negl. Trop. Dis. 10, e0004526 (2016).
Real, E. & Mancio-Silva, L. Single-cell views of the Plasmodium life cycle. Trends Parasitol. 38, 748–757 (2022).
Moon, R. W. et al. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc. Natl. Acad. Sci. U.S.A. 110, 531–536 (2013).
Sriprawat, K. et al. Effective and cheap removal of leukocytes and platelets from Plasmodium Vivax infected blood. Malar. J. 8, 115 (2009).
Kramer, K., Kan, S. & Siddiqi, W. A. Concentration of Plasmodium falciparum-Infected erythrocytes by density gradient centrifugation in Percoll. J. Parasitol. 68, 336–337 (1982).
Ribaut, C. et al. Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar. J. 7, 45 (2008).
Trang, D., Huy, N., Kariu, T., Tajima, K. & Kamei, K. One-step concentration of malarial parasite-infected red blood cells and removal of contaminating white blood cells. Malar. J. 3, 7 (2004).
Venkatesan, M. et al. Using CF11 cellulose columns to inexpensively and effectively remove human DNA from Plasmodium falciparum-infected whole blood samples. Malar. J. 11, 41 (2012).
De Meulenaere, K. et al. Band 3–mediated Plasmodium Vivax invasion is associated with transcriptional variation in PvTRAg genes. Front. Cell. Infect. Microbiol. 12, 1011692 (2022).
Moraes Barros, R. R., Gibson, T. J., Kite, W. A., Sá, J. M. & Wellems, T. E. Comparison of two methods for transformation of Plasmodium knowlesi: direct schizont electroporation and spontaneous plasmid uptake from plasmid-loaded red blood cells. Mol. Biochem. Parasitol. 218, 16–22 (2017).
Conte, M. I. & Fuentes-Trillo, A. Domínguez conde, C. Opportunities and tradeoffs in single-cell transcriptomic technologies. Trends Genet. 40, 83–93 (2024).
Fernández-Becerra, C. et al. Increased expression levels of the pvcrt-o and pvmdr1 genes in a patient with severe Plasmodium Vivax malaria. Malar. J. 8, 55 (2009).
Pava, Z. et al. Expression of Plasmodium Vivax crt-o is related to parasite stage but not Ex vivo chloroquine susceptibility. Antimicrob. Agents Chemother. 60, 361–367 (2015).
Chitnis, C. E. & Miller, L. H. Identification of the erythrocyte binding domains of Plasmodium Vivax and Plasmodium Knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 (1994).
Moon, R. W. et al. Normocyte-binding protein required for human erythrocyte invasion by the zoonotic malaria parasite Plasmodium knowlesi. Proc. Natl. Acad. Sci. 113, 7231–7236 (2016).
Ranjan, A. & Chitnis, C. E. Mapping regions containing binding residues within functional domains of Plasmodium vivax and Plasmodium knowlesi erythrocyte-binding proteins. Proc. Natl. Acad. Sci. 96, 14067–14072 (1999).
Obaldía, N. et al. Comparison of PvLAP5 and Pvs25 qRT-PCR assays for the detection of Plasmodium Vivax gametocytes in field samples preserved at ambient temperature from remote malaria endemic regions of Panama. PLoS Negl. Trop. Dis. 16, e0010327 (2022).
Bozdech, Z. et al. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 1, e5 (2003).
Reuling, I. J. et al. Concentration of Plasmodium falciparum gametocytes in whole blood samples by magnetic cell sorting enhances parasite infection rates in mosquito feeding assays. Malar. J. 16, 315 (2017).
Ramos, G. Q., Baia-da-Silva, D. C., Lacerda, M. V. G., Monteiro, W. M. & Lopes, S. C. P. Viability and infectivity of plasmodium Vivax gametocytes in Short-Term culture. Front. Cell. Infect. Microbiol. 11, 676276 (2021).
Deshmukh, S. S. et al. Multiparametric biophysical profiling of red blood cells in malaria infection. Commun. Biol. 4, 1–13 (2021).
Lew, V. L., Tiffert, T. & Ginsburg, H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum–infected red blood cells. Blood 101, 4189–4194 (2003).
Serebrennikova, Y. M., Patel, J., Milhous, W. K. & García-Rubio, L. H. Quantitative analysis of morphological alterations in Plasmodium falciparum infected red blood cells through theoretical interpretation of spectral measurements. J. Theor. Biol. 265, 493–500 (2010).
Oliveira, M. F. et al. On the pro-oxidant effects of haemozoin. FEBS Lett. 512, 139–144 (2002).
Stuart, T. et al. Comprehensive integration of Single-Cell data. Cell 177, 1888–1902e21 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods. 16, 1289–1296 (2019).
Aurrecoechea, C. et al. PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37, D539–D543 (2009).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587e29 (2021).
Wolock, S. L., Lopez, R., Klein, A. M. & Scrublet Computational identification of cell doublets in Single-Cell transcriptomic data. Cell. Syst. 8, 281–291e9 (2019).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open. Source Softw. 3, 861 (2018).
Kiselev, V. Y., Yiu, A. & Hemberg, M. Scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods. 15, 359–362 (2018).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 477 (2018).
Acknowledgements
We would like to thank Dr. Ellen Knuepfer for donating the P. knowlesi A1-H.1 parasite strain which was used in this study. This work was funded by the Department of Economy, Science and Innovation in Flanders (SOFI to ARU, 2020–2024) and (jPPP to ARU and RMB, 2022) and São Paulo Research Foundation (FAPESP) (Grant JP 2018/06219-8 to RMB, 2019–2024). The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation - Flanders (FWO) and the Flemish Government. ES received support from an FWO PhD-SB Fellowship from the Research Foundation Flanders (1SC5522N). The funders had no role in study design, data collection, analysis, and interpretation, manuscript preparation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
ARU, RBM, and ES conceived and designed the study. ES and PG completed the laboratory experiments. PM performed the bioinformatic analysis with input from ES and ARU. ES wrote the first draft of the manuscript with input from ARU and PM with the final version of the manuscript including edits from all authors. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sauve, E., Monsieurs, P., Guetens, P. et al. Novel single-cell preservation and RNA sequencing technology unlocks field studies for Plasmodium natural infections. Sci Rep 15, 24237 (2025). https://doi.org/10.1038/s41598-025-08765-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-08765-x
