
Abstract
Pancreatic adenocarcinoma (PAAD) is a highly lethal malignancy with limited effective prognostic biomarkers. In this study, 1,034 samples from TCGA-PAAD, GSE62452, GSE28735, GSE183795, and ICGC cohorts were systematically integrated to identify key programmed cell death-related genes (PCDRGs) associated with patient prognosis. Differential expression analysis and Univariate Cox regression analysis identified 17 candidate PCD-related genes significantly associated with overall survival. Using a comprehensive machine learning framework involving 117 algorithmic combinations under a Leave-one-out cross-validation (LOOCV) strategy, we identified the StepCox[both] + Ridge as the best algorithms composition to construct a prognostic model based on six PCDRGs, ITGA3, CDCP1, IL1RAP, CLU, PBK, and PLAU. The model was validated to have robust predictive performance. Risk scores were significantly correlated with clinical features, immune microenvironment characteristics, and chemotherapeutic sensitivity. High-risk patients exhibited worse prognosis and immunosuppressive infiltration patterns. Furthermore, consensus clustering identified two PAAD molecular subtypes with distinct PCDRGs expression patterns and survival outcomes. A nomogram integrating risk score and clinical variables exhibited strong prognostic accuracy for 1-, 3-, and 5-year survival prediction. In summary, we established and validated a PCD-related prognostic signature that effectively stratifies PAAD patients by clinical outcome, immune contexture, and therapeutic response, providing novel insights for personalized management strategies.
Similar content being viewed by others

Introduction
Pancreatic adenocarcinoma (PAAD), recognized as the third leading cause of cancer-related mortality worldwide, demonstrates a clinical prognosis that is closely linked to its pathological subtypes1. Among these, pancreatic ductal adenocarcinoma (PDAC) accounts for over 90% of diagnosed cases, constituting the predominant histological subtype of the disease2. Notably, PAAD exhibits a 5-year overall survival (OS) rate of approximately 10%, markedly lower than that of other common solid malignancies3. Furthermore, the global incidence and mortality rates of PAAD have shown a progressive annual increase, positioning this malignancy as a substantial public health threat. According to data released by the Global Cancer Observatory (GLOBOCAN) 2020, worldwide PAAD cases reached 495,773 newly diagnosed patients, with 466,003 reported deaths4. Compared to 2018 epidemiological data (458,918 new cases and 432,242 deaths), the incidence and mortality rates exhibited a marked increase of 8.03% and 7.81%, respectively, over the past two years5. This growth rate is significantly higher than that of most solid tumors. Smoking, alcohol consumption, obesity, type 2 diabetes mellitus, and trace element exposure are the main risk factors for the development of PAAD4,6. With the global prevalence of metabolic diseases and the exacerbation of an aging population, the disease burden of PAAD is expected to continue to increase over the next decade, especially in developing countries undergoing rapid socioeconomic transformation.
PAAD, due to its anatomically retroperitoneal location and insidious early symptoms, resulting in approximately 80–85% of patients being in the advanced stage at the time of initial diagnosis2. The clinical diagnosis currently predominantly depends on the detection of the serum biomarker Carbohydrate Antigen 19 − 9 (CA19-9); however, this modality exhibits substantial limitations in early-stage screening, with sensitivity and specificity ranges of 79-81% and 82–90%, respectively6,7. Although surgical resection of the primary tumor remains the sole curative intervention for PAAD detected at a very early stages, only 15-20% of patients have the opportunity to undergo such surgery, and the recurrence rate within 2 years after surgery is as high as 80%8. For patients with advanced PAAD, surgical intervention is unfeasible in most cases due to metastatic dissemination, leaving systemic chemotherapy regimens as the primary therapeutic modality. Phase III clinical trials have confirmed that the median OS time for the FOLFIRINOX regimen and the combination of gemcitabine and albumin-bound paclitaxel are 11.1 months and 8.5 months, respectively1 but the highly fibrotic tumor microenvironment and multiple drug resistance mechanisms significantly diminish the efficacy of the drugs9. It is worth noting that among patients with KRAS - mutated cancers, the incidence of PAAD is the highest, reaching 73.51%, but the therapeutic targets for its conversion treatment have not yet been achieved10. Therefore, the identification of novel therapeutic targets and chemotherapeutic drugs is of great significance for the treatment of PAAD and the extension of prognosis.
Programmed cell death (PCD) is an active cell demise process that is precisely regulated by genes. It plays a crucial role in maintaining tissue homeostasis, eliminating abnormal cells, and responding to pathological stimuli. Based on distinct molecular mechanisms and morphological characteristics, PCD can be classified into 21 subtypes, including necroptosis, pyroptosis, ferroptosis, entotic cell death, parthanatos, lysosome-dependent cell death, autophagy, alkaliptosis and oxeiptosis11 apoptosis, paraptosis12 cuproptosis, disulfidptosis13 Entosis, methuosis, necrosis, NETosis, mitoptosis14 Immunogenic cell death15 Anoikis16 and PANoptosis17. In recent years, studies have revealed that dysregulated PCD is not only a core mechanism in the occurrence and development of cancer, but also closely related to tumor drug resistance and microenvironment remodeling. For instance, cancer cells often evade apoptosis by upregulating anti-apoptotic proteins (e.g., BCL-2), downregulating pro-apoptotic proteins, or inducing mutations in death receptors and downstream signaling components, leading to uncontrolled cellular proliferation and therapeutic resistance18. Under hypoxia and nutrient deprivation stress conditions, autophagy contributes to the survival of tumor cells19. Furthermore, cuproptosis is considered to be associated with multiple cancer related signaling pathways, such as receptor tyrosine kinase (RTK), the phosphoinositide − 3 - kinase (PI3K) - AKT signaling pathway, and mitogen - activated protein kinase (MAPK) signaling pathway. These pathways not only promote tumor growth, migration, and proliferation, but also affect autophagy and metabolic pathways, thereby enhancing the survival and proliferation of cancer cells20. Therefore, elucidating the roles of PCD in PAAD holds significant clinical significance.
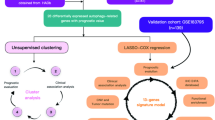
In this study, we employed a machine learning-based comprehensive algorithm and utilized various bioinformatics methods to screen for PCD-related prognostic signatures in PAAD, constructed a PCD-related prognostic model and revealed its clinicopathological correlations and immune microenvironment regulatory mechanisms. First, we integrated the transcriptome data of PAAD from the TCGA-PAAD, GSE62452, GSE28735, GSE183795, and ICGC cohorts. Through differential expression analysis and univariate Cox regression analysis, we identified 17 differentially expressed PCD genes that were significantly associated with patient survival, which were considered as potential prognostic genes. Subsequently, using the StepCox[both] + Ridge algorithm, we screening programmed cell death-related genes (PCDRGs) associated with PAAD prognosis and developed a nomogram for predicting the survival rate of PAAD patients. In addition, we also assessed the immune infiltration status of PAAD patients through bioinformatics analysis and predicted potential therapeutic targets. The PCD-related prognostic model demonstrated significant predictive accuracy for patient survival, while the integrated molecular signatures provide novel insights for personalized therapeutic strategies and drug repositioning of PAAD patients.
Results
Screening and identification of PCD related prognostic candidate genes in PAAD
To minimize batch effects across cohorts, batch effect correction was performed for the GSE183795, GSE62452, and GSE28735 cohorts, which were subsequently integrated into a combined GSE-merged cohort (Supplementary Fig. 1 A, B). Differential expression analysis identified 1,393 Differentially expressed genes (DEGs) in the TCGA-PAAD cohort and 900 DEGs in the GSE-merged cohort, resulting in a total of 2,129 unique DEGs. Among them, the TCGA-PAAD cohort contains 551 upregulated genes and 842 downregulated genes; whereas the GSE-merged cohort contains 583 upregulated genes and 317 downregulated genes, respectively (Fig. 1A, B). Through Venn plot analysis, 334 PCD-related DEGs shared between the TCGA-PAAD and GSE-merged cohorts were identified (Fig. 1C, Supplementary Table 1). Based on these genes, univariate Cox regression analysis was performed, resulting in the identification of 96 genes in the TCGA-PAAD cohort, 108 genes in the GSE-merged cohort and 73 genes in the ICGC cohort that were both differentially expressed and significantly associated with OS (P < 0.05). The 17 overlapping DEGs associated with OS across the three cohorts were considered potential prognostic candidate genes for PAAD (Supplementary Fig. 1 C, Supplementary Table 2). The 17 candidate genes’ univariate Cox analysis in the GSE-merged, TCGA-PAAD and ICGC cohorts were shown in Fig. 1D, Supplementary Fig. 1D and E, respectively. Subsequently, protein-protein interaction (PPI) network analysis was conducted to explore the interactions among the 17 prognostic candidate genes. Among them, FN1 exhibited the highest degree of connectivity with other genes (Fig. 1E). Heatmap revealed that CLU was downregulated in tumor samples from the GSE-merged cohort, while the remaining 16 genes were upregulated (Fig. 1G). In addition, copy number variation (CNV) analysis demonstrated frequent genomic alterations among these genes. Specifically, ITGA3 showed the most prominent CNV gain, whereas LAMC2, PLAU, SLC2A1, and CDH3 exhibited significant CNV loss (Fig. 1F).

Identification of prognostic candidate genes associated with 21 types of programmed cell death (PCD). (A, B) The volcano plots of differentially expressed genes (DEGs) in the TCGA-PAAD (A) and GSE-merged (B) cohorts. (C) Venn plot showing the overlapping between DEGs from the TCGA-PAAD and GSE-merged cohorts and the PCD-related genes. (D) Univariate Cox regression analysis of 17 prognostic candidate genes in the GSE-merged cohort. (E) Protein-protein interaction (PPI) network of 17 prognostic candidate genes. (F) Copy number variation (CNV) analysis of the 17 candidate genes. (G) Heatmap of expression levels of the 17 candidate genes in tumor and normal tissues from the GSE-merged cohort. (H-I) Gene Ontology (GO) (H) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (I) pathway enrichment analysis of the 17 candidate genes.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
GO and KEGG enrichment analyses of the PCD-related prognostic candidate genes revealed their involvement in multiple cellular signaling pathways, metabolic processes, and cancer-related pathways. GO enrichment indicated that these genes were primarily associated with biological processes (BP) such as regulation of transforming growth factor beta production, cellular components (CC) such as the basement membrane, and molecular functions (MF) such as extracellular matrix structural constituent (Fig. 1H). KEGG pathway analysis further highlighted significant enrichment in pathways including ECM-receptor interaction, PI3K-Akt signaling pathway, Central carbon metabolism in cancer, and HIF-1 signaling pathway (Fig. 1I).
Construction of a PCD related prognostic model using machine learning
We employed an integrated machine learning-based approach to develop a prognostic model using the expression profiles of 17 prognostic candidate genes. A total of 117 predictive models were fitted under a Leave-one-out cross-validation (LOOCV) framework, and the Harrell’s concordance index (C-index) was calculated for each model. Among them, the StepCox[both] + Ridge combination achieved the highest average C-index of 0.64 (Fig. 2A). Prior studies using RNA-based signatures have reported C-index ranging from 0.61-0.7121,22,23, suggesting that our model performs within an expected and acceptable range. Using this optimal algorithm, six PCDRGs, ITGA3, CDCP1, IL1RAP, CLU, PBK, and PLAU, were selected (Supplementary Table 3). A Circos plot was generated to visualize the chromosomal locations of these PCDRGs (Fig. 2B).

Integrated machine learning-based development and validation of a predictive model associated with 21 types of PCD. (A) Harrell’s concordance index (C-index) values of 117 algorithmic combinations were calculated to identify the optimal predictive model and algorithm. (B) Circos plot showing the chromosomal locations of the PCD-related genes (PCDRGs). (C) Gene Set Variation Analysis (GSVA) analysis revealed differences in enriched pathways between high- and low-risk groups in the GSE-merged cohort. (D) Boxplots showing expression differences of PCDRGs between normal and tumor tissues in the GSE-merged cohort. (E) Pearson correlation analysis among the PCDRGs. (F-K) Kaplan-Meier survival analyses of the six PCDRGs in the TCGA-PAAD cohort. (L-M) Waterfall plot showing the somatic mutation landscape of low-risk (L) and high-risk samples (M) in the TCGA-PAAD cohort. **: P < 0.01; ***: P < 0.001.
Subsequently, individual risk scores were calculated for each patient using the established risk score formula, and patients were stratified into high- and low-risk groups based on the median risk score. To explore the biological processes significantly enriched in each risk group, Gene Set Variation Analysis (GSVA) was performed. Results indicated that in the GSE-merged cohort, notable pathway enrichment differences were observed between the high- and low-risk groups. High risk samples were mainly enriched in cancer-related signaling pathways, including DNA repair, Wnt/β-catenin signaling, MYC targets V1, KRAS signaling dn, and Hedgehog signaling, while low risk samples were enriched in TNF-α Signaling via NF-κB, Angiogenesis, KRAS signaling up and Apoptosis, suggesting distinct underlying biological mechanisms between the two groups (Fig. 2C). In the TCGA-PAAD cohort, high risk samples showed enrichment in the Reactive oxygen species pathway, Oxidative phosphorylation and Inflammatory response, whereas low risk samples were primarily enriched in the p53 pathway and Wnt/β-catenin signaling (Supplementary Fig. 2 A).
Further analysis revealed significant differences in the expression levels of PCDRGs between normal and tumor tissues in the GSE-merged cohort. CLU was significantly downregulated in tumor samples, whereas the other five genes, ITGA3, CDCP1, IL1RAP, PBK, and PLAU, were significantly upregulated (P < 0.001) (Fig. 2D). Pearson correlation analysis showed a negative correlation between CLU and the other five genes, while ITGA3, CDCP1, IL1RAP, PBK, and PLAU exhibited positive correlations with each other (Fig. 2E). Kaplan-Meier survival analysis of the six PCDRGs in the TCGA-PAAD cohort revealed that patients with low CLU expression had poorer prognosis, while high expression of the remaining five genes was associated with worse prognosis (Fig. 2F-K). These results are consistent with their differential expression patterns in tumor samples. In addition, we analyzed the mutational landscape of patients in different risk groups. The high-risk group exhibited a markedly higher mutation frequency (95.12%) compared to the low-risk group (64.86%) (Fig. 2L, M). Notably, mutations in TGFBR2, ATM, and CHD6 were more frequent in the low-risk group, whereas RIMS2, TPO, and TNXB mutations were more prevalent in the high-risk group.
Evaluation of the clinical relevance of the PCDRGs prediction model
We validated the predictive performance of the risk model across five independent cohorts, TCGA-PAAD, GSE183795, GSE28735, GSE62452, and ICGC, and further explored the relationship between the risk score and clinical characteristics. As shown in Fig. 3A, patients in the high-risk group exhibited shorter OS time and poorer prognoses compared to those in the low-risk group across all cohorts. This finding was further supported by Kaplan-Meier survival analysis, which consistently demonstrated significantly shorter OS time in the high-risk group across all five cohorts (Fig. 3B). Principal component analysis (PCA) also revealed distinct distributions between high- and low-risk patients in each cohort (Fig. 3C).

Validation of the prognostic model and association between risk score and clinical characteristics. (A) Survival status of high- and low-risk patients across TCGA-PAAD, GSE183795, GSE28735, GSE62452, and ICGC cohorts. (B) Kaplan-Meier survival curves comparing high- and low-risk groups in the five cohorts. (C) PCA illustrating the distribution of high- and low-risk patients across the five cohorts. (D) Violin plots showing the distribution of risk scores between alive and dead patients in the five cohorts. (E-H) Violin plots comparing risk scores across different clinical subgroups in the TCGA-PAAD cohort: T (E), N (F), M (G), and Stage (H).
Violin plots showed that in the TCGA-PAAD, GSE183795, and ICGC cohorts, risk scores were significantly higher in dead patients compared to those who were alive (P < 0.05). Although no statistically significant differences were observed in the remaining two cohorts, dead patients still exhibited higher risk scores (Fig. 3D). Additionally, in the TCGA-PAAD cohort, the risk score was significantly associated with clinical staging. Patients with T3 + T4 tumors had significantly higher risk scores than those with T1 + T2 tumors, and patients at Stage II had higher scores compared to those at Stage I (P < 0.05). However, no significant differences in risk scores were observed across N and M stages (Fig. 3E-H).
Unsupervised consensus clustering analysis of prognostic PCDRGs
Unsupervised consensus clustering was performed to stratify PAAD patients and identify subgroups with distinct molecular characteristics. The optimal number of clusters was determined to be k = 2, and thus, patients in the TCGA-PAAD cohort were divided into two clusters (Fig. 4A, B). Subsequent Kaplan-Meier survival analysis revealed that patients in cluster C2 had significantly worse survival outcomes compared to those in cluster C1 (P = 0.01) (Fig. 4C). Moreover, a heatmap integrating clinical features and PCDRGs expression levels demonstrated marked differences in gene expression between the two clusters. Most patients in cluster C1 were characterized by early-stage disease and lower risk scores, whereas those in cluster C2 were predominantly in advanced stages and had higher risk scores (Fig. 4D).

Unsupervised clustering and construction of a nomogram model in the TCGA-PAAD cohort. (A) Unsupervised consensus clustering divided patients into two clusters. (B) The optimal number of clusters was determined to be k = 2. (C) Kaplan-Meier survival analysis comparing patients in clusters C1 and C2. (D) Heatmap showing the distribution of PCDRGs expression levels and clinicopathological characteristics across clusters. (E) Univariate Cox regression analysis of risk score and clinicopathological characteristics in the TCGA-PAAD cohort. (F) Multivariate Cox regression analysis of the same variables. (G) Nomogram constructed to predict 1-, 3-, and 5-year overall survival (OS) rate in PAAD patients. (H) Calibration curves evaluating the predictive accuracy of the nomogram. (I) Kaplan-Meier survival curves for patients stratified by nomogram scores. (J) Receiver operating characteristic (ROC) curves assessing the predictive performance of the nomogram for clinicopathological features. (K) ROC curves evaluating the predictive accuracy of the nomogram for 1-, 3-, and 5-year OS.
Establishment and assessment of the nomogram survival model
Univariate and multivariate Cox regression analyses were performed to evaluate the associations between OS and the risk score, along with clinicopathological characteristics in the TCGA-PAAD cohort (Fig. 4E, F). The results indicated that grade, T stage, N stage, tumor position, and risk score were all independent prognostic factors. Based on these variables, a nomogram was constructed to predict the 1-, 3-, and 5-year OS probabilities of PAAD patients (Fig. 4G). Calibration curves demonstrated good concordance between the predicted and observed survival rates at 1-, 3-, and 5-years (Fig. 4H). Kaplan-Meier survival analysis revealed that patients in the high nomogram score group had significantly worse prognosis (Fig. 4I). Receiver operating characteristic (ROC) curve analysis demonstrated the robust predictive performance of the model. The area under the ROC curve (AUC) for the risk score was 0.727, followed by grade (AUC = 0.656), stage (AUC = 0.679), T stage (AUC = 0.633), N stage (AUC = 0.752), state (AUC = 0.992), and cluster (AUC = 0.650). The AUCs for predicting 1-, 3-, and 5-year OS rate were 0.753, 0.776, and 0.727, respectively (Fig. 4J, K).
Immune landscape and its correlation with PCDRGs in PAAD patients
CIBERSORT analysis revealed that high-risk patients exhibited higher proportions of Macrophages M0 and Macrophages M2, whereas low-risk patients had higher proportions of T cells CD4 naïve, B cells naïve, and T cells CD8 (Fig. 5A). A heatmap of correlation analysis showed that PLAU was positively correlated with Macrophages M0, Macrophages M2 and Neutrophils, but negatively correlated with B cells naïve, Monocytes, NK cells activated, T cells CD4 memory resting and T cells CD8. In contrast, CLU was positively correlated with B cells naïve, Monocytes, and T cells CD8, but negatively correlated with Macrophages M0 (Fig. 5B). Significant associations were observed between the expression levels of PCDRGs and the abundance of 22 immune cell types. Specifically, CLU was negatively correlated with Macrophages M0, and PLAU was negatively correlated with T cells CD4 memory resting cells in both high- and low-risk groups (Fig. 5C-J). Furthermore, we analyzed the correlations between PCDRGs and tumor microenvironment (TME)-related gene sets and identified the top 21 TME-related gene sets with the strongest correlations. The results showed that PCDRGs were significantly positively correlated with multiple TME-related gene sets (Supplementary Fig. 2B). ESTIMATE algorithm analysis revealed that low-risk PAAD patients exhibited higher ImmuneScore and ESTIMATEScore, but lower tumor immune dysfunction and exclusion (TIDE) scores compared to the high-risk group (Fig. 5K, L). Moreover, immune checkpoint related genes, including BTN2A1, CD40, CEACAM1, PDCD1LG2, and TNFSF9, were highly expressed in the high-risk group (Fig. 5M).

Immune microenvironment analysis based on the risk score. (A) Boxplots showing differences in the relative proportions of 22 immune cell subtypes between high- and low-risk groups. (B) Heatmap illustrating correlations between PCDRGs and 22 immune cell populations. (C-J) Scatter plots displaying the correlations between PCDRGs expression levels and immune cell abundance. (K) Violin plots comparing StromalScore, ImmuneScore, and ESTIMATEScore between high- and low-risk groups. (L) Differences in TIDE scores between the two risk groups. (M) Boxplots showing the expression differences of 21 immune checkpoint-related genes between high- and low-risk groups. *: P < 0.05; **: P < 0.01; ***: P < 0.001.
Impact of PCDRGs on targeted therapy response in PAAD patients
To investigate the relationship between our prognostic model and drug sensitivity, we analyzed the half-maximal inhibitory concentration (IC50) values of several chemotherapeutic agents in PAAD samples using the Genomics of Drug Sensitivity in Cancer (GDSC) database. Boxplots illustrated the differences in IC50 values between the high- and low-risk groups for each drug (Fig. 6A). Interestingly, the IC50 values of Cisplatin, Niraparib, Olaparib and Oxaliplatin were significantly lower in the low-risk group (Fig. 6B-E), whereas the IC50 values of Afatinib, Sapitinib, SCH772984, and ULK1_4989 were lower in the high-risk group (Fig. 6F-I).

Predictive value of PCD-based risk model for chemotherapeutic drug sensitivity. (A) Boxplots showing differences in drug sensitivity between high- and low-risk groups across various chemotherapeutic agents. (B-I) Comparison of IC50 values for individual drugs between the high- and low-risk groups.
Validation of the identified PCDRGs
To validate the expression of the identified six PCDRGs in PAAD, we performed quantitative real-time PCR (qRT-PCR) on PAAD tissues and peritumoral PAAD tissues. The results showed that the mRNA expression levels of ITGA3, CDCP1, IL1RAP, CLU, PBK, and PLAU were significantly upregulated in PAAD tissues compared to peritumoral PAAD tissues (Supplementary Fig. 3A-F). Among them, the expression patterns of ITGA3, CDCP1, IL1RAP, PBK, and PLAU were consistent with the bioinformatics results from the GSE-merged cohort. These findings further confirmed the differential expression characteristics of these PCDRGs in PAAD and suggested their potential roles in tumor development and progression.
Discussion
PAAD is a highly lethal malignancy with a poor prognosis, primarily due to its asymptomatic nature in the early stages and the fact that most patients are diagnosed at advanced stages, resulting in limited treatment efficacy23. The high mortality associated with PAAD is closely linked to its complex molecular mechanisms, including changes in the tumor microenvironment, cellular heterogeneity, and resistance to therapeutic interventions24,25. In recent years, researchers have increasingly recognized the critical role of PCD in the initiation and progression of PAAD. PCD is not only an essential process for maintaining homeostasis but also serves as an escape mechanism for tumor cells under hostile conditions26. Various forms of PCD, such as pyroptosis27 cuproptosis28 and anoikis29 may all influence the growth, invasion, metastasis, and drug resistance of PAAD cells. In this study, we propose and validate a prognostic model based on PCDRGs, which has high clinical practical value. Our findings demonstrate that by integrating multiple large-scale cohorts, PCDRGs can provide more accurate prognostic information for PAAD, offering a theoretical foundation for the development of personalized treatment strategies.
In this study, we systematically evaluated the expression profiles and prognostic value of PCDRGs in PAAD by integrating multiple publicly available transcriptomic cohorts. Using a machine learning algorithm combination of StepCox[both] + Ridge, we identified six PCDRGs with potential prognostic significance: ITGA3, CDCP1, IL1RAP, CLU, PBK, and PLAU. Based on these PCDRGs, we developed a stable and reliable risk scoring model, which demonstrates strong prognostic value and suggests significant clinical application potential. It is noteworthy that although FN1 was identified as a central hub gene in the PPI network, it was not retained in the StepCox modeling process. This result suggests that topological centrality in network analysis does not always correspond to independent prognostic value in multivariate survival models. Nevertheless, FN1 may play a pivotal role in multiple cancer-related biological pathways, and its potential functional relevance remains noteworthy. It should be considered a priority candidate for future functional and translational research.
Integrin α−3 (ITGA3) is a member of the integrin family, primarily regulating cell adhesion, migration, and proliferation through interactions with the extracellular matrix30. In PAAD, the high expression of ITGA3 is closely associated with the invasiveness and migratory capacity of tumor cells31. CUB domain-containing protein 1 (CDCP1) is an effective oncogene predominantly located on the cell surface. It lies at the crossroads of several critical oncogenic and metastatic signaling pathways, including the PI3K/AKT, WNT, and RAS/ERK signal axes, plays a crucial role in cancer cell survival, growth, metastasis, and therapeutic resistance32. Interleukin-1 receptor accessory protein (IL1RAP) is a molecule involved in immune responses. It forms a receptor heterodimer with interleukin-1 receptor type I (IL1R1), mediating interleukin-1 (IL-1) activation of NFκB, which regulates inflammation and the tumor microenvironment, thus promoting tumor growth and metastasis33. Clusterin (CLU) is a secretory molecular chaperone with an important regulatory role in tumorigenesis. It modulates survival pathways in cancer cells and cancer stem cells (CSCs), enhancing tumor growth, metastasis, epithelial-mesenchymal transition (EMT), inflammation, and chemoresistance34. PDZ-binding kinase (PBK) is a kinase involved in cell cycle regulation and mitotic processes. Its overexpression allows tumor cells to bypass natural surveillance mechanisms associated with the G2/M checkpoint and lead to tumor abnormalities and mitosis by downregulating the tumor suppressor gene p53 and upregulating cyclin-dependent kinase inhibitor p2135. Plasminogen activator urokinase (PLAU) is an enzyme involved in fibrinolysis. It promotes cancer cell migration and invasion by degrading the extracellular matrix, releasing or activating various growth factors, and enhancing tumor cell proliferation through the activation of signaling pathways such as JAK-STAT, ERK, and MAPK36.
We further validated the prognostic model in high- and low-risk groups of patients from the TCGA-PAAD, GSE183795, GSE28735, GSE62452, and ICGC cohorts, and explored the relationship between risk scores and clinical features. The analysis revealed that patients in the high-risk group had significantly shorter survival times compared to those in the low-risk group, and exhibited poorer prognosis across all cohorts. Kaplan-Meier survival analysis further confirmed the prognostic value of the risk score. PCA demonstrated that the risk score effectively distinguished between different prognostic groups, indicating its broad applicability across various cohorts. Although the differences were not statistically significant in certain cohorts, the risk scores for dead patients were generally higher than those for alive, further supporting the reliability of the risk score as a prognostic tool. Notably, in the TCGA-PAAD cohort, the risk score was significantly associated with clinical stage, suggesting that it is not only an independent prognostic marker but also reflects tumor progression and invasiveness. Overall, this study highlights the potential clinical value of the model, especially its significance in tumor progression and prognosis assessment.
This study found that patients in the high-risk group not only had poorer survival outcomes but also exhibited distinct immune-suppressive features in their tumor microenvironment. Immune analysis revealed that the tumor microenvironment of the high-risk group was predominantly composed of Macrophages M0 and Macrophages M2, indicating an immunosuppressive state that may accelerate tumor progression and metastasis37. In contrast, the low-risk group exhibited a higher proportion of T cells CD4 naïve, B cells naïve, and T cells CD8, which play a key role in mediating the resistance to pathogens and the killing of malignant tumor cells, showing diverse immune regulatory functions38. These results suggest that immune microenvironment heterogeneity may play a significant role in the differences in prognosis. Furthermore, the TIDE score of the high-risk group was significantly higher than that of the low-risk group, indicating a higher likelihood of immune evasion and poorer response to immunotherapy in the high-risk patients39. Additionally, the expression of immune checkpoint genes such as PDCD1LG2, CD40, and TNFSF9 were elevated in the high-risk group, possibly correlating with enhanced immune evasion capabilities40. The immune microenvironment plays a crucial role in tumor initiation and progression, with increasing attention being given to the study of tumor immune evasion mechanisms. The immune-suppressive characteristics of high-risk patients may be linked to tumor immune evasion, providing new insights into the potential application of immunotherapy strategies in PAAD.
To investigate the relationship between our prognostic model and drug sensitivity, we utilized the GDSC database to assess the IC50 values of several chemotherapeutic agents in PAAD samples. The results revealed significant differences in drug sensitivity between the high- and low-risk groups of PAAD samples. Specifically, Cisplatin, Niraparib, Olaparib, and Oxaliplatin exhibited lower IC50 values in the low-risk group, suggesting enhanced sensitivity and potential therapeutic benefit in these patients. In contrast, drugs such as Afatinib, Sapitinib, SCH772984, and ULK1_4989 showed better therapeutic effects in the high-risk group, indicating their potential suitability for this subgroup. From a mechanistic perspective, the targets of these drugs may be functionally and mechanistically linked to our model genes. Afatinib, Sapitinib, and SCH772984 significantly downregulate CDCP1 expression by inhibiting the EGFR/MAPK/ERK and PI3K/AKT pathways41,42,43 since CDCP1 overexpression is highly dependent on the co‑activation of MAPK/ERK and AKT signaling pathways44. PBK mediates resistance to PARP inhibitors like Olaparib via the TRIM37/NF‑κB pathway45. Moreover, platinum agents such as Cisplatin and Oxaliplatin induce apoptosis by forming DNA adducts that block DNA synthesis and repair46 which may lead to increased expression of the stress‑response protein CLU47. These mechanistic links may account for the differential drug sensitivity observed between the risk subgroups and underscore the biological relevance of our six-gene signature. Collectively, our findings not only validate the risk score as a predictor of prognosis but also highlight its potential utility in guiding personalized therapeutic strategies for patients with PAAD.
Although this study provides a robust prognostic model based on PCDRGs and validates its clinical applicability in PAAD patients, there are still some limitations. First, while we integrated data from multiple public cohorts, the heterogeneity of these cohorts may affect the generalizability of the model. Samples from different data sources may differ in pathological features, treatment protocols, and experimental conditions, which could lead to less satisfactory validation results in external cohorts. Second, this study relies on existing gene expression data and lacks experimental validation from real clinical environments. Further validation, particularly in clinical samples, is still insufficient. Therefore, future studies should confirm the reliability of the model through prospective cohort studies or laboratory-based validation. Finally, although we employed various machine learning algorithms for model construction and optimization through cross-validation, the practical application of the model may still be limited by factors such as sample size, algorithm selection, and the diversity of clinical features, potentially impacting its widespread applicability in real-world clinical settings.
In summary, we developed and validated a robust six-gene prognostic model for PAAD based on programmed cell death related genes using integrated machine learning approaches. This model effectively predicts PAAD patient prognosis and was closely associated with immune features and drug sensitivity. Our findings provide a valuable tool for prognostic assessment and may help guide personalized therapeutic strategies in pancreatic cancer.
Materials and methods
Transcriptomic data collection and preprocessing
In total, transcriptomic data from 1,034 samples were collected for this study. Among them, the TCGA-PAAD cohort (4 normal and 179 tumor), along with associated clinicopathological information, was obtained from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) database. Additionally, three transcriptomic cohorts were retrieved from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database, including GSE62452 (61 normal and 69 tumor samples), GSE28735 (45 normal and 45 tumor samples), and GSE183795 (105 normal and 139 tumor samples). Furthermore, the ICGC cohort, consisting of 387 tumor samples, was obtained from the International Cancer Genome Consortium (ICGC, https://dcc.icgc.org/) database. Expression data from the three independent GEO cohorts, GSE62452, GSE28735, and GSE183795, were integrated, and batch effects were removed using the R package “sva”. Based on a 95% confidence interval, the GSE-merged cohort comprising 190 normal samples and 251 tumor samples was constructed.
To construct a comprehensive PCD-related gene set, we collected genes associated with 21 distinct types of PCD based on relevant review articles and manual curation. These included Apoptosis (n = 576), Pyroptosis (n = 50), Ferroptosis (n = 279), Autophagy (n = 366), Necroptosis (n = 101), Cuproptosis (n = 58), Parthanatos (n = 45), Entotic cell death (n = 33), PANoptosis (n = 32), Lysosome-dependent cell death (n = 218), Alkaliptosis (n = 23), Oxeiptosis (n = 23), NETosis (n = 128), Immunogenic cell death (n = 34), Anoikis (n = 333), Paraptosis (n = 65), Methuosis (n = 12), Entosis (n = 26), Disulfidptosis (n = 914), Necrosis (n = 118), and Mitoptosis (n = 29). After removing duplicates, a total of 2,684 unique PCD-related genes were retained for downstream analysis. The full list of genes is provided in Supplementary Table 4.
Screening of PCD related prognostic candidate genes and identification of variation levels
In this study, transcriptomic data from five independent cohorts, including TCGA-PAAD, GSE62452, GSE28735, GSE183795, and ICGC, were systematically integrated. The original expression data were normalized using log2(TPM + 1) transformation. Tumor samples with missing values or OS time less than 30 days were excluded from subsequent analyses. DEGs were identified in the TCGA-PAAD using the R package “Wilcoxon” and GSE-merged cohorts using the R package “limma”, respectively. Genes with |log2FC| > 0.585 and P < 0.05 were considered statistically significant. A Venn plot was used to identify overlapping PCD-related DEGs between the two cohorts. Subsequently, univariate Cox proportional risk regression analysis was performed on the PCD-related DEGs in the TCGA-PAAD, GSE-merged and ICGC cohorts using the R package “survival”. Genes significantly associated with OS (P < 0.05) were selected, and 17 intersection genes of these cohorts were selected as PCD-related prognostic candidates for downstream model construction. Notably, there was a lack of normal samples in the ICGC cohort, therefore, differential expression analysis was not performed for this cohort in the present study.
PPI networks of the prognostic candidate genes were analyzed using the STRING database (https://string-db.org/) and visualized with Cytoscape software (version 3.10). A heatmap was generated to illustrate the expression differences of the prognostic candidate genes between normal and tumor samples in the GSE-merged cohort. CNV data and single nucleotide variant (SNV) mutation data for the TCGA-PAAD cohort were downloaded from the website http://www.sxdyc.com/index. CNV alteration frequencies of the PCD-related prognostic candidate genes were calculated; a frequency greater than 0.2 was defined as “Gain,” while less than 0.2 was defined as “Loss.” A double value lollipop plot was then generated using the R package “ggpubr” to visualize the results. In addition, somatic mutation waterfall plots were generated separately for high- and low-risk groups using the R package “maftools”, visualizing the distribution differences of the top 15 most frequently mutated genes. Tumor mutation burden (TMB) scores were also calculated for each patient.
Functional and pathway enrichment analysis
Based on the candidate prognostic genes, GO and KEGG pathway enrichment analyses were performed using the R package “clusterProfiler”. The significantly enriched pathways were identified using a threshold of P < 0.05. Additionally, Hallmark gene sets were downloaded from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb), and GSVA was performed using the R package “GSVA”. Normalized Enrichment Score (t value) > 1 was used as a pathway significance screening criterion to identify highly enriched pathways in the high- and low-risk groups, respectively.
Selection of PCDRGs based on machine learning algorithms
Integrated machine learning algorithm was employed to construct a PAAD prognostic model, and 10 machine learning algorithms were integrated, including Stepwise Cox (StepCox), generalized boosted regression modeling (GBM), random survival forest (RSF), CoxBoost, elastic network (Enet), partial least squares regression for Cox (plsRcox), survival support vector machine (survival-SVM), supervised principal components (SuperPC), Lasso, and Ridge regression. A total of 117 algorithmic combinations were generated. The 17 candidate genes were selected for prognostic modeling. LOOCV was performed, using the ICGC cohort as the training set and the other four cohorts as the validation sets. Prognostic models were constructed based on all 117 algorithmic combinations. For each model, C-index was calculated, and the algorithm combination with the highest average C-index across the validation cohorts was selected as the optimal prognostic model for identifying PCDRGs. The chromosomal locations of the selected PCDRGs were obtained from the ENSEMBL database (https://asia.ensembl.org/index.html), and their genomic positions were visualized using the R package “RCircos”.
Expression and prognostic validation of PCDRGs
The Wilcoxon rank-sum test was used to evaluate the expression differences of PCDRGs between normal and tumor samples in the GSE-merged cohort. Pearson correlation coefficients were calculated to assess the correlations between the expression levels of PCDRGs. Furthermore, expression data of PCDRGs were extracted from the TCGA cohort and integrated with clinical information. Patients were divided into high and low expression groups based on the median expression value of each PCDRGs. Kaplan-Meier survival analysis was then conducted using the R packages “survival” and “survminer” to compare OS time between groups, and survival differences were assessed using the log-rank test.
Evaluation of the risk score as an independent prognostic factor in PAAD
The risk score for each patient was calculated using the following formula: RiskScore = \(\:{\sum\:}_{i=1}^{6}\beta\:i\times\:Ei\), where βi represents the coefficient of the ith gene obtained from Ridge regression under the optimal regularization parameter (Supplementary Table 5), and Ei denotes the expression level of the corresponding gene in each sample. The index i ranges from 1 to 6, corresponding to the six selected PCDRGs. Based on the median risk score, PAAD patients were divided into high-risk and low-risk groups. The relationships between risk score, survival time, and clinical events were visualized using the “plot” function in R, and Kaplan-Meier survival analysis was conducted to evaluate survival differences between the two groups. Additionally, PCA was performed using the “prcomp” function to explore the distribution characteristics of high- and low-risk group samples on the overall gene expression pattern.
To evaluate the stability and applicability of the risk scores across different cohorts, we collected survival information of PAAD patients from five independent cohorts: TCGA-PAAD, GSE62452, GSE28735, GSE183795 and ICGC. Violin plots were generated to visualize the distribution differences in risk scores between the Alive and Dead groups. Furthermore, based on clinical information from the TCGA-PAAD cohort, including T, N, M and Stage staging, the associations between the risk score and clinical characteristics were further assessed.
Unsupervised consensus clustering analysis
Consensus clustering analysis was performed using the R package “ConsensusClusterPlus” based on the expression profiles of PCDRGs. The cumulative distribution function (CDF) was calculated, and the CDF curve was plotted to determine the optimal number of clusters based on the area change and K-value stability under the CDF curve. Kaplan-Meier survival analysis was then conducted to assess survival differences among the identified clusters. Additionally, clinical information of PAAD patients from the TCGA cohort was collected, and a heatmap was generated to visually display the distribution of clinical characteristics and the expression patterns of PCDRGs across different risk groups and clusters.
Establishment and assessment of the prognostic nomogram
A nomogram model was constructed based on PCDRGs and clinical information to predict the OS rate of PAAD patients. First, clinical characteristics including age, gender, risk score, grade, N stage, T stage, and tumor position were collected from the TCGA-PAAD cohort. Univariate and multivariate Cox regression analyses were performed to identify clinical features significantly associated with patient survival. Subsequently, a nomogram prognostic model was developed based on the identified clinical variables and patient survival time. The predictive accuracy of the model was evaluated using calibration curves and ROC analysis. Furthermore, the “predict” function was used to calculate the nomogram score for each patient, and the patients were divided into high Nomogram score group and low Nomogram score group according to the median score. Kaplan-Meier survival analysis was performed to compare OS time between the two groups.
Assessment of the tumor immune microenvironment
The relative proportions of immune cell populations in each patient were estimated using the CIBERSORT algorithm. Differences in immune cell composition between high- and low-risk groups were assessed using the Mann-Whitney U test. In addition, Pearson correlation analysis was performed to construct a heatmap illustrating the correlations between PCDRGs and 22 types of immune cells or tumor microenvironment-related gene sets, aiming to reveal potential interactions. Furthermore, linear regression models were built using the “lm” function, with the expression levels of individual PCDRGs as independent variables and the relative abundance of immune cells as dependent variables, to explore the relationship between gene expression and immune cell infiltration. In addition, StromalScore, ImmuneScore, and ESTIMATEScore were calculated for each patient using the ESTIMATE algorithm. The “estimate” function was used to obtain TME scores, and Wilcoxon rank-sum tests were applied to compare the distribution of TME scores between high- and low-risk groups. TIDE scores were obtained from the TIDE database (http://tide.dfci.harvard.edu/) to assess immune evasion potential, and violin plots were used to visualize differences in TIDE scores between the two risk groups. Furthermore, based on previously published studies48 a panel of immune checkpoint-related genes was selected, and the Mann-Whitney U test was used to evaluate the differences in their expression levels between high- and low-risk groups.
Drug sensitivity prediction
The IC50 of each anticancer drug was estimated for all samples based on data from the GDSC database (http://www.cancerrxgene.org/). Drug sensitivity prediction was performed using the R package “oncoPredict” to evaluate the potential responses of high- and low-risk patients to various therapeutic agents. Differences in drug sensitivity between the two risk groups were assessed using the Wilcoxon rank-sum test, and violin plots were generated with the “ggplot2” package to visualize the results.
RNA extraction and qRT-PCR
A total of 18 pairs of PAAD tissues and matched peritumoral PAAD tissues obtained through surgical resection were included in this study. All samples were from PAAD patients who underwent surgical treatment in the Department of Hepatobiliary Pancreatic and Splentic Surgery of the Jingzhou Hospital Affiliated to Yangtze University from 2023 to 2024. All patients provided written informed consent form prior to surgery. This study has been approved by the Medical Ethics Committee of Jingzhou Hospital Affiliated to Yangtze University, and the ethics approval number is 2025-159-01.
Total RNA was extracted from each sample using TRIzol™ Reagent (Life Technologies, USA). The concentration and purity of RNA were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Total RNA was reverse transcribed into cDNA using the SweScript All-in-One RT SuperMix for qPCR kit (Servicebio Technology Co., Ltd., China). qRT-PCR was performed using the 2× Universal Blue SYBR Green qPCR Master Mix (Servicebio Technology Co., Ltd., China) on a QuantStudio™ 5 Real-Time PCR System (Applied Biosystems, USA). Gene expression levels were normalized to GAPDH and calculated using the 2^-ΔΔCt method. The primer sequences of PCDRGs are listed in Supplementary Table 6.
Statistical analysis
All statistical analyses were performed using R software (version 4.4.1). Differences in gene expression levels from qRT-PCR were analyzed using SPSS (version 26.0). Graphs were generated using GraphPad Prism (version 10.1.2). All statistical tests were two-sided, and a P < 0.05 was considered statistically significant. The normality of the data distribution was assessed using the Shapiro-Wilk test. For data with a normal distribution, Student’s t-test or one-way analysis of variance (ANOVA) was used for comparisons between two or more groups. For non-normally distributed data, the Wilcoxon rank-sum test or Mann-Whitney U test was applied for two-group or multi-group comparisons, respectively. Survival analysis was performed using Kaplan-Meier survival curves, and the log-rank test was applied to evaluate differences in OS time between high- and low-risk groups. In addition, univariate and multivariate Cox proportional hazards regression models were used to assess the independent prognostic value of clinical features and the risk score. Hazard ratios (HRs) and their corresponding 95% confidence intervals (CIs) were calculated.
Data availability
The TCGA-PAAD cohort, along with associated clinicopathological information, is publicly available in the TCGA (https://portal.gdc.cancer.gov/) database. The three transcriptomic cohorts, including GSE62452, GSE28735, and GSE183795, are retrieved from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database. The ICGC cohort is obtained from the ICGC (https://dcc.icgc.org/) database.
References
Del Chiaro, M., Sugawara, T., Karam, S. D. & Messersmith, W. A. Advances in the management of pancreatic cancer. BMJ 383, e073995. https://doi.org/10.1136/bmj-2022-073995 (2023).
Zhou, X. et al. Transcriptomics and molecular Docking reveal the potential mechanism of Lycorine against pancreatic cancer. Phytomedicine 122, 155128. https://doi.org/10.1016/j.phymed.2023.155128 (2024).
Grossberg, A. J. et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 70 (5), 375–403. https://doi.org/10.3322/caac.21626 (2020).
Cai, J. et al. Advances in the epidemiology of pancreatic cancer: trends, risk factors, screening, and prognosis. Cancer Lett. 520, 1–11. https://doi.org/10.1016/j.canlet.2021.06.027 (2021).
Rawla, P., Sunkara, T. & Gaduputi, V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol. 10 (1), 10–27. https://doi.org/10.14740/wjon1166 (2019).
Vlavcheski, F., O’Neill, E. J., Gagacev, F. & Tsiani, E. Effects of Berberine against pancreatitis and pancreatic cancer. Molecules 27 (23), 8630. https://doi.org/10.3390/molecules27238630 (2022).
Madadjim, R., An, T. & Cui, J. MicroRNAs in pancreatic cancer: advances in biomarker discovery and therapeutic implications. Int. J. Mol. Sci. 25 (7), 3914. https://doi.org/10.3390/ijms25073914 (2024).
Brozos-Vázquez, E. et al. Pancreatic cancer biomarkers: A pathway to advance in personalized treatment selection. Cancer Treat. Rev. 125, 102719. https://doi.org/10.1016/j.ctrv.2024.102719 (2024).
Chakkera, M., Foote, J. B., Farran, B. & Nagaraju, G. P. Breaking the stromal barrier in pancreatic cancer: advances and challenges. Biochim. Biophys. Acta Rev. Cancer. 1879 (1), 189065. https://doi.org/10.1016/j.bbcan.2023.189065 (2024).
Wu, L. et al. Pan-cancer analysis to character the clinicopathological and genomic features of KRAS-mutated patients in China. J. Cancer Res. Clin. Oncol. 151 (2), 94. https://doi.org/10.1007/s00432-025-06118-9 (2025).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell. Res. 29 (5), 347–364. https://doi.org/10.1038/s41422-019-0164-5 (2019).
Dai, X., Wang, D. & Zhang, J. Programmed cell death, redox imbalance, and cancer therapeutics. Apoptosis 26 (7–8), 385–414. https://doi.org/10.1007/s10495-021-01682-0 (2021).
Wang, S. et al. Machine learning reveals diverse cell death patterns in lung adenocarcinoma prognosis and therapy. NPJ Precis Oncol. 8 (1), 49. https://doi.org/10.1038/s41698-024-00538-5 (2024).
Park, W. et al. Diversity and complexity of cell death: a historical review. Exp. Mol. Med. 55 (8), 1573–1594. https://doi.org/10.1038/s12276-023-01078-x (2023).
Galluzzi, L., Buqué, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17 (2), 97–111. https://doi.org/10.1038/nri.2016.107 (2017).
Tower, J. Programmed cell death in aging. Ageing Res. Rev. 23(Pt A), 90–100. https://doi.org/10.1016/j.arr.2015.04.002 (2015).
Liu, J. et al. Programmed cell death tunes tumor immunity. Front. Immunol. 13, 847345. https://doi.org/10.3389/fimmu.2022.847345 (2022).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17 (7), 395–417. https://doi.org/10.1038/s41571-020-0341-y (2020).
Su, Z., Yang, Z., Xu, Y., Chen, Y. & Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer. 14, 48. https://doi.org/10.1186/s12943-015-0321-5 (2015).
Xie, J., Yang, Y., Gao, Y. & He, J. Cuproptosis: mechanisms and links with cancers. Mol. Cancer. 22 (1), 46. https://doi.org/10.1186/s12943-023-01732-y (2023).
Ge, J. et al. Machine learning-based identification of biomarkers and drugs in immunologically cold and hot pancreatic adenocarcinomas. J. Transl Med. 22 (1), 775. https://doi.org/10.1186/s12967-024-05590-0 (2024).
Zhu, W. et al. Integrated machine learning identifies epithelial cell marker genes for improving outcomes and immunotherapy in prostate cancer. J. Transl Med. 21 (1), 782. https://doi.org/10.1186/s12967-023-04633-2 (2023).
Wang, K. et al. The ufmylation modification of ribosomal protein L10 in the development of pancreatic adenocarcinoma. Cell. Death Dis. 14 (6), 350. https://doi.org/10.1038/s41419-023-05877-y (2023).
Hashimoto, A. & Hashimoto, S. Plasticity and tumor microenvironment in pancreatic cancer: genetic, metabolic, and immune perspectives. Cancers (Basel). 16 (23), 4094. https://doi.org/10.3390/cancers16234094 (2024).
Jiang, S. et al. Macrophage-organoid co-culture model for identifying treatment strategies against macrophage-related gemcitabine resistance. J. Exp. Clin. Cancer Res. 42 (1), 199. https://doi.org/10.1186/s13046-023-02756-4 (2023).
Wang, M., Yu, F., Zhang, Y. & Li, P. Programmed cell death in tumor immunity: mechanistic insights and clinical implications. Front. Immunol. 14, 1309635. https://doi.org/10.3389/fimmu.2023.1309635 (2023).
Yan, C., Niu, Y., Li, F., Zhao, W. & Ma, L. System analysis based on the pyroptosis-related genes identifies GSDMC as a novel therapy target for pancreatic adenocarcinoma. J. Transl Med. 20 (1), 455. https://doi.org/10.1186/s12967-022-03632-z (2022).
Chi, H. et al. Cuprotosis Programmed-Cell-Death-Related LncRNA signature predicts prognosis and immune landscape in PAAD patients. Cells 11 (21), 3436. https://doi.org/10.3390/cells11213436 (2022).
Song, W., Hu, H., Yuan, Z. & Yao, H. A prognostic model for anoikis-related genes in pancreatic cancer. Sci. Rep. 14 (1), 15200. https://doi.org/10.1038/s41598-024-65981-7 (2024).
Wang, R. Q. et al. ITGA3 participates in the pathogenesis of recurrent spontaneous abortion by downregulating ULK1-mediated autophagy to inhibiting trophoblast function. Am. J. Physiol. Cell. Physiol. https://doi.org/10.1152/ajpcell.00563.2024 (2024).
Li, R. et al. ITGA3 promotes pancreatic cancer progression through HIF1α- and c-Myc-driven Glycolysis in a collagen I-dependent autocrine manner. Cancer Gene Ther. 32 (2), 240–253. https://doi.org/10.1038/s41417-024-00864-7 (2025).
Khan, T., Kryza, T., Lyons, N. J., He, Y. & Hooper, J. D. The CDCP1 signaling hub: A target for Cancer detection and therapeutic intervention. Cancer Res. 81 (9), 2259–2269. https://doi.org/10.1158/0008-5472.Can-20-2978 (2021).
Hansen, N. et al. Blocking IL1RAP on cancer-associated fibroblasts in pancreatic ductal adenocarcinoma suppresses IL-1-induced neutrophil recruitment. J. Immunother Cancer. 12 (12), e009523. https://doi.org/10.1136/jitc-2024-009523 (2024).
Praharaj, P. P., Patra, S., Panigrahi, D. P., Patra, S. K. & Bhutia, S. K. Clusterin as modulator of carcinogenesis: A potential avenue for targeted cancer therapy. Biochim. Biophys. Acta Rev. Cancer. 1875 (2), 188500. https://doi.org/10.1016/j.bbcan.2020.188500 (2021).
Han, Z., Li, L., Huang, Y., Zhao, H. & Luo, Y. PBK/TOPK: A therapeutic target worthy of attention. Cells 10 (2), 371. https://doi.org/10.3390/cells10020371 (2021).
Wang, H., Lu, L., Liang, X. & Chen, Y. Identification of prognostic genes in the pancreatic adenocarcinoma immune microenvironment by integrated bioinformatics analysis. Cancer Immunol. Immunother. 71 (7), 1757–1769. https://doi.org/10.1007/s00262-021-03110-3 (2022).
Wang, Y. N. et al. Vinblastine resets tumor-associated macrophages toward M1 phenotype and promotes antitumor immune response. J. Immunother Cancer. 11 (8), e007253. https://doi.org/10.1136/jitc-2023-007253 (2023).
Sun, L., Su, Y., Jiao, A., Wang, X. & Zhang, B. T cells in health and disease. Signal. Transduct. Target. Ther. 8 (1), 235. https://doi.org/10.1038/s41392-023-01471-y (2023).
Tian, Y. et al. Exploring the tumor microenvironment of breast cancer to develop a prognostic model and predict immunotherapy responses. Sci. Rep. 15 (1), 12569. https://doi.org/10.1038/s41598-025-97784-9 (2025).
Guo, Y. et al. Novel immune checkpoint-related gene model to predict prognosis and treatment responsiveness in low-grade gliomas. Heliyon 9 (9), e20178. https://doi.org/10.1016/j.heliyon.2023.e20178 (2023).
Wang, Y. et al. Clinical efficacy and identification of factors confer resistance to Afatinib (tyrosine kinase inhibitor) in EGFR-overexpressing esophageal squamous cell carcinoma. Signal. Transduct. Target. Ther. 9 (1), 153. https://doi.org/10.1038/s41392-024-01875-4 (2024).
Pourjamal, N. et al. Comparison of trastuzumab emtansine, trastuzumab deruxtecan, and disitamab Vedotin in a multiresistant HER2-positive breast cancer lung metastasis model. Clin. Exp. Metastasis. 41 (2), 91–102. https://doi.org/10.1007/s10585-024-10278-2 (2024).
Long, Y. et al. FOXD1-dependent RalA-ANXA2-Src complex promotes CTC formation in breast cancer. J. Exp. Clin. Cancer Res. 41 (1), 301. https://doi.org/10.1186/s13046-022-02504-0 (2022).
Saponaro, M. et al. CDCP1 expression is frequently increased in aggressive urothelial carcinoma and promotes urothelial tumor progression. Sci. Rep. 13 (1), 73. https://doi.org/10.1038/s41598-022-26579-z (2023).
Ma, H. et al. PBK drives PARP inhibitor resistance through the TRIM37/NFκB axis in ovarian cancer. Exp. Mol. Med. 54 (7), 999–1010. https://doi.org/10.1038/s12276-022-00809-w (2022).
Mehmood, R. K. Review of cisplatin and oxaliplatin in current Immunogenic and monoclonal antibody treatments. Oncol. Rev. 8 (2), 256. https://doi.org/10.4081/oncol.2014.256 (2014).
Stein, T., Bitsina, C., Seiler, M., Schmugge, M. & Franzoso, F. D. Clusterin can mediate apoptosis-induced molecular mechanisms in immune thrombocytopenia. Blood Vessels Thromb. Hemost. 1 (3), 100012. https://doi.org/10.1016/j.bvth.2024.100012 (2024).
Hu, F. F., Liu, C. J., Liu, L. L., Zhang, Q. & Guo, A. Y. Expression profile of immune checkpoint genes and their roles in predicting immunotherapy response. Brief. Bioinform. 22 (3), 1–12. https://doi.org/10.1093/bib/bbaa176 (2021).
Funding
The present study was supported by the Natural Science Foundation of Hubei Province (2022CFB346).
Author information
Authors and Affiliations
Contributions
The experiment was conceived and designed by Bing Wang and Zhida Long. Bing Wang, Zhida Long, Xun Zou and Zhengang Sun conducted experiments, performed result processing and statistical analysis. Bing Wang wrote the manuscript and Yuanchu Xiao reviewed it. All authors agree to be responsible for all aspects of the work and to ensure its completeness and accuracy.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, B., Long, Z., Zou, X. et al. Machine learning-based construction of a programmed cell death-related model reveals prognosis and immune infiltration in pancreatic adenocarcinoma patients. Sci Rep 15, 25156 (2025). https://doi.org/10.1038/s41598-025-10847-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-10847-9
