
Abstract
The development of effective antidiabetic agents remains a critical challenge in diabetes management. In this study, we introduce novel 1,2,4-triazole-based derivatives designed as dual inhibitors of α-amylase and α-glucosidase, key enzymes in carbohydrate metabolism. Molecular docking identified six promising candidates, with compounds 4 and 10 showing the highest potency. Both compounds exhibited strong α-glucosidase inhibition (IC50 = 0.27 ± 0.01 µg/mL and 0.31 ± 0.01 μg/mL, respectively), surpassing acarbose, and also demonstrated potent α-amylase inhibition (IC50 = 0.19 ± 0.01 μg/mL and 0.26 ± 0.01 μg/mL, respectively). Structure–activity relationship analysis highlighted the crucial role of acetyl and bromo substituents in enhancing enzyme inhibition. These findings position triazole-based scaffolds as promising candidates for the development of next-generation antidiabetic therapies.
Similar content being viewed by others
Introduction
Diabetes mellitus is a widespread and increasingly prevalent metabolic disorder characterized by chronic hyperglycemia caused by insufficient insulin production, impaired insulin action, or both1. It has become a critical global health concern, with serious social and economic implications2. According to the International Diabetes Federation (IDF), an estimated 537 million adults worldwide were living with diabetes in 2021, and this number is projected to rise to 783 million by 20453. The condition is associated with high rates of morbidity and mortality due to its chronic complications, including cardiovascular diseases, retinopathy, nephropathy, and neuropathy4. These complications arise from sustained hyperglycemia and oxidative stress, which cause progressive damage to blood vessels and organ systems over time.
Type 2 diabetes mellitus (T2DM) accounts for 90–95% of all diabetes cases and typically manifests in adults, although its incidence is rising among younger populations due to sedentary lifestyles and poor dietary habits5. Unlike type 1 diabetes, which is primarily autoimmune in origin and results from the destruction of pancreatic β-cells, T2DM is characterized by insulin resistance combined with a relative deficiency in insulin secretion6. The primary therapeutic goals in managing T2DM include maintaining optimal blood glucose levels, preventing the onset or progression of complications, and improving patient quality of life. While numerous pharmacological agents are available for this purpose, including insulin sensitizers, insulin secretagogues, and enzyme inhibitors, none are free from side effects or long-term safety concerns.
In recent years, there has been an increasing focus on postprandial glucose regulation as a key aspect of glycemic control7. Elevated postprandial blood glucose levels have been strongly associated with the development of diabetic complications, and clinical studies suggest that targeting postprandial hyperglycemia is more effective in reducing cardiovascular risk than focusing solely on fasting glucose levels7. One promising strategy in this context involves the inhibition of carbohydrate-hydrolyzing enzymes such as α-amylase and α-glucosidase, which play critical roles in the digestion of complex carbohydrates.
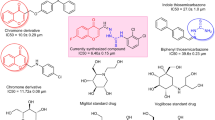
α-Amylase is responsible for catalyzing the hydrolysis of α-1,4-glycosidic bonds in starch, producing maltose and oligosaccharides, which are then further broken down by α-glucosidase into glucose for absorption through the intestinal epithelium8,9. Inhibiting these enzymes can effectively reduce the rate of glucose release into the bloodstream, thereby preventing sharp post-meal spikes in blood sugar levels. Several drugs targeting these enzymes, including acarbose, miglitol, and voglibose, have been approved for clinical use and have shown efficacy in lowering postprandial glucose levels Fig. 110,11. However, their application is often limited by adverse gastrointestinal effects such as flatulence, abdominal discomfort, and diarrhea, largely due to undigested carbohydrates undergoing fermentation by colonic bacteria10,11. These drawbacks underscore the urgent need to develop alternative enzyme inhibitors that retain efficacy while improving tolerability and patient adherence.

Acarbose, miglitol, voglibose, and some FDA approved bioactive agents containing 1,2,4-triazole ring structure.
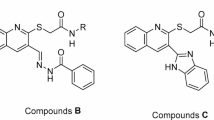
Among the diverse classes of heterocycles explored for antidiabetic drug discovery, the 1,2,4-triazole ring has gained considerable attention due to its versatile pharmacological profile and favorable physicochemical properties Fig. 112. The 1,2,4-triazole nucleus is a five-membered ring containing three nitrogen atoms, which imparts strong hydrogen bonding potential, metal ion coordination capability, and metabolic stability. These characteristics make triazoles attractive candidates for drug design, particularly in enzyme inhibition13. Previous studies have reported the efficacy of 1,2,4-triazole derivatives in a range of therapeutic areas, including anticancer13, antimicrobial14,15, antiviral16, and anti-inflammatory17 applications. The versatility of the 1,2,4-triazole ring arises from its ability to form hydrogen bonds and coordinate with metal ions, enabling strong interactions with various biological targets18,19. Notably, their potential as antidiabetic agents has also been explored, with promising findings demonstrating that certain triazole-based molecules exhibit inhibitory activity against α-glucosidase20,21. Despite this progress, there remains a relative paucity of triazole derivatives designed specifically for dual inhibition of both α-amylase and α-glucosidase22,23,24, a strategy that could offer synergistic benefits in controlling postprandial hyperglycemia.
To address this therapeutic gap, we designed and synthesized a new class of 1,2,4-triazole-based compounds incorporating a thioacetamide bridge and para-substituted phenyl rings Fig. 2. This hybrid molecular architecture was chosen to synergistically enhance biological activity by combining the enzyme-binding potential of the triazole scaffold with the flexibility and hydrogen bonding capacity of the thioacetamide moiety25,26,27. The thioacetamide linker was strategically introduced to facilitate interactions with polar residues in the enzyme active sites, while allowing structural adaptability to accommodate diverse binding pockets.

Structures of α-amylase and/or α-glucosidase inhibitors containing 1,2,4-triazole nucleus (I, II, III), α-glucosidase inhibitors containing thioacetamide linker (IV, V, VI) structures, and optimization strategy of designed molecules.
In the present study, our design strategy aimed to develop hybrid molecules bearing a 1,2,4-triazole nucleus connected via a thioacetamide bridge to various para-substituted phenyl rings Fig. 3. The substituents, including electron-withdrawing groups (such as acetyl and bromo) and electron-donating groups (such as methoxy), were chosen to modulate electronic distribution, hydrogen bonding capability, and lipophilicity28. These variations were expected to influence enzyme binding affinity and selectivity based on steric and electronic complementarity with the active sites of α-amylase and α-glucosidase29,30. Molecular modeling tools, including docking simulations and pharmacokinetic profiling, were employed to prioritize candidates for synthesis and biological evaluation. In particular, docking analyses guided the selection of substituents by predicting favorable interactions with key catalytic residues, such as TRP 59, ASP 197, and GLU 277, in the respective enzymes25,26,27. Additionally, SwissADME predictions ensured that lead compounds complied with key drug-likeness rules (e.g., Lipinski’s Rule of Five) and exhibited favorable absorption and solubility characteristics31,32,33,34.

Rational of the work.
Thus, the objective of this work was to design, synthesize, and characterize a new series of 1,2,4-triazole derivatives as potential dual inhibitors of α-amylase and α-glucosidase. Our approach integrates rational drug design, computational modeling, and biochemical validation to identify potent and selective antidiabetic agents. This study not only addresses the limitations of current enzyme inhibitors but also expands the structural diversity of small molecules available for the treatment of T2DM.
Results and discussion
Chemistry
The synthesis of the 1,2,4-triazole based thioacetamide target compounds is depicted in Scheme 1. The synthetic protocol depends on chemical modification of 2-(4-methoxyphenyl) acetohydrazide S1 and aniline derivatives S3 to 1,2,4-triazole-3-thiol derivative S2 and substituted 2-chloro-N-phenylacetamides S4, respectively, followed by base catalyzed-alkylation of the thiol group of intermediate S2 using the alkylating reagents S4. Initially, the intermediate, 5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazole-3-thiol (S2), was synthesized from the starting material 2-(4-methoxyphenyl) acetohydrazide S1 following previously reported procedures35,36. Next, the 2-chloro-N-phenylacetamide derivatives S4 were synthesized from aniline derivatives S3 using standard procedures37,38. Finally, the target 1,2,4-triazole compounds 1–10 were synthesized by alkylating the thiol group of intermediate S2 with the alkylating reagents S4 as a source of thioacetamide function. This reaction was carried out under basic conditions using sodium hydroxide in refluxing ethanol to afford the desired compounds 1–10 in moderate to high yield (54–93%), except for compound 8, which was synthesized smoothly at room temperature using triethylamine as mild catalytic base with yield (89%). The reaction progress was monitored via TLC using EtOAc/Petroleum Ether (1:1) as the solvent system, which confirmed the complete conversion of the starting materials. The structures of the synthesized target compounds were confirmed by IR, 1H NMR and 13C NMR analyses. For instance, compound 4 showed three stretching bands appeared at 3230, 1680 and 1520 cm−1 for NH, C=O, and C=N functions, respectively. Additionally, the absence of the thiol stretching band indicating successful alkylation. It’s 1H NMR spectrum, including three singlet characteristic signals at δ = 3.67, 3.89 and 4.13 ppm assigned for (CH3) of the p-methoxy group and the two methylene protons of the alkylated side chain and thioacetamide linker, respectively. The aromatic protons resonated in the range 6.71–7.92 ppm, while, the singlet proton of the (NH) of the thioacetamide linker appeared at δ 10.65 ppm. Furthermore, the 13C NMR spectrum of compound 1 showed three signals at δc = 29.89, 36.47 and 55.01 ppm for sp3 carbons, whereas, the thiol-linked carbon appeared at δc 149.9 ppm and the amidic carbonyl group at δc 166.3 ppm. The structure of the new 1,2,4-triazoles 2, 5 and 7 has been illustrated from their spectral data. For example, their IR spectra revealed the amidic (NH and C=O) and C=N functions at 3200–3229, 1641–1681, and 1490–1588 cm−1, respectively. The 1H NMR analysis of the triazole derivative 2 indicates the presence of two methyl, methoxy, two methylene, and amidic NH protons at δH 2.12, 2.22, 3.67, 3.91, 4.10 and 9.61 ppm as six singlets, respectively. The aryl protons appeared at δH 6.72, 6.80, 6.87, 7.06, 7.25 and (7.49–7.56) as five doublets and one multiplet. Its 13C NMR provided 22 signals at δC 17.31 (CH3), 20.71 (CH3), 29.88 (CH2), 36.40 (CH2-S), 54.98 (O-CH3), 113.7, 124.8, 125.8, 127.2, 127.5, 128.0, 129.4, 129.7, 129.9, 130.1, 132.8, 134.9, 135.7 (Ar–C), 150.0 (C-3), 154.8 (C-5), 157.9 (Ar–C) and 165.7 ppm (C=O) for sp3and sp2 carbons. The protons and carbons for the other compounds 5 and 7 are appeared at the expected field. Also, the chemical structure of compound 8 was elucidated from its spectral data. It was noted the presence of a broad band recorded in its IR spectrum at 1696 cm−1 that provided two (C=O) functions (ester and amide), beside amidic NH at 3220 cm−1. Furthermore, its 1H NMR assigned methoxy, two methylene, and NH-amide protons at δH 3.67, 3.89, 4.14 and 10.65 ppm as four independent singlets, respectively. While, the acetyl protons split as triplet and quartet at δH 1.30 and 4.27 ppm, respectively, with 3J = 6.8 Hz. Additionally, the aromatic protons are split at δH 6.71, 6.81, 7.25, 7.50, 7.67 and 7.91 ppm as six doublets. Another evidence for the synthesis of 1,2,4-triazole-based derivative 8 comes from its 13C NMR that demonstrated 21 signals at δC = 14.20 (CH3), 30.04 (CH2), 37.05 (CH2-S), 54.99 (O-CH3), 60.59 (O-CH2), for sp3 carbons and 113.7, 118.2, 123.9, 127.2, 127.5, 129.4, 129.7, 129.9, 130.3, 132.7, 143.0 (Ar–C), 149.8 (C-3), 154.9 (C-5), 157.9 (Ar–C), 165.2 (C=O, amide) and 166.2 ppm (C=O, ester) for sp2 carbons. Similar spectral patterns were observed for other derivatives, with variations depending on the substituents. The spectral data for all compounds corroborate their proposed structures and confirm the successful synthesis of the 1,2,4-triazole derivatives. These results validate the designed synthetic strategy and provide a solid foundation for further biological evaluation.

Pathway to the target 1,2,4-triazole new substances containing thioacetamide (1–10). Reagents and conditions: (I) Phenyl isothiocyanate, EtOH, reflux; (II) 10% NaOH, reflux; (III) HCl, r. t; (IV) 2-Chloroacetyl chloride, acetic acid, r. t; (V) Substituted 2-chloro-N-phenylacetamide, NaOH, EtOH, reflux for compounds (1–7) and (9–10); (VI) Substituted 2-chloro-N-phenylacetamide, triethylamine, EtOH, r. t. for compound 8.
Structure activity relationship
The inhibitory activities of the 1,2,4-triazole derivatives (3–10) were rationalized through structural analysis, focusing on substituent effects at the para-position of the phenyl ring Fig. 4. Key observations include:
-
1.
Electron-Withdrawing Groups (EWGs):
-
Acetyl (Compound 4): The carbonyl group’s hydrogen-bonding potential and moderate steric bulk optimized interactions with catalytic residues (e.g., ALA 106 in α-amylase; GLU 411 in α-glucosidase), as evidenced by docking scores (− 7.758 and − 8.116 kcal/mol, respectively).
-
Bromo (Compound 10): Halogen bonding with residues (e.g., ARG 315) and balanced lipophilicity enhanced binding affinity.
-
2.
Steric and Electronic Trade-offs:
-
Ester (Compound 8): The CH3CH2OCO group’s bulkiness reduced α-amylase inhibition but retained partial α-glucosidase activity due to flexibility in the latter’s active site.
-
Carboxyl (Compound 6): High polarity limited membrane permeability and enzyme access, explaining its weaker activity.
-
3.
Electron-Donating Groups (EDGs):
-
Methoxy (Compound 5): Lower activity compared to EWGs suggested that EDGs diminish charge stabilization in the enzyme’s catalytic pocket.
These SAR observations correlate with trends reported in Sect. 2.5 and are supported by molecular docking and MD simulation results. Together, they provide a coherent structural basis for understanding how substituent variation modulates dual inhibitory activity.

Summarized SAR of the produced 1,2,4-triazole containing thioacetamide.
These SAR observations correlate with trends reported in Sect. "Biological activity" and are supported by molecular docking and MD simulation results. Together, they provide a coherent structural basis for understanding how substituent variation modulates dual inhibitory activity.
Analysis of ADME characteristics of ligand
The most promising compounds, 4, 6, 8, and 10, were subjected to expected bioinformatics analysis to forecast their physicochemical and pharmacokinetic features, as well as if they may be bioavailable orally pharmaceutical agents relative to forxiga (Dapagliflozin) and acarbose. The Swiss ADME online tool was applied for assessing the drug-likeness of and the characteristic features of ADME of compounds. The test compounds had good oral bioavailability and excellent physicochemical properties, including size (MW between 150 and 500 g/mol), flexibility (maximum 10 rotatable bonds), polarity (TPSA between 20 and 140 Å), solubility (Log S less than 6), and lipophilicity (Log P between − 0.7 and + 5.0). It was determined that compounds varied in solubility from partially soluble to soluble, as opposed to compound 10, which is weakly soluble39,40. All studied compounds followed Lipinski’s rule of five31, M.wt. ≤ 500 g/mol except compounds 8 and 10, log P ≤ 5, TPSA ≤ 140 Å, HBA ≤ 10, and HBD < 5 are indicated32. As shown in Table 1, the designed compounds generally complied with Lipinski’s rule of five, exhibiting molecular weights below 510 g/mol, log P values under 533, and acceptable topological polar surface area (TPSA) values between 94.34 and 131.64 Å2. In contrast, acarbose violates multiple Lipinski parameters, including its high molecular weight (645.6 g/mol), excessive number of hydrogen bond donors (14), and TPSA (329.01 Å2), which contribute to its poor oral bioavailability. These findings support the favorable drug-likeness and absorption potential of the synthesized compounds. Compounds 4, 6, 8, and 10’s physicochemical characteristics and drug-likeness are presented in Table 1 in comparison to forxiga (Dapagliflozin) and acarbose. Furthermore, the ADME characteristics analysis demonstrated that drugs might be taken up by the intestine, with the white of the egg model34 demonstrating considerable gastrointestinal absorption for all substances. The investigation showed that the drugs could not be taken up passively across the blood–brain barrier, as proven by the egg yolk model34. Table 2 compares the ADME analysis results for compounds 4, 6, 8, and 10 to those for forxiga (Dapagliflozin) and acarbose. Generally, every ruling concept that support the conclusion that compounds 4, 6, 8, and 10 exhibit favorable drug-likeness, suggesting that these substances may fulfil the permeability and bioavailability criteria of the cell membrane.
Molecular docking
To achieve greater knowledge of how binding forces operate of the 1,2,4-triazole derivatives, a molecular docking analysis of four chosen substances was carried out towards the pancreatic α-amylase’s active regions and Saccharomyces cerevisiae α-glucosidase. Forxiga (Dapagliflozin) and acarbose were used to target α-amylase’s binding regions (PDB ID: 4X9Y), (PDB ID: 2QV4), (PDB ID: 1OSE) and (PDB ID: 5E0F) and α-glucosidase (PDB ID: 3A4A) and (PDB ID: 3AJ7). Compounds 1–10 exhibited stable docking poses in the α-amylase and α-glucosidase binding sites, generating multiple polar and non-polar interactions with the contained amino acid regions, as shown in Table 3S in supporting information file. The docked compounds 4, 6, 8, and 10 showed the best results compared with forxiga (Dapagliflozin) and acarbose as reference drugs.
Antidiabetic to the target enzyme α-amylase (2QV4)
Ligand interactions of forxiga (Dapagliflozin) and Acarbose as reference drugs
The molecular docking research on forxiga and acarbose revealed a variety of interactions with binding site residues. Forxiga (Dapagliflozin), the ASP 300, ASP 197, and HIS 299 residues produced a hydrogen bonding with the hydroxy group’s lone pair of oxygen, as illustrated in Fig. 5. Acarbose can hydrogen bond with GLU 233 (A) H-donor, ASP 197 (A) H-donor, and TRP 59, which are participating in the σ-H-interaction with the cyclic ring π-system.

Two- and three-dimensional representations for enzyme α-amylase (PDB ID: 2QV4) for forxiga and acarbose.
Ligand interactions of compound 4,6,8 and 10
Compound 4, the carbonyl group has the capability to bind with the ALA 106 with hydrogen bonding having a docking result of − 7.758 kcal/mol, while, compounds 6 and 10 are involved in a σ-H-interaction with the GLN 63 and LEU 162 with docking scores of − 7.396 and − 7.682 kcal/mol, correspondingly, as shown in Table 3 and Fig. 6.

Two- and three-dimensional representations for enzyme α-amylase (PDB ID: 2QV4).
Antidiabetic to the target enzyme α-glucosidase (3AJ7)
Ligand interactions of forxiga and acarbose as reference drugs
The molecular docking study of forxiga (Dapagliflozin) and acarbose demonstrated multiple types of interactions with the regions of the binding location. For forxiga (Dapagliflozin), the ASP 352, 4ASP 215 residues exhibited a hydrogen bonding interaction with the lone pair of oxygen of the hydroxy group and CH, as shown in Fig. 7. While the analysis of acarbose’s molecular docking is also able to hydrogen bond with more amino acids, as mentioned in Table 4.

Two- and three-dimensional representations for enzyme α-glucosidase (PDB ID: 3AJ7) for forxiga and acarbose.
Ligand interactions of compound 4,6,8, 9 and 10
All the examined compounds scored better than the commercial drug forxiga (Dapagliflozin). In compound 4, the sulfur atom is able to hydrogen bond with the GLU 411 and PRO 312 participated in the σ-H-interaction with the phenyl ring π-system with a docking result of − 8.116 Kcal/mol, while compound 6 is able to hydrogen bond with the TYR 158, GLU 277, GLU 277, HIS 280, and TYR 158 involved in a σ-H-interaction with the phenyl ring. The molecular docking studies of compounds 8 and 9 are also able to hydrogen bond with more amino acids, whereas compound 10 is able to bond with ARG315 by pi-H, as mentioned in Table 4, Fig. 8.

Two- and three-dimensional representations for enzyme α-glucosidase (PDB ID: 3AJ7).
Comparative analysis of docking interactions
Acarbose and forxiga (dapagliflozin), used as reference drugs, formed multiple hydrogen bonds and π-interactions with catalytic residues in α-amylase and α-glucosidase. Acarbose exhibited strong hydrogen bonding with GLU 233 and ASP 197, and π-stacking with TRP 59 in α-amylase, with a docking score of − 8.766 kcal/mol. In comparison, compound 4 formed hydrogen bonds with ALA 106 and π-interactions with PRO 312 in α-glucosidase, yielding a docking score of − 8.116 kcal/mol, while compound 10 engaged ARG 315 via halogen bonding and π-H interaction, with a comparable score of − 8.555 kcal/mol Table 5.
For α-amylase, forxiga interacted with ASP 197, ASP 300, and HIS 299, with a docking score of − 6.780 kcal/mol. Compound 4 achieved a stronger binding score (− 7.758 kcal/mol) through hydrogen bonding with ALA 106 and π-interactions. Compound 10 also surpassed forxiga, forming stable contacts with GLN 63 and LEU 162 Table 5.
These results suggest that the designed compounds not only mimic key interactions seen in reference drugs but also exploit additional binding pockets via halogen bonding and optimized hydrogen bond networks, enhancing affinity and selectivity toward both target enzymes. This comparative analysis supports the relevance of the docking predictions and highlights the improved binding performance of compounds 4 and 10 over existing clinical inhibitors. The comparative analysis confirms that our triazole derivatives achieve potent inhibition through focused interactions with catalytic residues, unlike acarbose’s nonspecific binding. This aligns with their superior in vitro activity (IC50 values 0.19–0.31 μg/mL vs. acarbose’s 0.34 μg/mL).
Biological activity
In vitro testing with α-amylase
The substances were examined for their potential to inhibit α-amylase in vitro. Acarbose is a widely used standard inhibitor of α-amylase. The superior inhibitory activity of compounds 4 and 10 over acarbose highlights their therapeutic potential and warrants further investigation into their pharmacokinetics and safety profiles. Two compounds, including 4 (IC50 = 0.19 ± 0.01 µg/mL) and 10 (IC50 = 0.26 ± 0.01 µg/mL), showed high activity relative to the reference acarbose. The presence of electron-withdrawing substituents (acetyl in compound 4 and bromo in compound 10 appears to enhance binding through electronic effects, stabilizing the inhibitor-enzyme complex. Compound 4’s stronger activity suggests that steric and electronic factors of the acetyl group are better aligned with the enzyme’s active site geometry.
Figure 9 shows a graphical depiction of the synthesized compounds and displays their IC50 values. The remaining four derivatives (3, 6, 8, and 9) demonstrated superb to less suppression with corresponding IC50 values of 0.37 ± 0.01, 1.24 ± 0.04, 1.45 ± 0.05, and 0.63 ± 0.02 µg/mL. Compound 3 showed moderate activity, likely due to a balance of steric and electronic effects that do not optimize binding. Compounds 6 and 8 showed weak activity, which can be attributed to unfavorable steric hindrance (in compound 8) or excessive polarity (in compound 6), both of which disrupt efficient binding. Detailed results for α-amylase activity are shown in Table 1S.

α-amylase activity of derivatives 3, 4, 6, 8, 9, and 10 compared to acarbose.
In vitro testing with α-glucosidase
The synthesized compounds were evaluated at concentrations ranging from 63 to 1000 µg/ml for their ability to block α-glucosidase.
The outcomes are presented in Fig. 10. The charts show that increasing concentration leads to higher activity levels. Yet, the IC50 value provided the most precise comparability. Compounds’ inhibitory effects on α-glucosidase were compared using their IC50 values. A total of six substances were explored for potential α-glucosidase inhibitory effects. The substances had a substantial inhibitory impact on α-glucosidase, as shown in the results.

α-Glucosidase activity of derivatives 3, 4, 6, 8, 9, and 10 compared to acarbose.
The results show that the compounds have substantial inhibitory action. Between these substances, 3, 4, and 10 possess the strongest inhibitory action, as demonstrated by IC50 values of 0.36 ± 0.01, 0.27 ± 0.01, and 0.31 ± 0.01 µg/mL, correspondingly, followed by 6, 8, and 9 with IC50 values of 0.83 ± 0.03, 0.67 ± 0.02, and 0.77 ± 0.02 µg/mL, correspondingly. Detailed results for α-glucosidase activity are shown in Table 2S.
Mechanism of Action, α-Glucosidase inhibitors delay carbohydrate breakdown into glucose, reducing postprandial blood sugar levels. The strong inhibition observed with compounds 3, 4, and 10 suggests they effectively block the enzyme’s active site, likely through well-placed functional groups that form stable enzyme-inhibitor complexes. Compounds 4 and 10 feature electron-withdrawing groups (acetyl and bromo, respectively) at the para-position, which enhance binding affinity by stabilizing interactions within the enzyme’s catalytic domain. In contrast, compounds 6, 8, and 9 have bulkier or less electron-attracting substituents that may introduce steric hindrance or suboptimal alignment within the binding pocket, leading to reduced efficacy.
Therapeutic Potential: The potency of compounds 3, 4, and 10 suggests they could serve as promising leads for developing α-glucosidase inhibitors. Their IC50 values outperform or rival existing drugs, highlighting their relevance for managing hyperglycemia in diabetes.
Molecular dynamics analysis
To evaluate the binding affinity of compounds 4 and 10 with the target proteins α-amylase (PDB ID: 2QV4) and α-glucosidase (PDB ID: 3AJ7), we conducted molecular dynamics simulations using GROMACS 2024.2. The 4:2QV4, 10:2QV4, 4:3AJ7, and 10:3AJ7 complexes were simulated over a period of 50 ns.
The results, illustrated in Figs. 11a–f and 12a–f, include various simulation plots depicting the behavior of the complexes. Root Mean Square Deviation (RMSD) analysis revealed that the RMSD values remained consistently below 1.7 nm, indicating stable binding throughout the simulation period. This suggests that the overall structures of the 4:2QV4, 10:2QV4, 4:3AJ7, and 10:3AJ7 complexes remained relatively unchanged, reflecting stability in the binding interactions. Root Mean Square Fluctuation (RMSF) analysis, which assesses the flexibility of individual residues, showed minimal fluctuations. This indicates that while the complexes were stable, the binding region exhibited limited mobility or flexibility, pointing to a robust and rigid interaction between the ligands and the proteins.

(a) Plot of protein backbone RMSD against time; (b) RMS fluctuation; (c) solvent-accessible surface area; (d) radius of gyration; (e) volume; and (f) density analysis for the complexes 3AJ7: compound 10 (pink) and 3AJ7: compound 4 (cyan).

(a) Plot of protein backbone RMSD against time; (b) RMS fluctuation; (c) solvent-accessible surface area; (d) radius of gyration; (e) volume; and (f) density analysis for the complexes 2QV4: compound 10 (orange) and 2QV4: compound 4 (green).
Solvent Accessible Surface Area (SASA) analysis reached a maximum value of approximately 270 nm2 for 3AJ7 and 205 nm2 for 2QV4, suggesting that the interaction surfaces between compound 4 or compound 10 and 2QV4 or 3AJ7 were sufficiently exposed to the solvent, consistent with expected binding dynamics. Radius of Gyration (Rg) analysis further supported the stability of the complexes, demonstrating that the binding of compounds 4 and 10 to 3AJ7 and 2QV4 remained stable throughout the simulation. The simulation results indicate that compounds 4 and 10 bind stably to the target proteins 2QV4 and 3AJ7, with consistent RMSD values and minimal RMSF fluctuations. These findings confirm the stability and robustness of the 4:2QV4, 10:2QV4, 4:3AJ7, and 10:3AJ7 complexes throughout the 50 ns simulation period.
Experimental
General information
Reagents, chemical solvents, and precursors were purchased from reliable vendors including Sigma-Aldrich, Loba, Merck, Acros, and El-Gomhoria. The melting points (°C) are unadjusted and were measured using open capillaries and a Stuart SMP10 melting point device. Using silica gel 60 GF254 (E-Merck) for thin layer chromatography, all reactions were tracked and visible under a UV lamp set at a wavelength (λ) of 254 nm. NMR spectra were acquired using dimethyl sulfoxide (DMSO)-d6 as the solvent and tetramethylsilane (TMS) as an internal standard on a Brüker high-performance digital FT-NMR spectrometer Advance 400 MHz for 1H NMR and 100 MHz for 13C NMR (see the supporting information for the spectra). In the 1H NMR spectrum, the letters ‘s’, ‘d’, ‘t’, ‘q’, and ‘m’ stand for singlet, doublet, triplet, quartet and multiplet, respectively. The coupling constants (J) and chemical shifts (δ) are reported in hertz (Hz) and parts per million (ppm), respectively. The Perkin-Elmer CHN-elemental analyzer was used for the microanalysis.
Chemistry
Standard synthetic method of compound (S2)
Compound S2 is a known intermediate and was synthesized according to previously reported methods35,36. A mixture of 2-(4-methoxyphenyl) acetohydrazide (1.0 equiv.) and phenyl isothiocyanate (1.1 equiv.) in ethanol was refluxed for 4 h. After cooling, 10% NaOH was added, and the mixture was refluxed for an additional 2 h. Acidification with HCl yielded the crude product, which was recrystallized from ethanol (Yield: 75%, m.p.: 180–182 °C).
Standard synthetic method of compounds (S4)
Compounds S4 are known intermediates and were synthesized according to previously reported methods37,38. Substituted aniline (1.0 equiv.) was reacted with 2-chloroacetyl chloride (1.2 equiv.) in acetic acid at 0 °C to r. t. for 3 h. The precipitate was filtered and recrystallized from ethanol/water (Yields: 70–85%).
Standard synthetic method of compounds (1–5) and compounds (7, 10)
A mixture of compound S2 (0.3 g, 1 mmol) and NaOH (0.06 g, 1.5 mmol) was refluxed with stirring in absolute ethanol (5 ml) for 0.5 h, then cooled to r. t. The appropriate 2-chloro-N-phenylacetamide derivative S4 (1.05 mmol) in absolute ethanol (5 ml) was dropped gradually to the above reaction mixture and then refluxed with stirring for an additional 1.5 h. Next, under lower pressure, the reaction mixture was evaporated until it was dry, yielding residue. After adding ice water to the residue, stir the mixture for an additional 20 min. In order to obtain the crude products, the precipitates were finally vacuum-filtered, completely cleaned with water, and dried. Recrystallization of the crude products from the suitable solvent gives the pure target compounds.
N-(2-chlorophenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (1)
White fluffy material (absolute ethanol: glacial acetic acid, 9: 1); yield: 72%; m.p.: 200–202 °C. I.R. (KBr, υcm−1): 3250 (NH), 1683 (C=O, amide), 1594 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.91 (s, 2H, CH2), 4.14 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.60 Hz, Ar-H), 6.80 (d, 2H, J = 8.60 Hz, Ar-H), 7.18 (t, 1H, J = 7.31 Hz, Ar-H), 7.26 (d, 1H, J = 6.32 Hz, Ar-H), 7.32 (t, 1H, J = 7.24 Hz, Ar-H), 7.47–7.54 (m, 5H, Ar-H), 7.76 (d, 1H, J = 7.88 Hz, Ar-H), 9.92 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 29.89 (CH2), 36.47 (CH2-S), 55.01 (O-CH3), 113.7, 125.1, 125.6, 126.2, 127.2, 127.5, 129.4, 129.5, 129.8, 130.0, 132.7, 134.6 (Ar-C), 149.9 (C-3), 154.9 (C-5), 157.9 (Ar-C) and 166.3 ppm (C=O). Anal. Calcd. for C24H21ClN4O2S (464.97): C, 62.00; H, 4.55; N, 12.05%. Found: C, 62.07; H, 4.56; N, 12.03%.
N-(2,5-dimethylphenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (2)
Off-white scales (absolute ethanol: glacial acetic acid, 9: 1); yield: 65%; m.p.: 168–170 °C. I.R. (KBr, υcm−1): 3229 (NH), 1681 (C=O, amide), 1530 (C=N). 1H NMR (DMSO-d6): δ = 2.12 (s, 3H, CH3), 2.22 (s, 3H, CH3), 3.67 (s, 3H, O-CH3), 3.91 (s, 2H, CH2), 4.10 (s, 2H, CH2-S), 6.72 (d, 2H, J = 8.56 Hz, Ar-H), 6.80 (d, 2H, J = 8.48 Hz, Ar-H), 6.87 (d, 1H, J = 7.60 Hz, Ar-H), 7.06 (d, 1H, J = 7.64 Hz, Ar-H), 7.25–7.28 (d, J = 7.64, 1H, Ar-H), 7.49–7.56 (m, 5H, Ar-H), 9.61 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 17.31 (CH3), 20.71 (CH3), 29.88 (CH2), 36.40 (CH2-S), 54.98 (O-CH3), 113.7, 124.8, 125.8, 127.2, 127.5, 128.0, 129.4, 129.7, 129.9, 130.1, 132.8, 134.9, 135.7 (Ar-C), 150.0 (C-3), 154.8 (C-5), 157.9 (Ar-C) and 165.7 ppm (C=O). Anal. Calcd. for C26H26N4O2S (458.58): C, 68.10; H, 5.72; N, 12.22%. Found: C, 68.05; H, 5.70; N, 12.24%.
2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-N-phenylacetamide (3)
White fluffy material (90% acetic acid); yield: 70%; m.p.: 183-185 °C. I.R. (KBr, υcm−1): 3210 (NH), 1665 (C=O, amide), 1591 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.90 (s, 2H, CH2), 4.10 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.52 Hz, Ar-H), 6.80 (d, 2H, J = 8.44 Hz, Ar-H), 7.05 (t, 1H, J = 7.24 Hz, Ar-H), 7.26–7.32 (m, 4H, Ar-H), 7.50–7.55 (m, 5H, Ar-H), 10.31 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 30.23 (CH2), 37.46 (CH2-S), 55.46 (O-CH3), 114.2, 119.5, 123.9, 127.7, 128.0, 129.2, 129.8, 130.2, 130.4, 133.2, 139.2 (Ar-C), 150.4 (C-3), 155.3 (C-5), 158.3 (Ar-C) and 166.0 ppm (C=O). Anal. Calcd. for C24H22N4O2S (430.53): C, 66.96; H, 5.15; N, 13.01%. Found: C, 66.91; H, 5.16; N, 13.04%.
N-(4-acetylphenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (4)
White powder (75% acetic acid); yield: 63%; m.p.: 186–188 °C. I.R. (KBr, υcm−1): 3230 (NH), 1680 (br, 2C=O, ketone, amide), 1520 (C=N). 1H NMR (DMSO-d6): δ = 2.52 (s, 3H, COCH3), 3.67 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.13 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.80 Hz, Ar-H), 6.79 (d, 2H, J = 8.40 Hz, Ar-H), 7.26 (d, 2H, J = 6.4 Hz, Ar-H), 7.51 (t, 3H, J = 7.6 Hz, Ar-H), 7.67 (d, 2H, J = 8.8 Hz, Ar-H), 7.92 (d, 2H, J = 8.8 Hz, Ar-H), 10.65 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 29.88 (COCH3), 30.35 (CH2), 36.99 (CH2-S), 54.99 (O-CH3), 113.7, 118.3, 127.2, 127.5, 129.4, 129.5, 129.7, 129.9, 131.9, 132.7, 143.0 (Ar-C), 149.8 (C-3), 154.9 (C-5), 157.9 (Ar-C), 166.2 (C=O, amide) and 196.5 ppm (COCH3). Anal. Calcd. for C26H24N4O3S (472.16): C, 66.08; H, 5.12; N, 11.86%. Found: C, 66.13; H, 5.10; N, 11.83%.
2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)-N-(4-methoxyphenyl)acetamide (5)
Faint violet fine material (80% acetic acid); yield: 54%; m.p.: 188–190 °C. I.R. (KBr, υcm−1): 3220 (NH), 1641 (C=O), 1490 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.71 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.06 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.48 Hz, Ar-H), 6.80 (d, 2H, J = 8.44 Hz, Ar-H), 6.86 (d, 2H, J = 8.84 Hz, Ar-H), 7.27 (d, 2H, J = 7.16 Hz, Ar-H), 7.43 (d, 2H, J = 8.8 Hz, Ar-H), 7.50–7.56 (m, 3H, Ar-H), 10.16 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 30.35 (CH2), 37.30 (CH2-S), 55.46 (CH3), 55.63 (CH3), 114.1, 114.4, 121.1, 127.7, 128.0, 129.8, 130.2, 130.4, 132.3, 133.2 (Ar-C), 150.4 (C-3), 155.3 (C-5), 155.8, 158.3 (Ar-C) and 165.4 ppm (C=O). Anal. Calcd. for C25H24N4O3S (460.55): C, 65.20; H, 5.25; N, 12.17%. Found: C, 65.26; H, 5.24; N, 12.21%.
N-(4-fluorophenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (7)
White fluffy material (90% acetic acid); yield: 89%; m.p.: 190–192 °C. I.R. (KBr, υcm−1): 3200 (NH), 1651 (C=O), 1588 (C=N). 1H NMR (DMSO-d6): δ = 3.66 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.08 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.56 Hz, Ar-H), 6.79 (d, 2H, J = 8.52 Hz, Ar-H), 7.14 (t, 2H, J = 8.8 Hz, Ar-H), 6.25 (d, 2H, J = 6.4 Hz, Ar-H), 7.50–7.57 (m, 5H, Ar-H), 10.37 ppm (s, 1H, NH); 13C NMR (DMSO-d6): δ = 29.64 (CH2), 36.97 (CH2-S), 55.14 (O-CH3), 113.8, 115.3, 121.0, 127.3, 127.6, 129.5, 129.9, 130.0, 132.8, 135.2 (Ar-C), 150.1 (C-3), 155.1 (C-5), 157.2, 158.0, 159.4 (Ar-C) and 165.5 ppm (C=O). Anal. Calcd. for C24H21FN4O2S (448.52): C, 64.27; H, 4.72; N, 12.49. Found: C, 64.32; H, 4.73; N, 12.45.
N-(4-bromophenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (10)
White fluffy material (90% acetic acid); yield: 88%; m.p.: 201–203 °C. I.R. (KBr, υcm−1): 3210 (NH), 1680 (C=O), 1599 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.90 (s, 2H, CH2), 4.10 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.0 Hz, Ar-H), 6.80 (d, 2H, J = 8.0 Hz, Ar-H), 7.26 (d, 2H, Ar-H), 7.48–7.54 (m, 7H, Ar-H), 10.45 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 29.77 (CH2), 37.05 (CH2-S), 55.00 (O-CH3), 113.5, 115.1, 121.0, 127.2, 127.5, 129.4, 129.7, 129.9, 131.6, 132.7, 138.1 (Ar-C), 149.8 (C-3), 154.8 (C-5), 157.9 (Ar-C) and 165.7 ppm (C=O). Anal. Calcd. for C24H21BrN4O2S (509.42): C, 56.59; H, 4.16; N, 11.00%. Found: C, 56.51; H, 4.18; N, 11.07%.
Synthetic method of compound (6)
A mixture of compound S2 (0.3 g, 1 mmol) and NaOH (0.06 g, 1.5 mmol) was refluxed with stirring in absolute ethanol (5 ml) for 0.5 h, then cooled to r. t. The appropriate 2-chloro-N-phenylacetamide derivative S4 (1.05 mmol) in absolute ethanol (5 ml) was dropped gradually to the above reaction mixture and then refluxed with stirring for an additional 1.5 h. Next, under lower pressure, the reaction mixture was evaporated until it was dry, yielding residue. After adding acidified ice water to the residue, stir the mixture for an additional 20 min. In order to obtain the crude product, the precipitate was finally vacuum-filtered, completely cleaned with water, and dried. Recrystallization of the crude product from the suitable solvent gives the pure target compound.
4-(2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamido)benzoic acid (6)
White powder (glacial acetic acid); yield: 93%; m.p.: 231–233 °C. I.R. (KBr, υcm−1): 3210 (OH and NH), 1681 (2C=O, acid and amide), 1509 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.13 (s, 2H, CH2-S), 6.71 (d, 2H, J = 8.8 Hz, Ar-H), 6.79 (d, 2H, J = 8.8 Hz, Ar-H), 7.26 (t, 3H, J = 6.4 Hz, Ar-H), 7.50 (d, 2H, J = 7.6 Hz, Ar-H), 7.65 (d, 2H, J = 8.8 Hz, Ar-H), 7.89 (d, 2H, J = 8.4 Hz, Ar-H), 10.62 (s, 1H, NH), 12.71 ppm (br, 1H, OH). 13C NMR (DMSO-d6): δ = 29.90 (CH2), 37.16 (CH2-S), 55.01 (O-CH3), 113.7, 118.4, 125.4, 127.2, 127.5, 129.4, 129.8, 130.0, 130.4, 132.8, 142.7 (Ar-C), 149.8 (C-3), 154.9 (C-5), 158.0 (Ar-C), 166.1 (C=O, amide) and 166.9 ppm (C=O, acid). Anal. Calcd. for C25H22N4O4S (474.54): C, 63.28; H, 4.67; N, 11.81%. Found: C, 63.22; H, 4.69; N, 11.84%.
Synthetic method of compound (8)
A mixture of compound S2 (0.3 g, 1 mmol) and triethylamine (0.21 ml, 1.5 mmol) was stirred at r. t. in absolute ethanol (5 ml) for 20 min. The appropriate 2-chloro-N-phenylacetamide derivative S4 (1.05 mmol) in absolute ethanol (5 ml) was dropped gradually to the above reaction mixture and then stirred for another 3 h. Next, under lower pressure, the reaction mixture was evaporated until it was dry, yielding residue. After adding ice water to the residue, stir the mixture for an additional 20 min. In order to obtain the crude product, the precipitate was finally vacuum-filtered, completely cleaned with water, and dried. Recrystallization of the crude product from the suitable solvent gives the pure target compound.
Ethyl 4-(2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamido)benzoate (8)
White fluffy material (absolute ethanol); yield: 89%; m.p.: 162–164 °C. I.R. (KBr, υcm−1): 3220 (NH), 1696 (br, 2C=O, ester and amide), 1513 (C=N). 1H NMR (DMSO-d6): δ = 1.30 (t, 3H, 3J = 6.8 Hz, CH2CH3), 3.67 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.14 (s, 2H, CH2-S), 4.27 (q, 2H, 3J = 6.8 Hz, CH2CH3), 6.71 (d, 2H, J = 8.0 Hz, Ar-H), 6.81 (d, 2H, J = 8.0 Hz, Ar-H), 7.25 (d, 2H, J = 6.4 Hz, Ar-H), 7.50 (d, 3H, J = 7.2 Hz, Ar-H), 7.67 (d, 2H, J = 8.4 Hz, Ar-H), 7.91 (d, 2H, J = 8.0 Hz, Ar-H), 10.65 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 14.20 (CH3), 30.04 (CH2), 37.05 (CH2-S), 54.99 (O-CH3), 60.59 (O-CH2), 113.7, 118.2, 123.9, 127.2, 127.5, 129.4, 129.7, 129.9, 130.3, 132.7, 143.0 (Ar-C), 149.8 (C-3), 154.9 (C-5), 157.9 (Ar-C), 165.2 (C=O, amide), 166.2 ppm (C=O, ester). Anal. Calcd. for C27H26N4O4S (502.59): C, 64.53; H, 5.21; N, 11.15%. Found: C, 64.57; H, 5.22; N, 11.11%.
Synthetic method of compound (9)
A mixture of compound S2 (0.3 g, 1 mmol) and NaOH (0.06 g, 1.5 mmol) was refluxed with stirring in absolute ethanol (5 ml) for 0.5 h, then cooled to r. t. The 2-chloro-N-phenylacetamide derivative S4 (1.05 mmol) in absolute ethanol (5 ml) was dropped gradually to the above reaction mixture and then refluxed with stirring for an additional 1.5 h. Next, under lower pressure, the reaction mixture was evaporated until it was dry, yielding residue. After adding acidified ice water to the residue, stir the mixture for an additional 20 min. The obtained crude product in the form of sticky material was completely cleaned with water, and dried. Recrystallization of the crude product from the suitable solvent gives the pure target compound.
N-(4-hydroxyphenyl)-2-((5-(4-methoxybenzyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetamide (9)
Brown fine material (glacial acetic acid); yield: 67%; m.p.: 227–229 °C. I.R. (KBr, υcm−1): 3470 (OH), 3200 (NH), 1667 (C=O), 1585 (C=N). 1H NMR (DMSO-d6): δ = 3.67 (s, 3H, O-CH3), 3.89 (s, 2H, CH2), 4.04 (s, 2H, CH2-S), 6.68–6.73 (m, 4H, Ar-H), 6.80 (d, 2H, J = 8.44 Hz, Ar-H), 7.25 (d, 2H, J = 6.88 Hz, Ar-H), 7.31 (d, 2H, J = 8.64 Hz, Ar-H), 7.50–7.55 (m, 3H, Ar-H), 9.22 (s, 1H, OH), 10.05 ppm (s, 1H, NH). 13C NMR (DMSO-d6): δ = 29.89 (CH2), 37.15 (CH2-S), 55.00 (O-CH3), 114.0, 115.0, 120.6, 127.2, 127.6, 129.4, 129.6, 129.9, 130.3, 132.7 (Ar-C), 149.9 (C-3), 153.5 (C-5), 154.8, 157.9 (Ar-C), 164.7 ppm (C=O). Anal. Calcd. for C24H22N4O3S (446.53): C, 64.56; H, 4.97; N, 12.55%. Found: C, 64.50; H, 4.98; N, 12.51%.
ADMET properties
Promising compounds 4, 6, 8, and 10 underwent in silico bioinformatic studies to predict their pharmacokinetics (liver metabolizing enzyme Cytochrome P450 metabolism, distribution, and GIT absorption) and physicochemical properties (size, lipophilicity, solubility, polarity, and flexibility). Additionally, in contrast to acarbose and forxiga (Dapagliflozin), if they are candidates for oral bioavailability (using Lipinski’s criterion). The compounds were assessed for drug-likeness and ADME qualities using the Swiss ADME online tool (www.SwissADME.ch/, accessed May 2, 2023)39.
Molecular docking
Molecular docking simulations were conducted with the Molecular Operating Environment (MOE) software 2022, as reported in the literature40,41. The MOE builder was used to generate ligands, which were 3D protonated and assigned partial charges before being energy-minimized using the MMFF94x force field. Forxiga (Dapagliflozin) and acarbose were used to target the binding regions of α-amylase (PDB ID: 4X9Y)42, (PDB ID: 2QV4)43, (PDB ID: 1OSE)44 and (PDB ID: 5E0F)45 and α-glucosidase (PDB ID: 3A4A)46 and (PDB ID: 3AJ7)47, which were got from the RCSB-Protein Data Bank. Proteins were prepared using traditional techniques such as 3D-protonation, automated atom type, and linkage correction. They were then potentially fixed. MOE’s alpha site finder was utilized to identify active regions. MOE’s induced fit docking technology was used. Validation typically involves the original ligand being re-docked against the active regions of 2QV4 and 3AJ7 with RMSD values < 2.0 Å. The triangle matcher approach and the London dG scoring function were used to investigate ligand interactions at active sites, with bond rotation as the initial scoring function. The GBVI/WSA dG forcefield-based scoring mechanism was employed as a secondary scoring function, yielding five poses out of thirty. Poses with higher S-values and lower RMSD were identified.
Biological assay
In vitro testing with α-amylase
Based on the manufacturer’s instructions as previously described48,49, the in vitro α-amylase suppression effect for the synthesized 1,2,4-triazole derivatives 3, 4, 6, 8, 9, and 10 was conducted using BioVision’s α-Amylase Inhibitor Screening Kit (Catalogue #K482-100) and a multi-well spectrophotometer (ELISA reader) using a 96-well microplate at (OD = 405 nm). The α-amylase inhibition assay was performed using a modified DNSA (3,5-dinitrosalicylic acid) method. Test compounds (dissolved in DMSO) were serially diluted (100, 10, 1, 0.1, and 0.01 µg/mL) and pre-incubated with porcine pancreatic α-amylase (0.5 U/mL in phosphate buffer, pH 6.8) at 37 °C for 10 min. Starch solution (1% w/v) was added, and the reaction proceeded for 30 min at 37 °C. The reaction was terminated by adding DNSA reagent, followed by heating at 85 °C for 10 min. Absorbance was measured at 540 nm using a microplate reader. Acarbose was used as a positive control. The percentage inhibition was calculated as: [(Acontrol–Asample)/Acontrol] × 100, where Acontrol represents enzyme activity without inhibitor. IC50 values were determined from dose-response curves using GraphPad Prism. Kinetic parameters (KActivity) were derived from ∆RFU (Relative Fluorescence Units) and slope values Table 1S.
In vitro testing with α-glucosidase
Using BioVision’s α-Glucosidase Inhibitor Screening Kit (Colorimetric) (Catalogue #K938-100) at (OD: 410 nm), the in vitro α-glucosidase suppression effect for the designed 1,2,4-triazole derivatives 3, 4, 6, 8, 9, and 10 was carried out. The results were analyzed on a multi-well spectrophotometer (ELISA reader) using a 96-well microplate in compliance with the manufacturer’s instructions as previously described49,50. The α-glucosidase inhibition assay was conducted using Saccharomyces cerevisiae α-glucosidase (0.5–1 U/mL in phosphate buffer, pH 6.8). Test compounds (63–1000 µg/mL) were pre-incubated with the enzyme at 37 °C for 10 min, followed by addition of pNPG (5 mM) and further incubation for 30 min. The reaction was stopped with 0.2 M Na2CO3, and absorbance was measured at 405 nm. Acarbose served as the positive control. Inhibition (%) was calculated as above, and IC50 values were derived from nonlinear regression analysis Table 2S.
Molecular dynamics analysis
Molecular dynamics (MD) simulations were conducted to investigate the interactions between α-amylase (PDB ID: 2QV4) and α-glucosidase (PDB ID: 3AJ7) with 1,2,4-triazole derivatives 4 and 10. All simulations were performed using GROMACS 2024.2, employing the CHARMM27 force field to describe atomic interactions. Protein structures were prepared by assigning the appropriate force field parameters and solvating them in TIP3P water molecules. Ligand structures were converted into a compatible format and positioned at the active site of each enzyme. The triclinic simulation box was defined with a minimum 1.0 nm buffer between the solute and the box edges. To neutralize the system, counterions were added by replacing an equivalent number of water molecules. Energy minimization was performed using the steepest descent algorithm to eliminate steric clashes and optimize the system’s initial configuration. The system was then equilibrated in two phases: first under constant volume and temperature (NVT) for 10 ns, where a velocity-rescaling thermostat was applied to regulate temperature, followed by constant pressure and temperature (NPT) for 10 ns, using the Parrinello-Rahman barostat to stabilize system density. Throughout equilibration, positional restraints were applied to the ligand to prevent large conformational fluctuations. Subsequently, production MD simulations were conducted in the NPT ensemble for 50 ns, maintaining biologically relevant temperature and pressure conditions. Post-simulation trajectory analysis included recentering and periodic boundary condition (PBC) corrections to remove system drifts and ensure accurate structural interpretation.
Conclusion
In this study, we designed and synthesized a novel series of 1,2,4-triazole derivatives incorporating a thioacetamide linker for dual inhibition of α-amylase and α-glucosidase. Among the synthesized compounds, compounds 4 and 10 demonstrated the most potent in vitro inhibitory activity, surpassing the reference drug acarbose. Molecular docking and dynamics simulations confirmed strong and stable interactions with key catalytic residues in both target enzymes. Furthermore, in silico ADMET profiling indicated favorable pharmacokinetic properties and oral drug-likeness for the top candidates. These findings support the therapeutic potential of triazole-based scaffolds in managing postprandial hyperglycemia and highlight a promising direction for the development of next-generation antidiabetic agents. Future work will focus on in vivo validation, toxicity assessment, and pharmacokinetic profiling of the most active candidates to advance them toward clinical development.
Data availability
Data is provided within the manuscript or supplementary information files.
Change history
15 August 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-15989-4
References
Marzouk, M. A. Pyrimidine derivatives as multifaceted antidiabetic agents: A comprehensive review of structure-activity relationships, mechanisms, and clinical potential. Eur. J. Med. Chem. 296, 117859. https://doi.org/10.1016/j.ejmech.2025.117859 (2025).
Ngoatle, C. & Mothiba, T. M. How is it to live with diabetes mellitus? The voices of the diabetes mellitus clients. Int. J. Environ. Res. Public Health 19, 9638. https://doi.org/10.3390/ijerph19159638 (2022).
Sun, H. et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119. https://doi.org/10.1016/j.diabres.2021.109119 (2022).
Sacchetta, L. et al. Synergistic effect of chronic kidney disease, neuropathy, and retinopathy on all-cause mortality in type 1 and type 2 diabetes: A 21-year longitudinal study. Cardiovasc. Diabetol. 21, 233. https://doi.org/10.1186/s12933-022-01675-6 (2022).
Forray, A. I. et al. The global burden of type 2 diabetes attributable to dietary risks: Insights from the global burden of disease study 2019. Nutrients 15, 4613. https://doi.org/10.3390/nu15214613 (2023).
Ahmad, E., Lim, S., Lamptey, R., Webb, D. R. & Davies, M. J. Type 2 diabetes. Lancet 400, 1803–1820. https://doi.org/10.1016/S0140-6736(22)01655-5 (2022).
Maguolo, A., Mazzuca, G., Smart, C. E. & Maffeis, C. Postprandial glucose metabolism in children and adolescents with type 1 diabetes mellitus: Potential targets for improvement. Eur. J. Clin. Nutr. 78, 79–86. https://doi.org/10.1038/s41430-023-01359-8 (2024).
Ur Rahman, S. et al. Novel acyl hydrazide derivatives of polyhydroquinoline as potent anti-diabetic and anti-glycating agents: Synthesis, in vitro α-amylase, α-glucosidase inhibition and anti-glycating activity with molecular docking insights. Bioorgan. Chem. 150, 107501. https://doi.org/10.1016/j.bioorg.2024.107501 (2024).
Attia, M. S. et al. Pharmaceutical polymers and p-glycoprotein: Current trends and possible outcomes in drug delivery. Mater. Today Commun. 34, 105318. https://doi.org/10.1016/j.mtcomm.2023.105318 (2023).
Khamees Thabet, H. et al. Discovery of new anti-diabetic potential agents based on paracetamol incorporating sulfa-drugs: Design, synthesis, α-amylase, and α-glucosidase inhibitors with molecular docking simulation. Eur. J. Med. Chem. 275, 116589. https://doi.org/10.1016/j.ejmech.2024.116589 (2024).
Liang, B., Xiao, D., Wang, S.-H. & Xu, X. Novel thiosemicarbazide-based β-carboline derivatives as α-glucosidase inhibitors: Synthesis and biological evaluation. Eur. J. Med. Chem. 275, 116595. https://doi.org/10.1016/j.ejmech.2024.116595 (2024).
Sadeghian, S. et al. 1,2,4-Triazole derivatives as novel and potent antifungal agents: Design, synthesis and biological evaluation. J. Mol. Struct. 1271, 134039. https://doi.org/10.1016/j.molstruc.2022.134039 (2023).
Fawzy, S. M. et al. Synthesis of 1,2,4-triazole and 1,3,4-oxadiazole derivatives as inhibitors for STAT3 enzymes of breast cancer. Arch. Pharm. 356, 2300345. https://doi.org/10.1002/ardp.202300345 (2023).
Strzelecka, M. & Świątek, P. 1,2,4-Triazoles as important antibacterial agents. Pharmaceuticals 14, 224. https://doi.org/10.3390/ph14030224 (2021).
Güzel, E. et al. Synthesis of benzimidazole-1,2,4-triazole derivatives as potential antifungal agents targeting 14α-demethylase. ACS Omega 8, 4369–4384. https://doi.org/10.1021/acsomega.2c07755 (2023).
Aly, A. A., Hassan, A. A., Makhlouf, M. M. & Bräse, S. Chemistry and biological activities of 1,2,4-triazolethiones—antiviral and anti-infective drugs. Molecules 25, 3036. https://doi.org/10.3390/molecules25133036 (2020).
Abo-Elmagd, M. I. et al. Design, synthesis and anti-inflammatory assessment of certain substituted 1,2,4-triazoles bearing tetrahydroisoquinoline scaffold as COX 1/2-inhibitors. Bioorgan. Chem. 150, 107577. https://doi.org/10.1016/j.bioorg.2024.107577 (2024).
Sharma, S., Mittal, N. & Banik, B. K. Chemistry and therapeutic aspect of triazole: Insight into the structure-activity relationship. Curr. Pharm. Des. 29, 2702–2720. https://doi.org/10.2174/0113816128271288231023045049 (2023).
Emami, L. et al. Design, synthesis and evaluation of novel 1,2,4-triazole derivatives as promising anticancer agents. BMC Chem. 16, 91. https://doi.org/10.1186/s13065-022-00887-x (2022).
Khan, I. et al. Synthesis and in vitro α-amylase and α-glucosidase dual inhibitory activities of 1,2,4-triazole-bearing bis-hydrazone derivatives and their molecular docking study. ACS Omega 8, 22508–22522. https://doi.org/10.1021/acsomega.3c00702 (2023).
Zhang, Y. et al. Design, synthesis and biological evaluation of novel betulinic acid derivatives containing 1,2,4-triazole-derived schiff bases as α-glucosidase inhibitors. J. Mol. Struct. 1315, 138889. https://doi.org/10.1016/j.molstruc.2024.138889 (2024).
Abchir, O. et al. Integrative approach for designing novel triazole derivatives as α-glucosidase inhibitors: QSAR molecular docking, ADMET, and molecular dynamics investigations. Pharmaceuticals 17, 261. https://doi.org/10.3390/ph17020261 (2024).
Sharma, A. et al. An insight on medicinal attributes of 1,2,3- and 1,2,4-triazole derivatives as alpha-amylase and alpha-glucosidase inhibitors. Mol. Divers. https://doi.org/10.1007/s11030-023-10728-1 (2023).
Dahmani, R., Manachou, M., Belaidi, S., Chtita, S. & Boughdiri, S. Structural characterization and QSAR modeling of 1,2,4-triazole derivatives as α-glucosidase inhibitors. New J. Chem. 45, 1253–1261. https://doi.org/10.1039/D0NJ05298A (2021).
Mohammadi-Khanaposhtani, M. et al. Design, synthesis, docking study, α-glucosidase inhibition, and cytotoxic activities of acridine linked to thioacetamides as novel agents in treatment of type 2 diabetes. Bioorgan. Chem. 80, 288–295. https://doi.org/10.1016/j.bioorg.2018.06.035 (2018).
Forozan, R. et al. Synthesis, in vitro inhibitor screening, structure–activity relationship, and molecular dynamic simulation studies of novel thioquinoline derivatives as potent α-glucosidase inhibitors. Sci. Rep. 13, 7819. https://doi.org/10.1038/s41598-023-35140-5 (2023).
Noori, M. et al. Design, synthesis, and in silico studies of quinoline-based-benzo[d]imidazole bearing different acetamide derivatives as potent α-glucosidase inhibitors. Sci. Rep. 12, 14019. https://doi.org/10.1038/s41598-022-18455-7 (2022).
Mahmoud, E. M. et al. Triphosgene/KF-mediated acyl fluoride synthesis via in situ fluoro (chloro) phosgene. Organ. Biomol. Chem. 23(25), 6116–6121. https://doi.org/10.1039/D5OB00735F (2025).
Khan, Y. et al. New quinoline-based triazole hybrid analogs as effective inhibitors of α-amylase and α-glucosidase: Preparation, in vitro evaluation, and molecular docking along with in silico studies. Front. Chem. 10, 995820. https://doi.org/10.3389/fchem.2022.995820 (2022).
Menteşe, E., Baltaş, N. & Bekircan, O. Synthesis and kinetics studies of N′ -(2-(3,5-disubstituted-4 H -1,2,4-triazol-4-yl)acetyl)-6/7/8-substituted-2-oxo-2 H -chromen-3-carbohydrazide derivatives as potent antidiabetic agents. Arch. Pharm. 352, 1900227. https://doi.org/10.1002/ardp.201900227 (2019).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25. https://doi.org/10.1016/S0169-409X(96)00423-1 (1997).
Ertl, P., Rohde, B. & Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43, 3714–3717. https://doi.org/10.1021/jm000942e (2000).
Daina, A., Michielin, O. & Zoete, V. iLOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 54, 3284–3301. https://doi.org/10.1021/ci500467k (2014).
Daina, A. & Zoete, V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem 11, 1117–1121. https://doi.org/10.1002/cmdc.201600182 (2016).
Rostom, S. A. F., Badr, M. H., Abd El Razik, H. A. & Ashour, H. M. A. Structure-based development of novel triazoles and related thiazolotriazoles as anticancer agents and Cdc25A/B phosphatase inhibitors. Synthesis, in vitro biological evaluation, molecular docking and in silico ADME-T studies. Eur. J. Med. Chem. 139, 263–279. https://doi.org/10.1016/j.ejmech.2017.07.053 (2017).
Hovsepyan, T. R., Dilanyan, S. V., Minasyan, N. S. & Melik-Ohanjanyan, R. G. Synthesis and reactions of new 4-methoxybenzyland 3-chloro-4-methoxybenzyl-substituted 1,2,4-triazoles and 1,3,4-thiadiazoles. Russ. J. Organ. Chem. 50, 736–741. https://doi.org/10.1134/S1070428014050194 (2014).
Zhu, X. et al. Design and synthesis of novel 4′-demethyl-4-deoxypodophyllotoxin derivatives as potential anticancer agents. Bioorgan. Med. Chem. Lett. 26, 1360–1364. https://doi.org/10.1016/j.bmcl.2015.06.089 (2016).
Abbas, S. E., Awadallah, F. M., Ibrahim, N. A. & Gouda, A. M. Novel substituted and fused pyrrolizine derivatives: Synthesis, anti-inflammatory and ulcerogenecity studies. Eur. J. Med. Chem. 45, 482–491. https://doi.org/10.1016/j.ejmech.2009.10.031 (2010).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717. https://doi.org/10.1038/srep42717 (2017).
Ali, J., Camilleri, P., Brown, M. B., Hutt, A. J. & Kirton, S. B. In silico prediction of aqueous solubility using simple QSPR models: The importance of phenol and phenol-like moieties. J. Chem. Inf. Model. 52, 2950–2957. https://doi.org/10.1021/ci300447c (2012).
Elsayed, D. A. et al. Bio-computational modeling, POM analysis and molecular dynamic simulation for novel synthetic quinolone and benzo[d][1,3]oxazine candidates as antimicrobial inhibitors. Sci. Rep. 14, 28709. https://doi.org/10.1038/s41598-024-73972-x (2024).
Das, K. et al. In silico studies and evaluation of in vitro antidiabetic activity of berberine from ethanol seed extract of Coscinium fenestratum (Gaertn.) Colebr. J. King Saud Univ. Sci. 35, 102666. https://doi.org/10.1016/j.jksus.2023.102666 (2023).
El Bakri, Y. et al. Potential antidiabetic activity and molecular docking studies of novel synthesized 3.6-dimethyl-5-oxo-pyrido [3, 4-f][1, 2, 4] triazepino [2, 3-a] benzimidazole and 10-amino-2-methyl-4-oxo pyrimido [1, 2-a] benzimidazole derivatives. J. Mol. Model. 24(7), 179. https://doi.org/10.1007/s00894-018-3705-9 (2018).
Toumi, A. et al. Synthesis, antidiabetic activity and molecular docking study of rhodanine-substitued spirooxindole pyrrolidine derivatives as novel α-amylase inhibitors. Bioorgan. Chem. 106, 104507. https://doi.org/10.1016/j.bioorg.2020.104507 (2021).
Chigurupati, S. et al. Molecular docking of phenolic compounds and screening of antioxidant and antidiabetic potential of Olea europaea L. Ethanolic leaves extract. Arab. J. Chem. 14, 103422. https://doi.org/10.1016/j.arabjc.2021.103422 (2021).
Karrouchi, K. et al. Synthesis, X-ray structure, vibrational spectroscopy, DFT, biological evaluation and molecular docking studies of (E)-N’-(4-(dimethylamino)benzylidene)-5-methyl-1H-pyrazole-3-carbohydrazide. J. Mol. Struct. 1219, 128541. https://doi.org/10.1016/j.molstruc.2020.128541 (2020).
Thao, T. T. P. et al. Isolation, semi-synthesis, docking-based prediction, and bioassay-based activity of Dolichandrone spathacea iridoids: New catalpol derivatives as glucosidase inhibitors. RSC Adv. 11, 11959–11975. https://doi.org/10.1039/D1RA00441G (2021).
Mphahlele, M. J., Magwaza, N. M., Malindisa, S. T. & Choong, Y. S. Biological evaluation the 2-aryl-2,3-dihydrobenzodiazaborinin-4(1H)-ones as potential dual α-glucosidase and α-amylase inhibitors with antioxidant properties. Chem. Biol. Drug Des. 98, 234–247. https://doi.org/10.1111/cbdd.13893 (2021).
Khamees Thabet, H. et al. Innovation of 6-sulfonamide-2 H -chromene derivatives as antidiabetic agents targeting α-amylase, α-glycosidase, and PPAR-γ inhibitors with in silico molecular docking simulation. RSC Adv. 14, 15691–15705. https://doi.org/10.1039/D4RA02143F (2024).
Marzouk, M. A. RVG-exosome-mediated delivery of GDNF mRNA: A non-invasive breakthrough for Parkinson’s disease. Med. Hypotheses 199, 111647. https://doi.org/10.1016/j.mehy.2025.111647 (2025).
Acknowledgements
G.E.K. would like to thank the Bibliotheca Alexandrina for providing computational resources through its High-Performance Computing (HPC) infrastructure (see hpc.bibalex.org).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). STDF (Science and Technology Development Fund).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.A.M.; Methodology, M.A.M. and E.M.M.; Software, D.A.E. and G.E.K.; Formal analysis, M.A.M., W.S.S., and D.A.E.; Investigation, M.A.M.; Resources, M.A.M., S.M.F., and M.A.A.; Data curation, M.A.M.; Writing-original draft, M.A.M., S.M.F., W.S.S., and D.A.E.; Writing—review & editing, M.A.M., W.S.S., S.M.M., D.A.E., and G.E.K.; Supervision, M.A.M. and W.S.S.; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors certify that none of the research provided in this paper has been impacted by any known financial or interpersonal conflicts.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the spelling of the author Elsayed M. Mahmoud which was incorrectly given as Mahmoud M. Elsayed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marzouk, M.A., Mahmoud, E.M., Shehab, W.S. et al. Dual α-amylase and α-glucosidase inhibition by 1,2,4-triazole derivatives for diabetes treatment. Sci Rep 15, 27172 (2025). https://doi.org/10.1038/s41598-025-11214-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11214-4
