
Abstract
There is a strong relationship between metabolic cell death (MCD) and neurodegenerative diseases. However, the involvement of metabolic cell death (MCD)-related genes (MCDRGs) in Parkinson’s disease (PD) pathogenesis remains poorly analyzed. Integrating PD-associated differentially expressed genes (DEGs) from GSE7621 with MCDRGs, we identified key biomarkers through protein-protein interaction networks and machine learning. Diagnostic performance was validated through nomogram analysis. Subsequent analyses included functional enrichment, immune profiling, drug prediction, and single-cell RNA sequencing. AKR1C2 and MAP1LC3A were identified as potential biomarkers. A nomogram with superior diagnostic performance was constructed. Gene set enrichment analysis indicated that both biomarkers were linked to the “Parkinson’s disease”. Further, immune infiltration revealed that AKR1C2 had the remarkably strongest positive correlation with M2 macrophages. Moreover, benzo[a]pyrene-1,6-dione, mestranol, and paraoxon-methyl might be potential therapeutic agents for PD. Single-cell RNA-seq analysis demonstrated endothelial-specific expression, with MAP1LC3A and AKR1C2 exhibiting distinct temporal regulation during differentiation. AKR1C2 and MAP1LC3A were identified as potential biomarkers associated with MCD in PD. These results offer fresh concepts for PD prevention and diagnosis.
Similar content being viewed by others


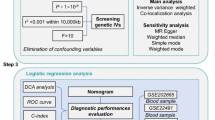
Introduction
Parkinson’s disease (PD) is a common and progressive neurodegenerative disorder affecting more than 6.1 million individuals worldwide. Its incidence increases significantly with age, making it one of the primary causes of neurological disability1,2. The main pathological characteristics of PD include the progressive degeneration of dopaminergic neurons in the substantia nigra and the formation of Lewy bodies3. Beyond typical motor dysfunction, patients often suffer from substantial non-motor symptoms, greatly impacting their quality of life. Although current treatments, such as dopamine replacement therapy, can relieve symptoms, they do not stop disease progression and come with notable side effects2. Consequently, research into PD pathogenesis and effective treatments is especially urgent, with a focus on identifying key genes or biomarkers that are essential for early diagnosis and understanding the underlying mechanisms.
Metabolic cell death (MCD) is a form of regulated cell death induced by intracellular metabolic imbalance and involves the precise regulation of multiple cellular signaling pathways4,5. In recent years, accumulating evidence has demonstrated a strong association between MCD and various neurodegenerative diseases, particularly PD4. MCD encompasses various forms, including ferroptosis, cuproptosis, disulfidoptosis, lysosomal zincoptosis, and alkaliptosis4, with ferroptosis receiving particular attention in PD research. Research has revealed that abnormalities in iron metabolism in PD patients often result in excessive iron accumulation within neurons, which is closely linked to the degeneration of dopaminergic neurons6. Iron-dependent cell death is thought to significantly contribute to the pathogenesis of PD by accelerating dopaminergic neuron loss in the substantia nigra through mechanisms involving lipid peroxidation and oxidative stress4,5,7. Although previous studies have indicated a close association between MCD and PD, the underlying molecular mechanisms at the genetic level remain poorly analyzed and warrant further investigation.
The Single-cell RNA sequencing technology provides the advantages of high resolution and throughput, allowing researchers to analyze the transcriptomic profiles of all genes at the single-cell level8,9. Its application enables comprehensive analysis of intercellular expression differences and offers clear insights into the distinct features of various cell subpopulations. For example, single-cell RNA sequencing has provided key cellular composition insights in cancer immune microenvironment studies, helping identify potential targets for immunotherapy9. Furthermore, this technology has revealed specific changes in different neuronal cell populations in neurodegenerative diseases, including PD, advancing the understanding of disease mechanisms8,10,11. The widespread application of single-cell RNA sequencing has significantly promoted the development of biomedical research, driving innovations in disease mechanisms, diagnosis, and therapeutic approaches8,9,10,11,12,13,14.
This study utilizes transcriptomic and single-cell data from public databases to identify potential biomarkers associated with MCD in PD and employs a series of bioinformatics approaches to investigate the molecular mechanisms of these biomarkers in PD. Through single-cell analysis, we annotated diverse cell types and systematically characterized the expression profiles of biomarkers across distinct cell populations, thereby identifying critical cell types that establish a strong theoretical basis for the clinical diagnosis and preventive treatment of PD.
Materials and methods
Data source
From the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/gds), transcriptomic datasets and single-cell dataset pertaining to PD were obtained. The GSE7621 dataset (GPL570 platform), which included samples from 16 PD patients and 9 normal brain substantia nigra tissues15were used as the training set for this study. Meanwhile, the GSE49036 dataset (GPL570 platform) consisting of brain substantia nigra tissue samples from 20 PD patients and 8 normals16 was used as the validation set (Supplementary Table S1–2). The single-cell dataset GSE157783 (GPL24676 platform) contained brain tissue samples from 5 PD patients and 6 controls. In addition, 295 metabolic cell death-related genes (MCDRGs) were obtained from previous literature17 and included in this study.
Identification of differentially expressed genes (DEGs)
The DEGs were identified using the limma package (v 3.58.1)18 (|log2fold change (FC)| > 0.5, and p < 0.05). Subsequently, ggplot2 package (v 3.4.4)19 and complexheatmap package (v 2.18.0)20 were applied to draw volcano plots and heatmaps, respectively, showcasing the top 10 upregulated and downregulated genes sorted by log2FC.
Identification and functional enrichment analysis of candidate genes
Candidate genes were obtained by taking the intersection of DEGs and MCDRGs using ggvenn package (v 0.1.9)21. After obtaining candidate genes, Gene Ontology (GO) analysis, which involved biological processes (BP), cellular components (CC), and molecular functions (MF), and Kyoto Encyclopedia of Genes and Genomes (KEGG)22,23,24, enrichment analyses were carried out using the clusterProfiler package (v 4.7.1.003)25 (p < 0.05). The top 5 most significantly enriched pathways in BP, CC, MF, and KEGG (count number order) were visualized using the ggplot2 package.
Construction of the protein–protein interaction (PPI) network
PPI analysis is mainly used to reveal the interaction network of target genes and identify other key genes or proteins associated with these genes. The candidate genes were introduced into Search Tool for the Retrieval of Interacting Genes (STRING, https://string-db.org) for PPI analysis (confidence interaction > 0.7). The PPI network was visualised using the igraph package (v 2.0.3), (http://igraph.org/) to obtain core genes through this network. Next, the chromosomal distribution of the core genes was analysed utilizing OmicCircos package (v 1.40.0) (https://doi.org/10.18129/B9.bioc.OmicCircos).
Recognition of feature genes using machine learning algorithms
LASSO (Least Absolute Shrinkage and Selection Operator) regression analysis incorporated an L1 regularization term into the regression model. This allowed LASSO regression to compress or even set unimportant features to zero, thereby achieving variable selection and model simplification. SVM-RFE (Support Vector Machine-Recursive Feature Elimination) was a method based on support vector machines for recursive feature elimination, which was commonly used in feature selection. LASSO and SVM-RFE were used to screen the feature genes. LASSO analysis was performed by the glmnet (v 4.1.4)26. The regularization parameter lambda was determined using 10-fold cross validation. In addition, SVM-RFE model was developed utilizing caret package (v 6.0-94)27. The genes obtained by the 2 algorithms were intersected and represented by constructing a Venn diagram using the ggvenn package, and the intersected genes were used as key feature genes.
Identification of biomarkers and construction of a nomogram
Further, the receiver operating characteristic (ROC) curves of key feature genes were created in GSE7621 and GSE49036 to assess their diagnostic value, and the genes with area under the curve (AUC) values greater than 0.7 were selected as candidate biomarkers. Then, differential expression of candidate biomarkers between PD and normal samples was compared utilizing Wilcoxon test (p < 0.05), and the genes with remarkable differential expression between PD and normal groups and consistent trends in GSE7621 and GSE49036 were selected as biomarkers.
To predict the risk of developing PD, rms (v 6.5-0)28 was employed to construct a nomogram containing biomarkers, and the probability of developing PD was assessed by calculating the total score. To evaluate the nomogram’s predictive performance, calibration curves were plotted by rms package, an ideal model would have calibration curves with a slope approaching 1, indicating high accuracy in prediction. In addition, ROC curves were created by pROC (v 1.18.5)29 to quantify the diagnostic effect of the nomogram (AUC > 0.7).
Gene set enrichment analysis (GSEA)
The correlation between the biomarkers and other genes was assessed utilizing psych package (v 2.4.3)30and the correlation coefficients were ranked from largest to smallest. Then the GSEA analysis for biomarkers was examined using the clusterProfiler to display top 5 significantly enriched pathways of the biomarkers (p.adj < 0.05 and normalized enrichment score |NES| > 1). The molecular signatures database (MSigDB) (http://software.broadinstitute.org/gsea/msigdb/) provided the reference gene set, “c2.cp.kegg.v7.5.1.symbols.gmt”.
Immune infiltration analysis
Firstly the abundance of 22 immune cells31 in normal and PD groups was explored by the CIBERSORT algorithm of Immuno-Oncology Biological Research (IOBR) package (v 0.99.9)32. Then, the level of infiltration of these immune cells across groups was compared using the Wilcoxon test (p < 0.05). Next, Mantel test was used to calculate the correlation between differential immune cells and the correlation between biomarkers and immune cells (p < 0.05). In order to explore the correlation between biomarkers and key cells, spearman correlation analysis was used to calculate the correlation coefficient between prognostic genes by using R package “psych”, and correlation scatter diagram was drawn by using R package “ggplot2” to explore the correlation between genes and cells.
Drug prediction and molecular networks
The potential therapeutic drugs targeted by the biomarkers were found by enrichR (v 3.2) (https://CRAN.R-project.org/package=enrichR). The top 10 drugs in terms of confidence were selected and further drug-mRNA mulberry diagram was constructed using the ggalluvial package (v 0.12.5) (http://corybrunson.github.io/ggalluvial/). The transcription factors (TFs) were predicted in the miRNet database (https://www.mirnet.ca/), followed by the construction of TF-mRNA network using Cytoscape package (v 3.10.1)33. Then, a co-expression network of biomarkers was created by the GeneMANIA (http://genemania.org).
Single-cell RNA sequencing (scRNA-seq) analysis
The Seurat package (v 5.0.1)34 was utilized to conduct the scRNA-seq analysis. Initial filtering removed cells expressing less than 200 genes and genes that were expressed in fewer than 3 cells. The quality control (QC) was performed by removing cells that were outliers by number of detected genes (≤ 200 or ≥ 5,000) and number of RNA counts (≤ 500 or ≥ 20,000). After filtering out the cells that did not meet the criteria, NormalizeData function was employed to normalize the data. Then the FindVariableFeatures function was used to extract the genes with higher coefficients of variation among cells, and the top 2000 highly variable genes (HVGs) with more obvious fluctuations were displayed for subsequent analysis. The LabelPoints function was used to visualize the results, identifying the top 10 genes with the highest variability. The ElbowPlot function was used to decide number of principal components (PCs).
Next, the resolution was set to 0.4, and the samples were clustered using the uniform manifold approximation and projection (UMAP) algorithm for dimensionality reduction. Subsequently, marker genes for each cell cluster were identified by the FindAllMarkers function. Then, cell clusters were annotated based on the cellular marker genes provided in published literature17. Expression patterns of the two previously identified biomarkers in cell types were further analyzed. Cell types with high expression of both biomarkers were selected as key cells. In addition, single-cell samples were functionally enriched using ReactomeGSA (https://github.com/reactome/ReactomeGSA) to explore the functions and pathways in which cell types were involved. Also, cell-type interactions were probed using Cellchat (v1.6.1)35. In addition, dimensionality reduction clustering of key cells was also performed. Lastly, Monocle (v 2.26.0)36 was employed to conduct cell pseudo-time analysis.
Statistical analysis
R software (v 4.2.2) was used for all analyses. The Wilcoxon test was used to evaluate group differences. Statistical significance was defined as p < 0.05.
MPTP-Induced parkinson’s mouse model
The MPTP (1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model was a commonly used experimental animal model for studying PD. MPTP, a neurotoxin, was injected into mice to selectively damage dopamine neurons in the substantia nigra, mimicking the pathological features of PD. This model was widely used to study the pathogenesis of PD, the efficacy of drug treatments, and their neuroprotective effects37. However, ten male C57BL/6J mice (6–8 weeks old, 20–25 g) were obtained from SPF (Beijing) Biotechnology Co., Ltd. The mice were housed in clean polypropylene cages under controlled conditions (temperature: 22 ± 1 °C, humidity: 50 ± 10%, 12-hour light/dark cycle) with ad libitum access to standard rodent chow and water. After a 7-day acclimatization period, the mice were randomly divided into MPTP and control groups (n = 5/group) using a random number table method. MPTP hydrochloride (MCE, Cat. No. HY-15608) was dissolved in 0.9% saline and administered intraperitoneally (30 mg/kg/day) for 7 consecutive days. Control mice received equivalent volumes of saline. Body weight and behavioral signs (e.g., mobility, grooming) were monitored daily throughout MPTP administration. Any animal that exhibited > 20% weight loss was euthanized immediately. Seven days after the final injection, the mice were deeply anesthetized with pentobarbital sodium (150 mg/kg, i.p.). Death was confirmed by pupillary dilation, cessation of breathing, and cardiac arrest, after which rapid decapitation was performed using a sharp guillotine, and their substantia nigra tissues were collected for subsequent analysis. The entire experiment was conducted by independent personnel for group assignment (Chao Zhang, Xueying Li), experimental implementation (Jia Fu, Jing Zhao), and data analysis (Na Mi, Xue Zhao), under strict blinding procedures. All procedures were approved by the Animal Care and Use Committee of Chifeng Municipal Hospital (Approval No: CFMH-LAEC-202405-01) and strictly adhered to the ARRIVE Guidelines 2.0 (https://arriveguidelines.org).
Ttotal RNA isolation and quantitative real-time PCR
Total RNA was isolated using TransZol Up reagent (Transgen, China) following the manufacturer’s instructions. The concentration and purity of RNA were determined using spectrophotometer (Multiskan Sky, Thermo Fisher, USA). Reverse transcription of total RNA was performed through the PerfectStart Uni RT & qPCR Kit (AUQ-01, Transgen, China). The reverse transcription reaction was carried out on a C1000 Touch Thermal Cycler (Bio-Rad, USA), and quantitative PCR was performed by the CFX96 Deep Well Real-Time System (Bio-Rad, USA). Primer sequences were designed based on NCBI references and are presented in Supplementary Table S3. GAPDH was employed as an internal reference gene for normalization of mRNA expression levels. Gene expression was quantified using the 2-ΔΔCT method, and the differences in gene expression between the MPTP and control groups were compared using an independent samples t-test (GraphPad Prism 9.0). Data normality was assessed using the Shapiro–Wilk test, and homogeneity of variances was checked using an F-test. If the data did not meet normality, the Mann–Whitney U test was used as an alternative.
Results
Identification and functional exploration of candidate genes
In the training set, differential expression analysis identified 1,718 DEGs between PD and normal samples, with 986 genes upregulated and 732 genes downregulated (Fig. 1a,b). Intersecting 1,718 DEGs and 295 MCDRGs yielded 34 candidate genes associated with MCD in PD (Fig. 1c). A total of 519 GO enrichment results were obtained, of which 405 were BP, 30 were CC, and 84 were MF (Fig. 1d). Among the BP entries enriched, candidate genes were significantly associated with functions such as “response to oxidative stress” and “fatty acid metabolic process”. Among the MF categories enriched, candidate genes were associated with “monooxygenase activity” and “DNA-binding transcription factor binding”, etc. Candidate genes were also enriched in CC pathways, such as “focal adhesion” and “cell-substrate junction”. Furthermore, the candidate genes were enriched to 22 KEGG pathways, such as “steroid hormone biosynthesis” and “folate biosynthesis” (Fig. 1e, Supplementary Table S4).

The relevant gene screening diagram (a) Differential gene volcano map, (b) Heat map of differential gene expression, (c) Candidate gene Venn diagram, (d) Candidate gene GO enrichment analysis (e) Candidate gene KEGG enrichment analysis.
Identification of core genes through PPI network
A PPI network included 16 genes, which were as core genes (Fig. 2a). Chromosomal localisation of the core genes revealed that FH was localised on chromosome 1, HSPB1 and DLD on chromosome 7, HSF1 on chromosome 8, AKR1C1, AKR1C2, AKR1C3, PDLIM1, and SCD on chromosome 10, FADS2 on chromosome 11, TXNRD1 and GABARAPL1 localised on chromosome 12. IQGAP1, ACTN4, MAP1LC3A and HMOX1 were localised on chromosomes 15, 19, 20 and 22, respectively (Fig. 2b).

Candidate gene PPI network and chromosomal localisation. (a) PPI network. (b) Chromosomal localisation of the core genes.
Machine learning identified 3 key feature genes
A total of 5 feature genes were screened using LASSO analysis (lambda = 0.1358) (Fig. 3a,b). Meanwhile, SVM-RFE algorithm screened 4 feature genes (Fig. 3c). Ultimately, the results of these 2 algorithms were combined to produce 3 key feature genes, AKR1C2, SCD and MAP1LC3A (Fig. 3d).

Candidate gene regression analysis plot (a) LASSO model, (b) Cross-validation results, (c) SVM-RFE model.
Identification of biomarkers of high diagnostic value
In order to further screen biomarkers and verify the expression levels of key characteristic genes and ROC, correlation analysis was performed in the validation set GSE49036 and training set GSE7621. MAP1LC3A and AKR1C2 had AUC values higher than 0.7, indicating that they had certain diagnostic value for PD patients (Fig. 4a,b). Moreover, their expression trends were consistent in both datasets, and AKR1C2 was remarkably highly expressed in the PD group, while MAP1LC3A was remarkably low-expressed (p < 0.05) (Fig. 4c,d). Therefore, AKR1C2 and MAP1LC3A were considered as biomarkers in this study.

biomarker screening (a) Training set ROC curve, (b) Verify the set ROC curve, (c) Raining set expression validation, (d) Validation set representation validation, (e) Nomogram model, (f) Nomogram model calibration curve, (g) Nomogram model ROC curve.
Next, a diagnostic nomogram for PD was constructed based on AKR1C2 and MAP1LC3A (Fig. 4e), and the calibration curve demonstrated the nomograms’ strong predictive accuracy for PD (Fig. 4f), while the ROC curve emphasized the plausible diagnostic value of the nomogram, with an AUC of 0.92 (Fig. 4g).
Functional analysis of biomarkers
GSEA results showed that biomarkers AKR1C2 and MAP1LC3A were enriched in 6 and 46 pathways, respectively. AKR1C2 was significantly enriched in pathways such as “parkinsons disease”, “oxidative phosphorylation” and “alzheimers disease”. MAP1LC3A was also enriched in the “parkinsons disease” and “oxidative phosphorylation” pathways (Fig. 5a,b). This suggested that AKR1C2 and MAP1LC3A were associated with multiple neurodegenerative diseases and energy metabolic pathways.

Functional of biomarkers (a) AKR1C2 gene set enrichment analysis, (b) Enrichment analysis of MAP1LC3A gene.
Association of biomarkers with the immune microenvironment of PD
The 22 immune cells’ abundance in PD and normal groups was explored (the CD8 T cells score was 0, and it was removed by subsequent analysis). And lower abundance of resting dendritic cells in PD samples were found (p < 0.05) (Fig. 6a). Moreover, resting dendritic cells were remarkably different between PD and normal, and resting dendritic cells was notably lower (Fig. 6b). In addition, AKR1C2 and M2 macrophages showed the most strikingly positive correlation (r = 0.14 and p < 0.05) (Fig. 6c). The scatter plot results of AKR1C2 and M2 macrophage marker genes such as CD206 were shown (Supplementary Fig. S1).

Immunoinfiltration analysis (a) CIBERSORT Stack diagram, (b) Box diagram of immune cell differences, (c) Correlation analysis of biomarkers and immune cells.
Drug forecasting and TF-mRNA regulatory network construction
Further, 2 biomarker-associated drugs, such as benzo[a]pyrene-1,6-dione, mestranol, and paraoxon-methyl, were predicted, suggesting that these drugs might be of potential value for PD treatment (Fig. 7a). In addition, TFs of predictive biomarkers revealed that TWIST1 and TWIST2 regulated AKR1C2, whereas E2F1 regulated MAP1LC3A (Fig. 7b). A co-expression network of biomarkers was obtained by GeneMANIA. The network consisted of 2 biomarkers and 20 peripheral predictor genes including AKR1C1, ATG3, SNCA, etc. (Fig. 7c) Together they were involved in 60 biological functions such as “cellular response to nitrogen levels”, “autophagosome”, and “vacuole organisation”. These processes involved cellular response to nutrients and intracellular degradation mechanisms, reflecting cellular adaptive mechanisms in response to environmental changes.

Molecular regulatory network (a) Biomarker—drug network Sankey map, (b) Biomarker (mRNA)—TFs network, (c) The GeneMANIA Network.
Screening of key cells
There were shown pre- and post-QC results of the single-cell dataset (Supplementary Fig. S2b). The top 2000 HVGs and 10 PCs were selected for cell clustering (Supplementary Fig. S2c,e) UMAP clustering classified the cells into 15 cell clusters (Fig. 8a), which were annotated by marker genes into 8 cell types (Fig. 8b,c), namely oligodendrocytes, microglia, astrocytes, oligodendrocyte precursor cells (OPCs), excitatory neuronal, endothelial cell, ependymal, and GABAergic. Moreover, both AKR1C2 and MAP1LC3A had the highest expression in endothelial cell (Fig. 9a,b) and therefore endothelial cell was considered as key cell. Endothelial cell was mainly enriched to biological functions such as “metabolism of ingested MeSeO2H into MeSeH”, “biosynthesis of DPAn-6 SPMs”, “synthesis of hepoxilins (HX) and trioxilins (TrX)”, “TWIK related potassium channel (TREK)”, and “SDK interactions”. These signalling pathways played important roles in immune response, cell survival and stress response (Fig. 9c).

cell clustering analysis (a) UMAP plot of clustering analysis of all sample cells, (b) Bubble map of Marker gene expression, (c) UMAP plot of clustering analysis of different grouped cells.

Distribution of biomarker expression in each cell cluster (a) UMAP plot of biomarker distribution in cells, (b) Bubble plots of biomarker expression in different cell clusters, (c) Heatmap of key cell function analysis.
Cellular communication and pseudo-time analysis
The cellular communication of different cell types in PD and normal samples was analyzed. In the PD group, key cells endothelial cell and astrocytes had strong communication relationship, which was mainly through the interaction of PTN-PTPRZ1 receptor (Fig. 10a–c).

Cellular communication interactions map (a) The size of the various coloured circles around the periphery indicates the number of cells, (b) Probability intensity values for interactions, (c) Bubble diagram of receptor-ligand pair interactions.
Further, pseudo-time analysis was performed on different subtypes of key cells. Endothelial cell was divided into 9 cellular subtypes (Fig. 11a). Subsequently, developmental and differentiation trajectories of cells were constructed. Cells differentiated over time from lower right to upper left. endothelial cell differentiates into a total of 9 cellular subtypes with 4 different states of differentiation (Fig. 11b–d). In addition, in the early stages of differentiation, MAP1LC3A was highly expressed, while AKR1C2 was highly expressed in the late stages (Fig. 11e).

The key cell cluster ndothelial was analysed for proposed temporal trajectories. (a) Endothelia subgroup clustering UMAP plot; (b-d) Pseudo-temporal analysis of key cells, with dark blue in-dicating the early stages of differentiation and light blue representing the later stages of differen-tiation, where state 1 is the earliest period of differentiation; (e) Dynamic map of biomarker expression in proposed time series; top: AKR1C2, bottom: MAP1LC3A.
qRT-PCR validation
To validate the expression of the predicted biomarker genes in PD, we established an MPTP-induced mouse model and extracted RNA samples from the substantia nigra tissue of the mouse midbrain for analysis. This region is closely related to motor control, postural stability, and other functions, and it has an important link with neurodegenerative diseases such as PD. qRT-PCR was performed to confirm the expression patterns of the two biomarker genes. The results revealed that the mouse homolog of AKR1C2, Akr1c21, was significantly upregulated in PD, whereas Map1lc3a was significantly downregulated, which is consistent with our predictions (Fig. 12a,b).

Key gene expression (a) Akr1c21 and (b) Map1lc3a.
Discussion
PD is a neurodegenerative disease impacting millions globally, marked by the loss of dopaminergic neurons and the emergence of various motor and non-motor symptoms1,2,3. Although studies have demonstrated a correlation between MCD and PD4,5,7the precise underlying mechanisms remain poorly analyzed. This research integrates transcriptomic and single-cell data from public databases, utilizing bioinformatics approaches to explore the role of MCD-related genes in PD and identify AKR1C2 and MAP1LC3A as key therapeutic targets. Investigating the biological pathways and regulatory mechanisms of these genes offers new avenues for developing innovative therapies for PD.
Aldo-keto reductase family 1 member C2 (AKR1C2) is a crucial cytoplasmic enzyme, responsible for reduction of aldehyde and ketone compounds38. Studies have shown that AKR1C2 plays a significant role in cellular detoxification and antioxidative stress, especially in processing reactive aldehydes and lipid peroxides, both of which are crucial to cellular injury and ferroptosis7,39,40,41. Notably, AKR1C2 has been reported to be upregulated in neurodegenerative conditions such as amyotrophic lateral sclerosis and Alzheimer’s disease38,42,43underscoring its importance in the pathogenesis of neurodegenerative disorders. In our study, we observed a significant upregulation of AKR1C2 in PD samples, which may represent a compensatory mechanism to counteract oxidative stress and iron overload. This adaptive response likely enhances cellular resilience and antioxidative capacity, thereby mitigating neuronal degeneration44,45. Recognizing this mechanism offers fresh perspectives on understanding the pathogenesis of PD. In contrast to AKR1C2, microtubule-associated protein 1 light chain 3 alpha (MAP1LC3A) plays an essential role in microtubule-cytoskeleton interactions and serves as a pivotal protein in autophagy. MAP1LC3A is engaged in various stages, such as autophagosome formation and maturation46,47,48. Immunoblotting or immunofluorescence detection of LC3 conversion is now a reliable technique for assessing autophagy and related processes49. LC3A plays a critical protective role in neurons by aiding in the cellular stress response, clearance of damaged organelles, and removal of protein aggregates50,51,52. Our gene expression analysis confirmed the downregulation of MAP1LC3A in PD samples. Consistent with previous studies53,54,55. Our findings suggest that alterations in MAP1LC3A expression are closely associated with the neurodegenerative pathology of PD, underscoring its pivotal role in disease progression and highlighting its potential as a therapeutic target for neurodegenerative disorders.
A diagnostic model was developed, and a corresponding nomogram was constructed based on the biomarkers AKR1C2 and MAP1LC3A. This model showed exceptional diagnostic performance in both the training and external validation datasets, emphasizing the potential of the two biomarkers in PD diagnosis and providing significant insights into the mechanisms underlying PD.
Through GSEA analysis, we identified that the biomarker genes AKR1C2 and MAP1LC3A were significantly enriched in 6 and 46 pathways, respectively, including various neurodegenerative disease-related pathways and oxidative phosphorylation (OXPHOS). This finding suggests that these genes may play critical roles in the pathogenesis and progression of neurodegenerative diseases. OXPHOS is a fundamental and highly conserved cellular bioenergetic process primarily occurring in mitochondria56,57. It serves as the principal mechanism by which cells generate adenosine triphosphate (ATP), the universal energy currency, through a series of redox reactions in the electron transport chain56,57. This process is essential for maintaining cellular energy homeostasis and is implicated in numerous physiological functions57. Extensive prior research has demonstrated that mitochondrial dysfunction, characterized by energy depletion and elevated oxidative stress, is a central pathogenic mechanism underlying neuronal cell death in various neurodegenerative diseases, including PD57,58,59. During OXPHOS, reactive oxygen species (ROS) are inevitably generated as byproducts60. Under pathological conditions in PD and other neurodegenerative disorders, excessive production of ROS overwhelms cellular antioxidant defense mechanisms58. This leads to oxidative damage of lipids, proteins, and DNA, which disrupts cellular functions and exacerbates neuronal degeneration56,57. Consequently, these processes drive the progressive worsening of the disease. Based on this, we hypothesize that the antioxidative properties of AKR1C2 may confer it a potentially critical role in alleviating oxidative stress. By neutralizing excessive ROS, AKR1C2 could effectively reduce oxidative damage to neurons40,41thereby protecting them from the adverse effects of oxidative stress during the progression of PD. Furthermore, elevated oxidative stress not only exacerbates mitochondrial damage but also impairs autophagy, a critical process for maintaining cellular homeostasis56. As a key protein in autophagy, loss of MAP1LC3A function may hinder the effective clearance of damaged mitochondria and accumulated misfolded proteins. This leads to the accumulation of cellular damage, further exacerbating neuronal injury. Additionally, defective autophagy may exacerbate OXPHOS-induced mitochondrial dysfunction, disrupting cellular energy metabolism and accelerating PD progression56. In summary, research on these biomarkers provides new insights into PD pathogenesis and offers potential therapeutic targets for treatment.
Immunoinfiltration analysis indicates a marked decrease in resting dendritic cells in PD. As vital antigen-presenting cells, dendritic cells play an essential role in the nervous system61. They participate in immune responses and impact T cell differentiation by modulating dopamine production and release41,62,63. This decrease may indicate that the composition and function of the immune microenvironment are impacted in PD patients. Furthermore, we found a notable correlation between AKR1C2 and M2 macrophages. Studies have reported that multiple immune cells are in an activated state in PD, with a higher presence of M2 macrophages64. Systemic inflammation in PD could result in alterations in immune cell ratios65and AKR1C2 expression might be associated with this inflammation, implying a potential role in immune regulation. This study shows that the correlation coefficient between AKR1C2 and M2 macrophages is r = 0.14, indicating a weak association. AKR1C2 has a certain influence on the occurrence and development of PD, possibly due to factors such as a small sample size or sample heterogeneity. To further validate this correlation, more experiments or independent datasets with a reasonable sample size are needed.
Using single-cell RNA sequencing data, we identified endothelial cells as key players. Functional analysis indicates that endothelial cells primarily engage in signaling pathways related to immune response, cell survival, and stress response in PD. Analysis of cell-cell communication demonstrates a strong interaction between endothelial cells and astrocytes mediated through the PTN-PTPRZ1 ligand-receptor pair. Endothelial and astrocyte cells are key components of the neurovascular unit, with their pathological alterations closely linked to neuroinflammation and neurodegeneration66,67,68,69. Research have identified that the breakdown of endothelial cells in brain capillaries is an essential pathological characteristic in PD patients70,71. Endothelial cell dysfunction and blood-brain barrier (BBB) disruption allow immune cells and plasma proteins to infiltrate brain tissue more readily, thereby aggravating neuroinflammation71,72,73,74. Furthermore, α-synuclein (αSyn) expression in endothelial cells is closely related to PD progression, with endothelial cell dysfunction exacerbating this process69,75. These pathological alterations underscore the crucial role of endothelial cells in the PD pathological process, with significant implications for disease progression and neuroprotective mechanisms.
This research successfully identified PD-associated biomarkers, AKR1C2 and MAP1LC3A, suggesting their potential impact on PD onset through modulation of immune and metabolic pathways. These findings provide a crucial foundation for understanding PD pathogenesis and for improving clinical diagnosis and treatment strategies. However, the study has certain limitations, such as a small sample size and the need for further clinical trials are needed to validate the identified biomarkers, and we will further expand the sample size. Additionally, the proposed drugs must undergo extensive in vivo and in vitro testing to confirm their efficacy and safety. Future studies will aim to further investigate these mechanisms and their potential impact on PD therapy.
Conclution
This research used PD transcriptome and single-cell RNA sequencing data from public databases to explore the role of MCD in PD. The study identified MCD-related biomarkers AKR1C2 and MAP1LC3A, and examined their expression patterns and associations with neurodegenerative pathology in PD. These discoveries offer new potential targets for the early diagnosis and treatment of PD, with future research aimed at validating their clinical applications and therapeutic potential.
Data availability
The datasets (GSE7621, GSE49036, GSE157783) analysed during the current study are available in the Gene Expression Omnibus (GEO) repository, (https://www.ncbi.nlm.nih.gov/gds). The (MCDRGs) from the previous literature.
References
Tolosa, E., Garrido, A., Scholz, S. W. & Poewe, W. Challenges in the diagnosis of parkinson’s disease. Lancet Neurol. 20, 385–397. https://doi.org/10.1016/s1474-4422(21)00030-2 (2021).
Di Luca, D. G., Reyes, N. G. D. & Fox, S. H. Newly approved and investigational drugs for motor symptom control in parkinson’s disease. Drugs. 82, 1027–1053. https://doi.org/10.1007/s40265-022-01747-7 (2022).
Tansey, M. G. et al. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 22, 657–673. https://doi.org/10.1038/s41577-022-00684-6 (2022).
Mao, C., Wang, M., Zhuang, L. & Gan, B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 15, 642–660. https://doi.org/10.1093/procel/pwae003 (2024).
Guo, D., Liu, Z., Zhou, J., Ke, C. & Li, D. Significance of programmed cell death pathways in neurodegenerative diseases. Int. J. Mol. Sci. 25 https://doi.org/10.3390/ijms25189947 (2024).
Ding, X. S. et al. Ferroptosis in Parkinson’s disease: Molecular mechanisms and therapeutic potential. Ageing Res. Rev. 91 https://doi.org/10.1016/j.arr.2023.102077 (2023).
Lin, K. J. et al. Iron brain menace: the involvement of ferroptosis in Parkinson disease. Cells. 11 https://doi.org/10.3390/cells11233829 (2022).
Cuevas-Diaz Duran, R., González-Orozco, J. C., Velasco, I. & Wu, J. Q. Single-cell and single-nuclei RNA sequencing as powerful tools to Decipher cellular heterogeneity and dysregulation in neurodegenerative diseases. Front. Cell. Dev. Biol. 10, 884748. https://doi.org/10.3389/fcell.2022.884748 (2022).
Zhang, Y. et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. CR. 40, 81. https://doi.org/10.1186/s13046-021-01874-1 (2021).
Ma, S. X. & Lim, S. B. Single-cell RNA sequencing in Parkinson’s disease. Biomedicines. 9 https://doi.org/10.3390/biomedicines9040368 (2021).
Xiong, L. L. et al. Single-cell RNA sequencing reveals peripheral immunological features in Parkinson’s disease. NPJ Parkinson’s Dis.. 10, 185. https://doi.org/10.1038/s41531-024-00790-3 (2024).
Hu, Z., Zu, S. & Liu, J. S. SIMPLEs: a single-cell RNA sequencing imputation strategy preserving gene modules and cell clusters variation. NAR Genom. Bioinf. 2, lqaa077. https://doi.org/10.1093/nargab/lqaa077 (2020).
Ranzoni, A. M. et al. Integrative Single-Cell RNA-Seq and ATAC-Seq analysis of human developmental hematopoiesis. Cell. Stem Cell. 28, 472–487e477. https://doi.org/10.1016/j.stem.2020.11.015 (2021).
Yu, X., Xu, X., Zhang, J. & Li, X. Batch alignment of single-cell transcriptomics data using deep metric learning. Nat. Commun. 14, 960. https://doi.org/10.1038/s41467-023-36635-5 (2023).
Lesnick, T. G. et al. A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet. 3, e98. https://doi.org/10.1371/journal.pgen.0030098 (2007).
Dijkstra, A. A. et al. Evidence for immune response, axonal dysfunction and reduced endocytosis in the substantia Nigra in early stage parkinson’s disease. PLoS One. 10, e0128651. https://doi.org/10.1371/journal.pone.0128651 (2015).
Smajić, S. et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain J. Neurol. 145, 964–978. https://doi.org/10.1093/brain/awab446 (2022).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. Ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinf. (Oxford England). 38, 3844–3846. https://doi.org/10.1093/bioinformatics/btac409 (2022).
Gu, Z. & Hübschmann, D. Make interactive complex heatmaps. Bioinf. (Oxford England). 38, 1460–1462. https://doi.org/10.1093/bioinformatics/btab806 (2022).
Zheng, Y. et al. Ferroptosis and autophagy-related genes in the pathogenesis of ischemic cardiomyopathy. Front. Cardiovasc. Med. 9, 906753. https://doi.org/10.3389/fcvm.2022.906753 (2022).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. Publ. Protein Soc. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Li, Y., Lu, F. & Yin, Y. Applying logistic LASSO regression for the diagnosis of atypical crohn’s disease. Sci. Rep. 12, 11340. https://doi.org/10.1038/s41598-022-15609-5 (2022).
Zhang, Z., Zhao, Y., Canes, A., Steinberg, D. & Lyashevska, O. Predictive analytics with gradient boosting in clinical medicine. Ann. Transl. Med. 7, 152. https://doi.org/10.21037/atm.2019.03.29 (2019).
Sachs, M. C. & plotROC: A tool for plotting ROC curves. J. Stat. Softw. 79 https://doi.org/10.18637/jss.v079.c02 (2017).
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77. https://doi.org/10.1186/1471-2105-12-77 (2011).
Correction to Lancet Psych 2022; 9: 938. Lancet. Psychiatry 10, e10. https://doi.org/10.1016/s2215-0366(22)00411-4 (2023).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. (Clifton N J). 1711, 243–259. https://doi.org/10.1007/978-1-4939-7493-1_12 (2018).
Zeng, D. et al. Multi-Omics Immuno-Oncology biological research to Decode tumor microenvironment and signatures. Front. Immunol. 12 https://doi.org/10.3389/fimmu.2021.687975 (2021).
Liu, P., Xu, H., Shi, Y., Deng, L. & Chen, X. Potential molecular mechanisms of plantain in the treatment of gout and hyperuricemia based on network Pharmacology. Evid.-Based Complement. Altern. Med. eCAM. 2020 (3023127). https://doi.org/10.1155/2020/3023127 (2020).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502. https://doi.org/10.1038/nbt.3192 (2015).
Fang, Z. et al. Single-cell transcriptomics of proliferative phase endometrium: systems analysis of cell-cell communication network using cellchat. Front. Cell. Dev. Biol. 10, 919731. https://doi.org/10.3389/fcell.2022.919731 (2022).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 566, 496–502. https://doi.org/10.1038/s41586-019-0969-x (2019).
Jackson-Lewis, V. & Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2, 141–151. https://doi.org/10.1038/nprot.2006.342 (2007).
Zhao, H. et al. Identification ferroptosis-related hub genes and diagnostic model in Alzheimer’s disease. Front. Mol. Neurosci. 16 https://doi.org/10.3389/fnmol.2023.1280639 (2023).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125. https://doi.org/10.1038/s41422-020-00441-1 (2021).
Rossner, P. et al. Toxic effects of the major components of diesel exhaust in human alveolar basal epithelial cells (A549). Int. J. Mol. Sci. 17 https://doi.org/10.3390/ijms17091393 (2016).
Pacheco, R., Contreras, F. & Zouali, M. The dopaminergic system in autoimmune diseases. Front. Immunol. 5, 117. https://doi.org/10.3389/fimmu.2014.00117 (2014).
Luchetti, S. et al. Neurosteroid biosynthetic pathways changes in prefrontal cortex in alzheimer’s disease. Neurobiol. Aging. 32, 1964–1976. https://doi.org/10.1016/j.neurobiolaging.2009.12.014 (2011).
Zhang, Q. et al. The classification and prediction of ferroptosis-related genes in ALS: A pilot study. Front. Genet. 13 https://doi.org/10.3389/fgene.2022.919188 (2022).
Liu, W., Zhang, F., Yang, K. & Yan, Y. Comprehensive analysis regarding the prognostic significance of downregulated ferroptosis-related gene AKR1C2 in gastric cancer and its underlying roles in immune response. PloS One. 18, e0280989. https://doi.org/10.1371/journal.pone.0280989 (2023).
Ide, H. et al. Modulation of AKR1C2 by Curcumin decreases testosterone production in prostate cancer. Cancer Sci. 109, 1230–1238. https://doi.org/10.1111/cas.13517 (2018).
Ghavami, S. et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49. https://doi.org/10.1016/j.pneurobio.2013.10.004 (2014).
Xu, Y. & Wan, W. Acetylation in the regulation of autophagy. Autophagy. 19, 379–387. https://doi.org/10.1080/15548627.2022.2062112 (2023).
Cheng, X. et al. Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy. 12, 1330–1339. https://doi.org/10.1080/15548627.2016.1185590 (2016).
Rakovic, A. et al. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 26, 1428–1441. https://doi.org/10.1038/s41418-018-0219-z (2019).
Mei, L. et al. Tethering ATG16L1 or LC3 induces targeted autophagic degradation of protein aggregates and mitochondria. Autophagy. 19, 2997–3013. https://doi.org/10.1080/15548627.2023.2234797 (2023).
Tu, H. Y. et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell. 20, e13522. https://doi.org/10.1111/acel.13522 (2021).
Yao, R. Q., Ren, C., Xia, Z. F. & Yao, Y. M. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. 17, 385–401. https://doi.org/10.1080/15548627.2020.1725377 (2021).
Chen, X. et al. LC3-associated phagocytosis and human diseases: insights from mechanisms to therapeutic potential. FASEB J. 38, e70130. https://doi.org/10.1096/fj.202402126R (2024).
Wu, Y., Jiao, Z., Wan, Z. & Qu, S. Role of autophagy and oxidative stress to astrocytes in fenpropathrin-induced Parkinson-like damage. Neurochem. Int. 145, 105000. https://doi.org/10.1016/j.neuint.2021.105000 (2021).
Abou-Hany, H. O., El-Sherbiny, M., Elshaer, S., Said, E. & Moustafa, T. Neuro-modulatory impact of felodipine against experimentally-induced Parkinson’s disease: possible contribution of PINK1-Parkin mitophagy pathway. Neuropharmacology. 250, 109909. https://doi.org/10.1016/j.neuropharm.2024.109909 (2024).
Anandhan, A. et al. Metabolic dysfunction in parkinson’s disease: bioenergetics, redox homeostasis and central carbon metabolism. Brain Res. Bull. 133, 12–30. https://doi.org/10.1016/j.brainresbull.2017.03.009 (2017).
Belarbi, K., Cuvelier, E., Destée, A., Gressier, B. & Chartier-Harlin, M. C. NADPH oxidases in Parkinson’s disease: a systematic review. Mol. Neurodegener. 12, 84. https://doi.org/10.1186/s13024-017-0225-5 (2017).
Cobb, C. A. & Cole, M. P. Oxidative and nitrative stress in neurodegeneration. Neurobiol. Dis. 84, 4–21. https://doi.org/10.1016/j.nbd.2015.04.020 (2015).
Martin, L. J. Biology of mitochondria in neurodegenerative diseases. Prog. Mol. Biol. Transl. Sci. 107, 355–415. https://doi.org/10.1016/b978-0-12-385883-2.00005-9 (2012).
Okoye, C. N., Koren, S. A. & Wojtovich, A. P. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. 67, 102926. https://doi.org/10.1016/j.redox.2023.102926 (2023).
Pajares, M., I. R., A., Manda, G., Boscá, L. & Cuadrado, A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 9 https://doi.org/10.3390/cells9071687 (2020).
Alarcón, B., Mestre, D. & Martínez-Martín, N. The immunological synapse: a cause or consequence of T-cell receptor triggering? Immunology. 133, 420–425. https://doi.org/10.1111/j.1365-2567.2011.03458.x (2011).
Nakano, K. et al. Dopamine released by dendritic cells polarizes Th2 differentiation. Int. Immunol. 21, 645–654. https://doi.org/10.1093/intimm/dxp033 (2009).
Lei, C. et al. Identification of necroptosis-related genes in Parkinson’s disease by integrated bioinformatics analysis and experimental validation. Front. NeuroSci. 17 https://doi.org/10.3389/fnins.2023.1097293 (2023).
Chen, Y. et al. Integrated bioinformatics analysis for revealing CBL is a potential diagnosing biomarker and related immune infiltration in parkinson’s disease. Int. J. Gen. Med. 17, 2371–2386. https://doi.org/10.2147/ijgm.S456942 (2024).
Al-Bachari, S., Naish, J. H., Parker, G. J. M., Emsley, H. C. A. & Parkes, L. M. Blood–Brain barrier leakage is increased in Parkinson’s disease. Front. Physiol. 11, 593026. https://doi.org/10.3389/fphys.2020.593026 (2020).
Lange, C., Storkebaum, E., de Almodóvar, C. R., Dewerchin, M. & Carmeliet, P. Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat. Rev. Neurol. 12, 439–454. https://doi.org/10.1038/nrneurol.2016.88 (2016).
Ouellette, J. & Lacoste, B. From neurodevelopmental to neurodegenerative disorders: the vascular continuum. Front. Aging Neurosci. 13, 749026. https://doi.org/10.3389/fnagi.2021.749026 (2021).
Florey. The endothelial cell. Br. Med. J. 2, 487–490. https://doi.org/10.1136/bmj.2.5512.487 (1966).
Farkas, E., De Jong, G. I., de Vos, R. A., Steur, J., Luiten, P. G. & E. N. & Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and parkinson’s disease. Acta Neuropathol. 100, 395–402. https://doi.org/10.1007/s004010000195 (2000).
Yang, P. et al. String vessel formation is increased in the brain of Parkinson disease. J. Parkinson’s Dis. 5, 821–836. https://doi.org/10.3233/jpd-140454 (2015).
McConnell, H. L., Li, Z., Woltjer, R. L. & Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: correlation or causation? Neurochem. Int. 128, 70–84. https://doi.org/10.1016/j.neuint.2019.04.005 (2019).
Nagatsu, T., Mogi, M., Ichinose, H. & Togari, A. Cytokines in Parkinson’s disease. J. Neural Trans. Suppl. 143–151 (2000).
Dolgacheva, L. P., Zinchenko, V. P. & Goncharov, N. V. Molecular and cellular interactions in pathogenesis of sporadic Parkinson disease. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms232113043 (2022).
Guan, J. et al. Vascular degeneration in Parkinson’s disease. Brain Pathol. 23, 154–164. https://doi.org/10.1111/j.1750-3639.2012.00628.x (2013).
Acknowledgements
We would like to express our sincere gratitude to all individuals and organizations who supported and assisted us throughout this research. We also extend our heartfelt thanks to the following funding agencies for their generous support: the National Natural Science Foundation of China, the Heilongjiang Provincial Natural Science Foundation, and the Science and Technology Program of the Joint Fund of Scientific Research for the Public Hospitals of Inner Mongolia Academy of Medical Sciences. Without their financial support, this research would not have been possible.
Funding
This work was supported by National Natural Science Foundation of China (NO.82471259), Heilongjiang Provincial Natural Science Foundation (NO. ZD2023H004), Science and Technology Program of the Joint Fund of Scientific Research for the Public Hospitals of Inner Mongolia Academy of Medical Sciences (2023090101, 2024GLLH0988).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and data collection were conducted by C.Z., X.L., L.W., and L.H., while data analysis was carried out by J.F., J.Z., N.M., and X.Z. The initial draft of the manuscript was prepared by J.F. and J.Z., with all authors providing critical feedback on subsequent versions. L.Y. and Y.Z. oversaw manuscript editing and review. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was reviewed and approved by the Experimental Animal Ethics Committee of Chifeng Municipal Hospital (Date: 1 May 2024; No: CFMH-LAEC-202405-01). We confirm that all animal experiments adhered to international ethical standards.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, J., Zhao, J., Zhao, X. et al. Exploring the mechanism of metabolic cell death-related genes AKR1C2 and MAP1LC3A as biomarkers in Parkinson’s disease. Sci Rep 15, 25610 (2025). https://doi.org/10.1038/s41598-025-11481-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11481-1
