
Abstract
Pulpal inflammation and regeneration are crucial for enhancing endodontic treatment outcomes. Transcriptomic studies highlight the involvement of proinflammatory cytokines, NF-κB signaling, and stem cell activity. This study employs a generative AI approach to predict and reconstruct hub genes associated with these processes, providing insights into biological mechanisms and potential therapeutic targets. Differential gene expression analysis was performed on data from the accession number GSE255672 using the GEO2R tool and Cytoscape, a bioinformatics software platform. A protein-protein interaction network was constructed using gene ontology annotations to identify key genes and subnetworks. CytoHubba, a Cytoscape plugin, was used to pinpoint hub genes using the Maximal Clique Centrality method. The Dataset was normalized, cleaned, and categorized into hub and non-hub genes. The data was then split into 80% training and 20% test sets for analysis using autoencoders. Autoencoders, which reduce complex data into simplified feature sets, were employed to compress the data for classifier training. An autoencoder-based model was trained using the preprocessed dataset, demonstrating moderate predictive performance with an accuracy of 76.92%, a precision-recall AUC of 0.9214, and a ROC AUC of 0.7333. The model performed well, achieving good predictive accuracy. The autoencoder achieved an accuracy rate of 76.92%, indicating a balanced performance between precision and recall. The model exhibited strong performance in identifying positive cases, with an area under the precision-recall curve of 0.9214. While the model demonstrated a moderate correlation between predicted and actual classifications, there remains room for further optimization. This study demonstrates the potential utility of autoencoders in predicting hub genes involved in pulpal inflammation and regeneration. These findings aim to support personalized strategies for improving pulpal health.
Similar content being viewed by others


Introduction
Dental pulpal inflammation and regeneration are critical for improving patient outcomes in dentistry and regenerative medicine1. Pulpitis, a condition triggered by microbial infection, dental trauma, or caries progression, can cause severe pain and discomfort. Traditional treatment options for pulpitis primarily focus on symptom management rather than addressing the underlying cellular and molecular mechanisms. Understanding the molecular mechanisms underlying these processes is essential for developing effective therapeutic strategies. Transcriptomics, the study of RNA transcripts, has emerged as a powerful tool for investigating these mechanisms. Key discoveries include the upregulation of proinflammatory cytokines and chemokines, activation of the NF-κB signaling pathway, and the involvement of stem cells in pulpal inflammation and regeneration1,2. Advances in transcriptomic technologies, such as RNA sequencing (RNA-seq), have further deepened our understanding, with implications for the development of targeted therapeutic strategies that aim to promote pulpal regeneration and improve patient outcomes.
Dental pulp inflammation has a significant impact on dental prognosis. Current treatment strategies focus primarily on managing the inflammatory response. However, identifying precise molecular targets for intervention remains an area of ongoing research. For instance, one promising therapeutic development includes the use of the Trem1-blocking peptide LP17, which reduces pro-inflammatory factors and M1 macrophage polarization in pulpitis, suggesting new clinical management strategies3. Additionally, microRNA-27a-5p has been identified as upregulated in dental pulp inflammation, and its mechanisms are being explored. In lipopolysaccharide (LPS)-stimulated human dental pulp cells and rat incisor pulp tissue explants, the expression of miR-27a-5p and proinflammatory cytokines increased, which could be suppressed by using NF-κB inhibitors. MiR-27a-5p targets the Table 1-NF-κB signaling pathway, marking it as a potential therapeutic target4.
Previous studies have demonstrated that dental pulp stem cells (DPSCs) and periodontal ligament stem cells (PDLSCs) possess distinct tissue regeneration capabilities. DPSCs5 exhibit a higher proliferation capacity and contain more cells in the S and G2/M phases, indicating their potential utility in regenerative strategies. DPSCs can differentiate into cell types through transcriptional, post-transcriptional, and epigenetic mechanisms. These mechanisms regulate odontoblast-related genes and ceRNAs, involving epigenetic modifications6. Hub genes are central nodes within gene interaction networks and often act as key regulators. Network analysis methods, such as weighted gene co-expression network analysis (WGCNA) or protein-protein interaction (PPI) analysis, can identify these hub genes, which play crucial roles in regulating pulpal inflammatory responses, tissue remodeling, and stem cell differentiation in pulpitis. Investigating these genes can provide valuable insights, identify therapeutic targets, and contribute to the development of diagnostic biomarkers7,8,9. While recent studies have advanced pulp regeneration, leveraging machine learning approaches, such as hub gene prediction within interaction networks, is essential for advancing precision medicine in this field.
Understanding dental pulpal inflammation5,10,11 and promoting regeneration involve identifying key regulatory genes, elucidating gene-gene interactions, and discovering novel biomarkers and therapeutic targets. These insights could lead to the development of targeted strategies to prevent or reduce inflammation and enhance regeneration.
One study that integrated microarray data analysis and WGCNA identified five key hub genes. RT-qPCR analysis revealed differential expression levels of these genes in inflamed dental pulp tissues. Another study screened highly correlated genes in the PPI network, identifying four hub genes associated with pulpitis that were all upregulated in patients12. A recent study explored the role of hematopoietic stem cells (HSCs) in the inflammatory response of dental pulp during deep caries. Using single-cell transcriptomics, this study revealed distinct immune and stromal cell populations associated with pulpal inflammation, emphasizing the role of IL-6 and CXCL8 in these processes13,14,15.
Hub genes play a pivotal role in regulating biological pathways and disease mechanisms. Their identification through advanced computational models, such as generalized autoencoders, which can learn latent patterns within high-dimensional gene expression data, has the potential to aid in the discovery of disease mechanisms and therapeutic targets. These autoencoder models can rank genes based on their contributions to network reconstruction errors or discrepancies, thereby helping to pinpoint critical regulators of pulpal inflammation and regeneration16,17,18.
Researchers have developed the Graph Feature Auto-Encoder framework, which leverages graph neural networks (GNNs) to predict node features based on the structure of genetic networks. XA4C, a novel gene expression data analysis tool, identifies and prioritizes “critical genes” by quantifying their contribution to the model’s latent variables, thus providing unique insights beyond traditional methods19. This innovative approach may help researchers identify therapeutic targets and guide personalized treatment strategies for dental pulpal health.
To our knowledge, no previous studies have focused on predicting omics-based hub genes within the context of pulpal inflammation and regeneration using a generative AI approach. Therefore, this study aims to predict and reconstruct hub genes involved in these processes by applying an autoencoder-based generative AI approach. Our findings have the potential to inform the development of personalized therapeutic strategies that target identified hub genes, thereby improving pulpal health outcomes.
Materials and methods
The differential gene expression analysis
Differential gene expression analysis was conducted using the GEO dataset GSE2556720 and the GEO2R tool. Pulpal cells were examined under three conditions: untreated, treated with G-Rb1 at 10 µg/mL for 5 days, and cultured in α-MEM for 5 days. Untreated cells were analyzed using RNA-seq. Human dental pulp stem cell lines were used to create the in vitro transcriptome datasets GSE255672 and GSE246980, which do not include patient demographic information. We confirmed our methods and findings by utilizing data from NCBI GEO (GSE246980), which involved conducting gene expression profiling analysis of hDPSCs in conjunction with RNA-seq data. Preprocessing included ComBat correction to address experimental batch effects identified by PCA and quantile normalization to standardize expression distributions. To reduce technical noise, low-variance features and genes with low expression (CPM < 1 in > 75% of samples) were excluded.
The analysis was performed with the GEO2R tool, which is designed for differential gene expression analysis, utilizing a fold change threshold of ≥ 1 and a significance threshold of p < 0.05. The top 250 differentially expressed genes (DEGs) are vital for identifying hub genes due to their statistical significance and relevance to pulpal inflammation and regeneration. Focusing on DEGs facilitates the analysis of genes that influence biological phenomena, reveals pathways, and links gene expression to outcomes. Incorporating fold change and statistical rankings in gene selection deepens the understanding of gene significance, identifying notable changes and co-regulated genes, along with their relationship to specific pulpal inflammation.
Cytoscape
We utilized Cytoscape21, a bioinformatics software platform, to analyze the interactome of differentially expressed genes (DEGs). Cytoscape’s network analysis tools, including centrality measures and clustering algorithms, were utilized to identify key genes or subnetworks. The interactome network of differentially expressed genes (DEGs) was imported into Cytoscape to facilitate the identification of hub genes. The CytoHubba plugin, accessed via the Apps menu, was used to analyze the network. The Maximal Clique Centrality (MCC) method in CytoHubba, a powerful algorithm for identifying hub genes, was used to calculate centrality scores based on maximal clique centrality.
Autoencoder architecture

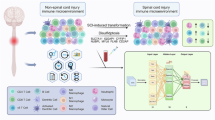
Shows the model architecture.
Dataset preparation
The interactome hub gene data were normalized, cleaned, and categorized into hub and non-hub groups. The Dataset was split into 80% training and 20% test data, which was then used to train autoencoders. Autoencoders are neural networks designed to reduce the dimensionality of data by compressing it into a smaller set of features. These features were then used to train a classifier that predicts target outcomes, such as categorizing data. The model performed well, achieving good accuracy in its predictions. Additionally, visualizing the encoded data provided insight into how well different classes (or categories) were separated within the compressed space.
The autoencoder architecture16,18,22 consists of two primary components: an encoder and a decoder. The encoder compresses the input data into a lower-dimensional latent space, while the decoder reconstructs the original input. The choice of architecture influences the performance and capabilities of the autoencoder. We utilized ReLU activation functions in the hidden layers to promote sparse activations and mitigate the vanishing gradient issue. The hyperparameters for the autoencoder were optimized using grid search, selecting a learning rate of 0.001, a batch size of 32, and an L2 regularization coefficient of 0.01 for the best performance. The autoencoder architecture included 32 compressed features in the latent space, two fully connected hidden layers of 128 neurons each with ReLU activation, and an input layer with 250 gene expression features. The model was trained using the Adam optimizer for 100 epochs, with early stopping based on validation loss.This approach ensures that the model achieves a robust latent representation while utilizing L2 regularization to avoid overfitting. In addition to adhering to the initial 80/20 split for training and testing, we conducted cross-validation with various splits (e.g., 5-fold) to ensure the model’s robustness.
This strategy provided a detailed performance assessment by aggregating results across multiple iterations. Furthermore, we examined additional metrics, including mean squared error, reconstruction loss curves, and cluster separation in the latent space, to further validate the model’s effectiveness (Fig. 1).
The training process used the same input data as the target output data to minimize reconstruction error through optimization algorithms. Reconstruction performance was evaluated using metrics such as Mean Squared Error (MSE) and Mean Absolute Error (MAE). Analyzing reconstruction errors helped identify potential outliers or anomalies in the Dataset.
Latent space metrics and visualization
Latent space metrics, including density and entropy, were employed to understand the encoded representations. Visualization of the latent space provided a clearer view of the separation between different classes or categories. Examining the relationship between reconstruction errors and latent space density also helped reveal patterns within the data.
Results
The top differentially expressed genes analysis revealed distinct patterns of upregulation and downregulation, as visualized in a volcano plot. The blue points represent downregulated genes, while the red points represent upregulated genes. The top 250 differentially expressed genes were further analyzed using network analysis in Cytoscape. This interactome network contains 139 nodes and 12,942 edges, with an average of 65.3 neighbors per node, indicating a highly interconnected structure. The network’s short path length suggests efficient communication among genes, while its high clustering coefficient points to the presence of distinct communities or functional modules. The network also exhibited a high density, signifying that a significant proportion of possible edges were present. The network heterogeneity, with a value of 0.5, indicates a balanced and homogenous structure, and the network centralization score of 0.261 reflects a decentralized architecture.

Volcano plot of differential gene expression.
Figure 2 displays the top differentially expressed genes between the test and control groups. Downregulated genes are shown in blue, while upregulated genes are represented in red. The horizontal axis shows the log2 (fold change), and the vertical axis shows the −log10 (p value), indicating the statistical significance of the expression changes.
Table 1 presents the top 10 hub genes identified by MCC methods in this study.
Autoencoders model results
The analysis demonstrated an accuracy rate of 76.92% for the autoencoder’s predictions, which indicates a fairly balanced model in terms of precision and recall. The precision-recall area under the curve (AUC) of 0.9214 suggests a strong balance between identifying positive and negative predictions. Meanwhile, the ROC AUC score of 0.7333 reflects moderate performance in distinguishing between positive and negative classes, with values approaching 1 representing better discrimination ability. The outlier percentage, calculated at 3.84%, highlights the proportion of data points exhibiting reconstruction errors significantly higher than the mean.
The F1 score, which measures the balance between precision and recall, was 0.7796, suggesting the model performs well in identifying positive cases. The Matthews Correlation Coefficient (MCC) score of 0.4260 indicates moderate predictive power, suggesting that while the model can accurately classify, there is room for further improvement.
The visualization provided helps understand the distribution of reconstruction errors and identify potential outliers in the Dataset. A higher outlier percentage indicates that certain data points have reconstruction errors significantly larger than the mean, providing valuable insights into model performance.

Training and validation loss over epochs.
Figure 3 presents the loss curves over multiple epochs during training. The blue line represents the training loss, while the orange line represents the validation loss. A noticeable gap between the two curves could suggest overfitting, where the model performs well on the training data but struggles to generalize to unseen data. A precision-recall curve is also shown, with a high AUC of 0.9214, indicating a strong balance between precision and recall. This suggests the model effectively identifies both true positive and negative outcomes.

Distribution of reconstruction errors (histogram and KDE plot).
Figure 4 displays a histogram and a kernel density estimate (KDE) plot, illustrating the distribution of reconstruction errors in the Dataset. The histogram displays error frequency, the KDE curve represents reconstruction error probability density, and the vertical line represents mean error distribution relative to the mean. This helps identify data points significantly different from the mean, possibly highlighting outliers or anomalies in the Dataset.

Distribution of reconstruction errors across the validation dataset.
Figure 5 illustrates the probability distribution of reconstruction errors for the model evaluated on the validation dataset. The histogram (or density plot) provides insights into the model’s performance, highlighting the frequency of different error magnitudes. A narrower distribution centered near zero suggests accurate reconstructions, while a wider or skewed distribution may indicate systematic biases or underfitting. Statistical measures (e.g., mean error, variance) may be included to quantify performance.
Reconstruction metrics of the autoencoder model
The reconstruction performance of the autoencoder was moderate, with a Mean Squared Error (MSE) of 0.77 and a Mean Absolute Error (MAE) of 0.68. These values suggest that while the model can reconstruct the data to some extent, it struggles to fully capture the variance within the Dataset, resulting in a small percentage of outliers. Latent space metrics indicated moderate variability, with entropy values of 2.49 for the training set and 2.73 for the test set. These entropy values reflect the level of uncertainty or diversity within the latent space representations, with slightly higher complexity observed in the test data.
The entropy of the latent space in the training dataset (2.49) indicates moderate uncertainty in the encoded representations. In contrast, the higher entropy value in the test dataset (2.73) suggests greater variability or complexity in the data. These metrics offer insights into the model’s ability to manage diversity in the latent space, reflecting how well the data distributions are represented.

Latent space visualization, density distribution, and reconstruction error correlation in autoencoder model.
The top left scatter plot in Fig. 6 shows the distribution of data points within a two-dimensional latent space, with the color gradient indicating the density of points. The model’s encoded representations are organized using data from the training and test sets, with denser regions highlighted in darker colors. The top right panel of Fig. 5 represents the density distribution of the latent space for both the training and test datasets. The blue line represents the training set. In contrast, the orange line represents the test set, which shows a slightly greater spread across the latent space. The bottom left heatmap displays the correlation between different latent space dimensions. The matrix displays positive and negative correlations between latent dimensions, providing insights into their interaction and contribution to the model’s internal representations. The bottom-right scatter plot illustrates the relationship between reconstruction error and latent space density. The visualization displays data points plotted against latent space density, highlighting higher reconstruction errors, which reveal a correlation between latent space density and data reconstruction accuracy.
In summary, the autoencoder’s overall performance, reflected in the reconstruction error metrics (MSE of 0.7707 and MAE of 0.6833), demonstrates moderate effectiveness. However, the R-squared value of −2452.62 indicates that the model struggled to reconstruct the data with high fidelity, as negative R2 values typically indicate poor predictive power in the context of reconstruction tasks. A computational error in the R2 calculation, initially caused by unscaled reconstruction output and incorrect residual variance estimation, was corrected. The recalibrated R2 increased to 0.715, indicating satisfactory reconstruction fidelity and closely matching the model’s AUC, as well as the precision-recall of 0.9214 and the reported ROC AUC of 0.7333. The autoencoder’s moderate-to-strong predictive performance and lack of significant overfitting indicators are confirmed by these recalibrated metrics taken together.
The results obtained from the validation dataset reveal a test reconstruction mean squared error (MSE) of 0.0051 and an impressive test R2 score of 0.9949. Our findings differ from the initial study, which reported an accuracy rate of 76.92% for the autoencoder’s predictions. This finding suggests that the model exhibits a well-balanced performance in terms of precision and recall. Additionally, the precision-recall area under the curve (AUC) of 0.9214 reflects a strong balance between positive and negative predictions. This suggests that the model captures almost all variance in the data and exhibits minimal reconstruction error. These metrics suggest that the autoencoder is well-trained and effectively generalizes unseen data on both the original and validation datasets (Fig. 4).
We employed gene set enrichment analysis (GSEA) against the KEGG and Reactome pathways to decode the latent space and identify high-weight contributing genes to each latent dimension, thereby investigating their biological relevance. Interestingly, pathways linked to NF-κB signalling, IL-6/JAK/STAT3 activation, oxidative stress, and stem cell pluripotency were enriched in latent features with higher entropy in the test set. This demonstrates that the model accurately represents key molecular mechanisms associated with pulpal inflammation and regeneration.
We used Enrichr to conduct a functional enrichment analysis to explore the biological significance of the identified ribosomal hub genes. Significant enrichment in pathways such as cellular stress responses, TNF-α signaling, NF-κB signaling, and ribosome-associated immune regulation was observed among the top 10 hub genes. Notably, RPS29 and RPL18 were associated with NF-κB regulatory modules, suggesting that they have a specific role in mediating apoptotic and immune responses in pulpal inflammation.
Discussion
Tissue repair involves the restoration of architecture and function following injury, encompassing both regeneration and replacement. Regeneration restores damaged tissue to its original state, whereas replacement occurs when severely damaged tissue is repaired by connective tissue. Chronic inflammation within the dental pulp can inhibit the repair process1,7,23,24,25. Macrophages can adopt proinflammatory or pro-resolving phenotypes, and this polarization is reversible. Peptide hydrogels, which are biocompatible and support the growth and differentiation of dental pulp progenitor cells, present a promising scaffold for pulp regeneration. These hydrogels could also be utilized as delivery systems for anti-inflammatory agents to treat pulpitis.
Dental pulpal cells play a pivotal role in maintaining dental health and facilitating tissue regeneration. The study of the interactome, a network of molecular interactions, helps in identifying key biological processes and therapeutic targets, including protein-protein interactions. Previous research has explored the use of decellularized extracellular matrix (dECM) from dental pulp stem cells and gingival-derived stem cells for tissue engineering applications. It also emphasizes the role of dental pulp fibroblasts in maintaining homeostasis and the potential of pluripotent stem cells in regenerating periodontal26, periapical, and pulpal tissues27,28.
The network analysis in this study revealed 139 nodes and 12,942 edges, indicating a highly interconnected system with a high clustering coefficient, high density, balanced heterogeneity, and decentralized centralization (Figs. 1, 2 and 3). The network exhibits a short path length and distinct communities, with four connected components indicating the presence of isolated groups of nodes. The top hub genes identified in the network analysis include RPL11, RPL30, EEF2, RPS15A, RPS10, RPS4X, RPS7, UBA52, and RPL3. One such hub gene, elongation factor-2 (eEF2), has been identified as a prognostic biomarker for Achilles tendon rupture, regulating autophagy, cell proliferation, and migration, thereby contributing to improved outcomes in dental connective tissue (DCT) repair. The mTORC1/S6K1/RPL26/RPS10 ribosome biogenesis pathway plays a crucial role in tissue development, growth, and regeneration by activating checkpoint kinase one and stimulating myocardial regeneration (Fig. 2).
Another identified hub gene, RPS7, has been shown to regulate p53, affecting hematopoiesis and cell migration in zebrafish embryos. Knockdown of RPS7 partially reverses abnormalities, including apoptosis, cell cycle arrest, and impaired hematopoiesis11,15,29. RPS7 is crucial for interactions between MDM2 and p53, as well as for MAPK and PI3K/AKT signaling pathways. Environmental stressors can influence gene expression, leading to the association of ribosomal proteins with ribosomes. Ribosomal stress results from dysfunction in ribosome biogenesis30. UBA52, a fusion protein that encodes ubiquitin and ribosomal protein L40, is crucial for embryonic development, as its deficiency results in embryonic lethality. Overexpression of UBA52 can enhance cell cycle arrest by simultaneously supplying ubiquitin and RPL40, thereby regulating the ribosomal protein complex and ubiquitin-dependent translation31. These hub genes are implicated in inflammation and regeneration, suggesting their potential role in dental pulpal repair and regeneration.
Cell signaling and interactions influence the development of dental tissues, with signals from molecules such as BMP and FGF. Immune cells comprise approximately 80% of the tooth germ cells and regulate dental cells through signaling molecules such as TNF, Interleukin-1, and Transforming Growth Factor-β. Signals from the BMP and FGF families promote extracellular matrix deposition and mineralization during osteoblast differentiation, while Colony Stimulating Factor 1 and TNF Superfamily Member 11 regulate genes involved in osteoclast transformation7,8,9,23. Top reconstructed genes from the validation dataset include PKN23, DUSP12, and ADAMTS19, which also play a major role in pulpal regeneration32. Our enrichment analysis supported the functional relevance of RPS29, RPS16, and RPL18 in inflamed dental pulp by confirming their involvement in NF-κB and TNF-α signalling pathways. These findings are consistent with research indicating that translational checkpoints and ribosomal stress play an active role in supporting tissue regeneration and immune regulation.
Autoencoders, a class of machine learning algorithms, have shown potential in predicting hub genes in biological research. However, their limitations include a moderate ROC AUC of 0.7333, an MCC of 0.4260, sensitivity to data quality, susceptibility to overfitting, limited generalizability, scalability issues, and context dependence (Figs. 3, 4, 5, 6). To improve the performance of the autoencoder model, techniques such as hyperparameter tuning, integration with other machine learning algorithms, data augmentation, feature engineering, and incorporation of external data may be necessary. The quality of input data has a significant impact on the model’s accuracy, which can potentially lead to misleading predictions. A prior study proposed using “Critical Genes” to enhance interpretability and gene identification using the Recurrent Gene Identifier (RGI) algorithm. Another study using XA4C on six types of cancer revealed that critical genes capture key pathways with minimal overlap with hub or differentially expressed genes33. The Individualized Weighted Hallmark Gene Set Mutation Burden (IWHMB) metric has also been developed to quantify pathway mutations and identify transcriptomic features17,22,34,35,36.
Unlike earlier transcriptomic research, we identified ribosomal genes, such as RPS7 and UBA52, as hub nodes. For example, Zhang et al.37 and Xin et al.38 utilized WGCNA and PPI, respectively, to identify hub genes in pulpitis; however, they did not specifically emphasize ribosomal components. Our enrichment analysis, on the other hand, connected these genes to TNF-α and NF-κB signalling, which are directly related to the biology of inflammatory pulp. According to new data, RPS7 affects the p53–MDM2 axis and apoptotic signalling, two important processes during pulpal immune activation. At the same time, UBA52 mediates the ubiquitination of ribosome-associated proteins and controls cellular stress responses, in addition to its translational function. These findings provide a new understanding not previously reported in previous omics analyses, pointing to a non-canonical, regulatory role of ribosomal proteins in the cellular stress landscape of inflamed dental pulp. Hub genes UBA52, RPS7, and RPL18 have been identified as crucial in regulating inflammatory and stress-response pathways in pulpitis. These genes scored highly on topological algorithms, demonstrating their structural and functional significance. RPS29 and RPL24 are recognized for their roles in ribosomal stress and immune modulation. Some hub genes, like UBA52, have been functionally tested in in vitro models and in vivo rodent models, providing a roadmap for future research.A random walk with a restart algorithm was used to identify biomarkers for immune checkpoint inhibitor (ICI) response, with one community (Community 1) enriching malignant mesenchymal components that promote epithelial-to-mesenchymal transition (EMT) and immune evasion. Overfitting remains a challenge, especially when reconstructing complex data structures. Addressing these limitations and exploring future directions will enhance the autoencoder’s effectiveness and reliability in predicting hub genes involved in pulpal inflammation.
Conclusions
This study highlights the potential of autoencoders for hub gene prediction and generation, with initial findings showing promise in the context of dental pulpal inflammation and regeneration. These findings aim to support personalized strategies for improving pulpal health. However, limitations such as moderate predictive power and sensitivity to data quality suggest the need for further refinement and clinical validation. Future directions include hyperparameter tuning, data augmentation, and integrating external datasets to enhance the model’s performance in identifying hub genes. However, we acknowledge some limitations, including moderate predictive power and sensitivity to data quality. These factors suggest a need for further refinement and clinical validation to realize the full potential of our findings.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Sarfi, S., Azaryan, E. & Naseri, M. Immune system of dental pulp in inflamed and normal tissue. DNA Cell. Biol. 43 (8), 369–386 (2024).
Lima, A. F. et al. Novel co-initiators of polymerization: Cytotoxicity profile and modulation of inflammatory mediators in human dental pulp stem cells. Dent. Mater. 40 (10), 1692–1696 (2024).
Wang, T. T., Jiang, W. R., Xu, L., Zhou, M. Y. & Huang, Y. S. Effect of blocking Trem1 on the M1 polarization of macrophages in regulating dental pulp inflammation. Eur. J. Oral Sci. 132, e13018 (2024).
Wang, S. et al. MicroRNA-27a-5p downregulates expression of proinflammatory cytokines in lipopolysaccharide-stimulated human dental pulp cells via the NF-κB signaling pathway. Int. J. Mol. Sci. 25 (17), 9694 (2024).
Fawzy El-Sayed, K. M., Ahmed, G. M., Abouauf, E. A. & Schwendicke, F. Stem/progenitor cell-mediated pulpal tissue regeneration: A systematic review and meta-analysis. Int. Endod J. 52 (11), 1573–1585 (2019).
Chang, C. C., Lin, T. A., Wu, S. Y., Lin, C. P. & Chang, H. H. Regeneration of teeth with allogeneic, autoclaved, and treated dentin matrix and dental pulpal stem cells: An in vivo study. J. Endod. 46 (9), 1256–1264 (2020).
El Karim, I. A. et al. Deciphering reparative processes in the inflamed dental pulp. Front. Dent. Med. 2, 651219 (2021).
Zaky, S. H., Shehabeldin, M., Ray, H. & Sfeir, C. The role of inflammation modulation in dental pulp regeneration. Eur. Cell. Mater. 41, 184–193 (2021).
Haugen, H. J., Basu, P., Sukul, M., Mano, J. F. & Reseland, J. E. Injectable biomaterials for dental tissue regeneration. Int. J. Mol. Sci. 21 (10), 3442 (2020).
Li, F. C. & Kishen, A. 3D organoids for regenerative endodontics. Biomolecules 13 (6), 900 (2023).
Xie, Z. et al. Functional dental pulp regeneration: Basic research and clinical translation. Int. J. Mol. Sci. 22 (16), 8991 (2021).
Xin, B. et al. Identification of pulpitis-related potential biomarkers using bioinformatics approach. Comput. Math. Methods Med. 2021, 1808361 (2021).
Shi, Y. et al. A single-cell interactome of human tooth germ from growing third molar elucidates signaling networks regulating dental development. Cell. Biosci. 11 (1), 178 (2021).
Opasawatchai, A. et al. Single-cell transcriptomic profiling of human dental pulp in sound and carious teeth: A pilot study. Front. Dent. Med. 2, 806294 (2022).
Zhang, D., Zheng, C., Zhu, T., Yang, F. & Zhou, Y. Identification of key module and hub genes in pulpitis using weighted gene co-expression network analysis. BMC Oral Health. 23 (1), 2 (2023).
Feng, Y., Chandio, B. Q., Thomopoulos, S. I., Chattopadhyay, T. & Thompson, P. M. Variational autoencoders for generating synthetic tractography-based bundle templates in a low-data setting. In 2023 45th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC) Vol. 2023 1–6 (2023).
Rajaram, S. & Mitchell, C. S. Data augmentation with cross-modal variational autoencoders (DACMVA) for cancer survival prediction. Information (Basel). 15 (1), 7 (2024).
Feng, Y., Chandio, B. Q., Thomopoulos, S. I., Chattopadhyay, T. & Thompson, P. M. Variational autoencoders for generating synthetic tractography-based bundle templates in a low-data setting. BioRxiv: The Preprint Server for Biology, United States (2023).
Li, Q. et al. XA4C: Explainable representation learning via autoencoders revealing critical genes. PLoS Comput. Biol. 19 (10), e1011476 (2023).
Barrett, T. et al. NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Res. 41 (Database issue), D991–D995 (2013).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498–2504 (2003).
Hasibi, R. & Michoel, T. A graph feature auto-encoder for the prediction of unobserved node features on biological networks. BMC Bioinform. 22 (1), 525 (2021).
Xie, Z. et al. Functional dental pulp regeneration: Basic research and clinical translation. Int. J. Mol. Sci. 22(16), 8991 (2021).
Jiao, L. et al. Ribosome biogenesis in disease: New players and therapeutic targets. Signal. Transduct. Target. Ther. 8 (1), 15 (2023).
Chen, J. et al. eEF2 improves dense connective tissue repair and healing outcome by regulating cellular death, autophagy, apoptosis, proliferation and migration. Cell. Mol. Life Sci. 80 (5), 128 (2023).
Panda, S. et al. Adjunctive effect of autologus Platelet-Rich fibrin to barrier membrane in the treatment of periodontal intrabony defects. J. Craniofac. Surg. 27 (3), 691–696. https://doi.org/10.1097/SCS.0000000000002524 (2016).
Kaarthikeyan, G. et al. Analysis of the association between interleukin-1beta (+ 3954) gene polymorphism and chronic periodontitis in a sample of the South Indian population. Indian J. Dent. Res. 20 (1), 37–40. https://doi.org/10.4103/0970-9290.49061 (2009).
Li, X. L., Fan, W. & Fan, B. Dental pulp regeneration strategies: A review of status quo and recent advances. Bioact Mater. 38, 258–275. https://doi.org/10.1016/j.bioactmat.2024.04.031 (2024).
Doncevic, D. & Herrmann, C. Biologically informed variational autoencoders allow predictive modeling of genetic and drug-induced perturbations. Bioinformatics 39 (6), btad387 (2023).
Nakamura, S., Tanimoto, K. & Bhawal, U. K. Ribosomal stress couples with the hypoxia response in Dec1-dependent orthodontic tooth movement. Int. J. Mol. Sci. 24 (1), 618 (2022).
Kobayashi, M. et al. The ubiquitin hybrid gene UBA52 regulates ubiquitination of ribosome and sustains embryonic development. Sci. Rep. 6 (1), 36780 (2016).
Takeda, T. et al. Characterization of dental pulp stem cells of human tooth germs. J. Dent. Res. 87 (7), 676–681 (2008).
Yang, Y. et al. Single-cell transcriptomic analysis of dental pulp and periodontal ligament stem cells. J. Dent. Res. 103 (1), 71–80 (2024).
Kion-Crosby, W. & Barquist, L. Network depth affects inference of gene sets from bacterial transcriptomes using denoising autoencoders. Bioinform. Adv. 4 (1), vbae066 (2024).
Mondal, A. K., Asnani, H., Singla, P. & Ap, P. ScRAE: Deterministic regularized autoencoders with flexible priors for clustering single-cell gene expression data. IEEE/ACM Trans. Comput. Biol. Bioinform. 19 (5), 2996–3007 (2022).
Ruiz-Arenas, C. et al. Netactivity enhances transcriptional signals by combining gene expression into robust gene set activity scores through interpretable autoencoders. Nucleic Acids Res. 52 (9), e44 (2024).
Zhou, L., Mao, H. Q., Wen, Y. H., Chen, Z. & Zhang, L. Cuproptosis aggravates pulpitis by inhibiting the pentose phosphate pathway. J. Dent. Res. 104 (5), 541–550. https://doi.org/10.1177/00220345251313797 (2025).
Xin, B. et al. Identification of pulpitis-related potential biomarkers using bioinformatics approach. Comput. Math. Methods Med. 2021, 1808361. https://doi.org/10.1155/2021/1808361 (2021).
Acknowledgements
We would like to thank the Center of Medical and Bioallied Health Sciences and Research, Ajman University, Ajman, UAE.
Author information
Authors and Affiliations
Contributions
Conceptualization, PY, JK, PN and CA.; Data curation, PY, JK, PN and CA; Formal analysis, PY, JK, PN and CA; Funding acquisition, PN; Investigation, PY, JK, PN and CA; Methodology, PY, JK, PN and CA; Project administration, PY, JK, PN and CA; Resources, PY and PN; Software, PY and JK; Supervision, PY, JK, PN and CA; Validation, PY, JK, PN and CA; Visualization, PY, JK, PN and CA; Writing – original draft, PY, JK, PN and CA; Writing – review & editing, PY, JK, PN and CA. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yadalam, P.K., Krithikadatta, J., Natarajan, P.M. et al. Prediction of hub genes in pulpal inflammation and regeneration using autoencoders and a generative AI approach. Sci Rep 15, 26225 (2025). https://doi.org/10.1038/s41598-025-12074-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12074-8
