
Abstract
Cutaneous leishmaniasis, a neglected tropical disease (NTD) caused by Leishmania tropica, Leishmania major and other members of the same species, poses significant challenges in the public health sector, especially in developing countries. The limitations of current treatments, including toxicity, resistance, availability, and cost effectiveness, necessitate the development of novel therapeutics. This study investigated the leishmanicidal potential of five Thiadiazine thione derivatives (THTT) against L. tropica, at a dose range of 25–426 µM using an MTT assay. Compounds T9, T24, T25 (tris-THTT) and PG (mono-THTT) exhibited notable IC50 values of 20.01, 26.94, 27.71, and 82.79 µM, respectively. Moreover, the same compounds demonstrated promising docking scores against critical leishmanial enzymes, such as Trypanothione reductase, Trypanothione synthetase, Leishmanolysin, and C24-sterol methyl transferase. Hemolytic assay revealed that all the compounds are non-toxic at lower concentrations. Mono-THTTs (PG and BG) showed higher CC50 values i.e., 401.65, 375.68 as compared to tris-THTTs (T9, T24, T25) which is 104.61, 104.51 and 89.73 µM respectively. All derivatives showed high selectivity indices (SI) compared to the standard drug Amphotericin-B. Furthermore, in-silico investigations targeting COX1 and COX2 enzymes unveiled a high affinity of the tested compounds towards these enzymes, indicating their potential involvement as anti-inflammatory agents. Subsequent in-vitro HRBC membrane stabilization and egg albumin denaturation assays confirmed the anti-inflammatory potential of all the compounds. These findings validate THTT derivatives as an effective and novel class of anti-leishmanial agents against L. tropica with a favorable safety profile and potential anti-inflammatory properties. Further research is recommended to elucidate their mechanisms of action and in-vivo efficacy.
Similar content being viewed by others
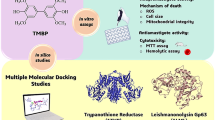

Introduction
Parasitic diseases affect hundreds of millions of people worldwide and remain a major public health concern1. These diseases include trypanosomiasis, pediculosis, trichomoniasis, malaria, giardiasis, and leishmaniasis, all caused by different Protozoans2. Leishmaniasis, an overlooked disease that is more prevalent in tropical, subtropical regions and Southern Europe3. The disease is caused by infection with protozoan parasites belonging to the genus Leishmania, and is spread by the bite of a vector, the phlebotomine sand fly4,5. There are 12 million instances of the disease every year, and 350 million individuals are in danger because of its predominance in 98 countries6. Over one billion people are at risk of these diseases in virtually all continents7. Larger increases in the incidence of Cutaneous leishmaniasis have been observed over the past few years. The 3.9 million prevalent cases from the 2015 GBD study represent only twice the sum of 11 years of reported incidence from WHO (2005–2015) and only 3 times the previously estimated annual global incidence8. These diseases debilitate large numbers of people, keeping them from full, productive lives. The primary clinical manifestations include cutaneous, mucocutaneous, and visceral leishmaniasis9,10. First-line treatment with pentavalent antimonials has been practiced for a considerable period of time11. Amphotericin B or pentamidine, recognized as pivotal agents in the secondary line of treatment12. However, both the first-line and second-line drugs are associated with significant drawbacks, including toxicity, instances of resistance, and high expenses13. The first oral remedy used for visceral leishmaniasis was Miltefosine, introduced in 200214.
Despite its notable efficacy, Miltefosine is associated with teratogenic, hepatotoxic, and nephrotoxic effects, thereby exacerbating the challenges faced by patients15,16,17. Among the Leishmania species, L. tropica stands out for its ability to manifest all clinical manifestations of leishmaniasis, thereby playing a pivotal role in understanding the disease’s epidemiology18. Diffuse cutaneous leishmaniasis, a grave public health concern characterized by extensive skin lesions, inflicts profound suffering upon patients and remains unyielding to current therapeutic options. This condition often arises due to an inadequate immune response against the parasite18,19. Hence, the quest for novel chemical entities with reduced adverse effects for treating leishmaniasis gained significant importance. In the situation of visceral leishmaniasis, inflammation emerges as a crucial factor contributing to patient mortality. Following the invasion of macrophages and other immune cells by Leishmania parasites, an immune reaction arises, marked by the secretion of pro-inflammatory cytokines and chemokines20. Inflammation also involves the lysis of lysosomes, which release their component enzymes, producing a variety of disorders. Non-steroidal anti-inflammatory drugs (NSAIDs) exert their beneficial effects by either inhibiting the release of lysosomal enzymes or by stabilizing the lysosomal membranes. The erythrocyte membrane is similar to the lysosomal membrane, which is why the HRBC membrane stabilization method was chosen for the in-vitro assessment of the anti-inflammatory property21. Computational analysis helps in predicting the binding behavior of compounds against any particular biological target22,23. This binding analysis demonstrates the mechanism of inhibition by drug-like molecules. Therefore, computational analysis of test compounds was conducted to understand their mode of binding with COX1 and COX2, which are involved in causing inflammation; thus, inhibiting these enzymes can be a pivotal24. Trypanosomatidae parasites differ genetically from other eukaryotes by using the specific Trypanothione Reductase (Try-R) and Trypanothione Synthetase (Try-S) system for trypanothione metabolism instead of Glutathione reductase. Protozoan survival directly depends on this essential trypanothione system25,26. Leishmanolysin, previously identified as a potential target, is involved in cell signaling and virulence.
The development of future inhibitors targeting Gp63 protein has possible benefits to reduce both the pathogenic behavior of L. donovani and its harmful invasion of mammalian macrophages27,28. The sterol biosynthetic pathway functions as a primary target when developing new drug candidates against Leishmania. Potential drug development requires targets that are present exclusively in parasites. The previous investigators chose LdSMT because scientists find it unique to the parasite in order to develop new therapeutic drugs29. Thus, computational analysis of these Leishmania specific enzymes such as Trypanothione reductase (Try-R), Trypanothione synthetase (Try-S), C24-Sterol methyl transferase (SMT) and Leishmanolysin (GP63) that are critical for the survival of Leishmania was conducted in current study as its inhibition may also play a significant role in the management of the disease. The regulation of inflammation and modulation of the immune response emerge as pivotal therapeutic considerations in managing leishmaniasis, aiming to reduce mortality rates and enhance patient outcomes tolerance.
Recently, derivatives of tetrahydro-2H-1,3,5-thiadiazine-2-thione (THTT) have gained significant interest due to their pharmacological potential, particularly their anti-parasitic and anti-inflammatory properties30. Early investigations into these compounds underscored the importance of the substituent’s composition at the N-3 position regarding antimicrobial activity, while the impact of substitutions at N-5 on toxicity has also been elucidated31. Thiadiazine-thione derivatives (THTT) have emerged as promising candidates for medicinal applications, exhibiting a spectrum of pharmacological effects that encompass antibacterial, antifungal, anthelminthic, antiprotozoal, tuberculostatic, herbicidal, and antioxidant activities32. In recent years, efforts to enhance the antiprotozoal efficacy have led to the incorporation of two THTT rings within the same molecular structure, linked via their N-3 atom through a linear or branched aliphatic backbone, with alkyl or carboxyalkyl residues at N-5 (bis-THTT)33.
These distinctive characteristics, combined with the ability to append various structurally diverse substituents to the heterocyclic ring, impacting the biological and physicochemical properties of these compounds, led to the selection of the THTT ring as a foundational framework in our ongoing research endeavors aimed at developing novel anti-parasitic agents. In the present study, we performed a comprehensive screening of five THTT derivatives to evaluate their efficacy against leishmaniasis, followed by a detailed exploration into their anti-inflammatory properties and cytotoxic effects. Our study particularly focus on promising compounds, encompassing two mono and three tris-THTT derivatives.
Materials and methods
Tested thiadiazine thione derivatives
The present study utilized a series of synthetic Thiadiazine thione derivatives (THTT) provided by the Institute of Chemical Sciences (ICS) at the University of Peshawar, Khyber Pakhtunkhwa, Pakistan. A total of five derivatives, two mono-THTTs (BG and PG) and three tris-THTTs (T9, T24, and T25) were selected for investigation in this research work.
Compounds BG and PG were synthesized by the pot domino method as follows, 20% aqueous KOH solution was treated with butyl amine (BG) and propyl amine (PG), followed by the addition of 20 mmol CS2 (Scheme 1). The mixture was stirred for four hours. Subsequently, 37% formaldehyde solution (40 mmol) was added dropwise to a suspension of glycine (20 mmol) in phosphate buffer (pH 7.8), and the mixture was stirred for one hour. The reaction mixture was then acidified to a pH of 2.0 with hydrochloric acid at 0–5 °C, resulting in the formation of a precipitate. The precipitates were washed, dried, and purified via recrystallization from ethanol.

Synthesis of compounds BG and PG.
Compounds T9, T24, and T25 were synthesized by reacting tris (2-aminoethyl) amine with 20 mmol CS2 in 15% aqueous KOH solution for five hours. The reaction mixture was then treated with 37% formaldehyde solution (40 mmol) and added to phenylalanine, para-ethyl aniline, or 2,4,6-trimethyl aniline (R2), respectively (Scheme 2).

Synthesis of compounds T9, T24, and T25.
In-silico study
Target proteins selection
Choosing an appropriate protein for testing compounds is the first step in molecular docking analysis34. The enzymes were selected through a literature survey on the basis of their importance, uniqueness, and significant role in the infection26. The 3D structures of compounds were drawn through ChemDraw Professional 15.0 and stored in a database created by MOE software (version 2019.01). Crystallographic structures of receptors were downloaded from protein data bank (PDB). For optimal results, the receptors and ligands were individually prepared e.g., protonation issues addressed, partial charges calculated, and energy minimization and geometry optimization performed using the structure preparation wizard. The unwanted moiety i.e., cofactors and water molecules, was removed from the enzymes35.
Docking protocol validation
Docking analysis was carried out to study the binding orientation and binding affinity of the compounds in the active site of enzymes. Before docking the compounds in question, the docking protocol is validated by re-docking the co-crystalized protoporphyrin IX containing CO compound with the COX2 enzyme (PDB code 5IKR). The co-crystalized and re-docked ligands in complex with COX2 were superimposed to investigate the RMSD, which was found to be 0.2 Å, indicating that the MOE docking protocol can reproduce the correct pose and can be used for our docking study36.
Molecular docking assay
The docking experiment on the enzymes was perfourmed by placing the energy-minimized ligand into the binding site through their respective grid map dimensions. The Binding sites of enzymes were identified based on literature reviews. The docking protocol implemented in MOE (version 2019.01) with default parameters was used for the molecular docking study. Settings included placement method (Triangle Matcher), London DG and GBVI/WSA ΔG scoring functions, and induced-fit refinement option. A flexible ligand model was used in the docking and subsequent optimization scheme to find the correct conformations with minimum energy. The apex pose of each compound was selected based on the docking score (S) for further analysis. Subsequently, the interaction and docking output were examined. At the end of docking, the best conformations based on score were analyzed for hydrogen bonding37. Computational technique was employed to calculate the binding affinities of the Thiadiazine thione derivatives (ligands) with the key residues in the active sites of enzymes (receptors) i.e., Trypanathione reductase (PDB ID: 2JK6)38, Trypanathione synthase (PDB ID: 2VPM)39, Leishmanolysin GP63 (PDB ID: 1LML)40 and homology model of C24-sterol methyl transferase SMT (UniProt ID: E9AT15)41 respectively. These binding potential insights are pivotal for evaluating the test compound as an inhibitor of the enzymatic activity of the subject proteins. Similarly, THTT derivatives were evaluated for their anti-inflammatory properties and mode of action by a docking study using the COX1 and COX2 enzymes. The X-ray crystal structures of these cyclooxygenases, COX1 (PDB ID: 6Y3C)42 and COX2 (PDB ID: 5IKR)43 were downloaded from the protein data bank. The compounds were docked in the active sites of these enzymes. The docking study predicts the binding affinity, binding energies, binding modes, and orientation of ligands (THTTs) at the active site of COX1 and COX2.
ADMET prediction of THTT derivatives
ADMET analysis constitutes a drug’s pharmacokinetic profile, including absorption, distribution, metabolism, excretion, and toxicity. In this study Swiss ADME server tool was used44,45. In this server, the import icon in the molecular sketcher was selected. In a new window, already prepared structures of THTT derivatives were selected and presented on the molecular sketcher and then transferred to the SMILES format. The format was run to offer the ADMET parameters and related values.
Biological activities
Parasite culture
Leishmania tropica, designated as KWH23 previously reported by Islam et al. (2019), was used in this study46. Isolates were cultured in RPMI-1640 medium to which 10% fetal bovine serum (FBS) and 1% antibiotic solution of streptomycin and penicillin was added. The culture was stored at a temperature of 25 ± 1 °C. Following a 4-day incubation period, the culture was examined by using an inverted microscope manufactured by Olympus®. Subsequently, the Leishmania was subcultured for further growth47.
Promastigotes proliferation assessment by MTT assay
The promastigotes of the L. tropica species were inoculated at a density of 1 × 106 cells per well in a microtiter plate sourced from Sigma-Aldrich. Thiadiazine thione derivatives were introduced into the wells at a dose range of (25–426 µM) each, alongside a negative control consisting of 0.1% DMSO and a positive control of amphotericin B. The plates were incubated for 48 h at 24 ± 1 °C.
Following the incubation, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), which serves as an indicator of cell viability. The optical density was recorded at 570 nm by using a multi-well microtiter plate reader, Bio-Tek ELx-800 model. To enhance the reliability, the assay was conducted in triplicate48,49. Percent inhibition was calculated using the following formula, while IC50 values were calculated via linear interpolation method.
Toxicological evaluations
Blood sample collection
Blood samples were obtained through venous puncture from healthy individuals with no recent history of infectious diseases for the studies mentioned below. Sample collection was conducted by a trained professional. Informed consent was obtained from all participants, adhering to the principles outlined in the Helsinki Declaration of 1975 (revised 1997). Ethical clearance for the current study was obtained from the bioethical committee of the Department of Biotechnology, Abdul Wali Khan University Mardan, Pakistan.
Cytotoxicity assay (Hemolytic assay)
THTT derivatives were evaluated for their toxicity to human blood erythrocytes at a dose range (25–426 µM). Compounds were incubated with 5% suspension of blood erythrocytes (1 mL) at 37 °C for 3 h. 0.1% DMSO was used as a negative control, and Triton X-100 (0.5%) as the positive control. Following the incubation, samples were centrifuged at 1000 × g for 10 min to separate the cellular components effectively. After centrifugation, the extent of cell lysis indicated by hemoglobin release, quantified by using a high precision spectrophotometer (Agilent-DAD, 8453, Agilent Tech., Germany) at 576 nm50.
Hemolytic percentage was measured by using the formula below, while CC50 value were calculated via linear interpolation method.
Genotoxicity evaluation by DNA laddering assay
The assessment of genotoxicity involved conducting a DNA laddering assay. This procedure, also referred to as DNA fragmentation analysis, following the protocol outlined by51. Human DNA was isolated from blood and then quantified by using the spectrophotometer. An equal amount of DNA (3 µg) was dispensed into 96-well plates and treated with the different concentrations (25 to 426 µM) of the selected THTT derivatives. For a reference, human DNA was exposed to 200 µM of hydrogen peroxide to induce complete damage, while untreated DNA and DNA treated with the vehicle were employed as negative controls. The treatments were then incubated at 37 °C for 30 min to analyze the effects of THTT derivatives on human DNA integrity. Following the incubation, gel electrophoresis as performed on 2% agarose gel, stained with ethidium bromide (0.5 µg mL−1)52.
In-vitro evaluation of anti-inflammatory effect
Blood sample preparation for the membrane stabilization assay
The Human red blood cell (HRBC) membrane stabilization assay is a well-established procedure for evaluating in-vitro anti-inflammatory potential53. Blood suspension was prepared by adding an equal volume of Alsvere solution (0.8% sodium citrate, 2% dextrose, 0.42% NaCl, and 0.5% citric acid) to the blood. Subsequently, the blood samples were centrifuged for 5min at 2500 rpm to separate the cellular components, and the supernatant was discarded. Cell suspension was washed multiple times with normal saline (0.9% w/v NaCl) to eliminate any residual impurities until the supernatant appeared clear. Blood suspension 40% (v/v) was prepared by adding sterile saline solution (0.9% w/v NaCl) to the cellular part.
Hypotonicity-induced hemolysis
The compounds were tested at a dose range of 25–426 µM (25, 50, and 100 ppm). HRBC suspension (0.5 mL) and hyposaline (2 mL) were added to the samples. The resulting mixture underwent incubation at 37 °C for 30 min and then centrifuged at 3000 rpm for 20 min to separate the supernatant from cellular debris. Subsequently, the supernatant was analyzed for hemoglobin content spectrophotometrically at a wavelength 560 nm, indicating the degree of hemolysis or membrane stabilization induced by the test compounds. Aspirin (100 µg mL−1 or 555.09 µM) was utilized as a reference drug. Additionally, a negative control was prepared by excluding the test compounds to establish the baseline measurement54.
The percentage of membrane stabilization or inhibition of hemolysis was measured by the following formula.
where OD1 = Optical density of control sample, OD2 = optical density of compounds in hypotonic medium.
Egg albumin protein denaturation assay
A reaction mixture of 5 mL was prepared for each test compound by adding 0.2 mL albumin protein, which was extracted from a hen’s egg, 2.8 mL of Phosphate buffer saline (PBS) having a pH of 6.4, and 2mL of the test compounds. The compounds were tested at a dose range of 25–426 µM (25, 50, and 100 ppm). Double-distilled water was used as a control, and Diclofenac sodium was utilized as a standard drug (200 µg mL−1 or 628.6 µM). Subsequently, the reaction mixture was incubated in a BOD incubator from Labline Technologies at 37 ± 2 °C for 15 min. After incubation, the mixture was subjected to heat shock at a temperature of 70 °C for 5 min to initiate protein denaturation. Following the heat shock, optical density was measured at 660nm using a multi-well microtiter plate reader, Bio-Tek ELx-800 model55. The formula used to evaluate the percentage inhibition of protein denaturation is;
where Vt = absorbance of compounds, Vc = absorbance of control sample.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4 software. To assess the significance of difference, one way analysis variance test (ANOVA) was employed, followed by student T test. All experiments were conducted in triplicate and the graphs were constructed by using the mean values, with error bars illustrating the standard deviation (SD) among them. A P-vaue of < 0.05 was considered statistically significant.
Results and discussion
Drug repurposing is an intriguing field of medicinal chemistry that involves investigating the effectiveness of compounds with established activity for their efficacy against neglected diseases. In this context, Thiadiazine thione derivatives have been recognized for their diverse range of bioactivities, including anti- proliferative56, antibacterial57, antiepileptic58, antifungal59, anti-leishmanial59, antimalarial60, anti-tubercular61, trypanocidal62, antioxidant63, and herbicidal64 properties. The notable biological potentials exhibited by THTT derivatives, coupled with the growing interest in prodrug therapy, have spurred numerous researchers to explore novel derivatives of Thiadiazine. THTT prodrugs offer enhanced pharmacokinetic profiles, facilitating greater accessibility of the active drug form to its therapeutic target, thereby necessitating lower dosages and minimizing adverse effects65.
This investigation assessed the in-vitro and in-silico leishmanicidal potential, cytotoxicity, genotoxicity, and anti-inflammatory potential of five previously prepared THTT derivatives (BG, PG, T9, T24, and T25) reported by Ali et al.66. The IUPAC names and chemical formulas of the compounds are given below in Fig. 1.

Chemical structures of THTT derivatives 2-(5-Butyl-6-thioxo-1, 3, 5-thiadiazinan-3-yl) acetic acid (BG), 2-(5-propyl-6-thioxo-1,3,5-thiadiazinan-3-yl)acetic acid (PG), 2,2′,2″-(5,5′,5″-(nitrilotris(ethane-2,1-diyl))tris(6-thioxo-1,3,5 thiadiazinane-5,3-diyl))tris(3-phenylpropanoic acid) (T9), 5,5′,5″-(nitrilotris(ethane-2,1-diyl))tris(3-(4-ethylphenyl)-1,3,5-thiadiazinane-2-thione) (T24) and 3,3′,3″-(nitrilotris(ethane-2,1-diyl))tris(5-mesityl-1,3,5-thiadiazinane-2-thione) with code (T25).
Molecular docking studies
Docking study of THTT derivatives with Leishmania-specific enzymes
The molecular docking study revealed the binding modes of two distinct groups of compounds; monocyclic (PG and BG) and tricyclic (T09, T24, and T25) with target proteins, Try-R and Try-S. The docking procedure involved generating 30 conformations for each compound, and the top-ranked conformations were selected for detailed protein–ligand interaction profile studies. The results showed that all compounds exhibited favorable orientations within the active sites, establishing key interactions with essential residues such as Cys364, Thr335, Tyr198, Lys60, Lys61, Cys57, Cys52 and Glu652, Glu650, Arg375, Asp124, Phe41, His39 respectively38,39, contributing to binding stability or creating binding patches, as illustrated in Fig. 2a,b). For TryR, compound T9 displayed the best docking score, forming five hydrogen bonds with active site residues Ser178, Arg287, Thr335, Gly15, and Tyr178. In contrast, compound PG formed two hydrogen bonds with Cys52 and Ser14, while T24 interacted with Cys57 and Lys61. Notably, the presence of hydrogen bond donor/acceptor groups in the compounds contributed to these interactions. In TryS, compounds T9 and PG exhibited distinct PL interaction profiles. Compound PG formed hydrogen bonds with Gln58, Asp38, and Glu65, whereas compound T9 interacted with Asp124, Gly376, and Asp651. These differences in residue interactions may be attributed to the unique structural features of each compound. Inhibiting these target proteins, Trypanothione reductase and Trypanothione synthetase, could disrupt the redox balance, impede DNA repair and replication, and attenuate antioxidant defenses, rendering the parasites susceptible to oxidative stress and compromising their survival67. Depletion of trypanothione, which is vital for detoxification and energy metabolism, might lead to genomic instability and reduced metabolic activity. While inhibition of Try-R and Try-S presents a promising therapeutic approach against Trypanosoma spp.-related diseases67.

Represents the protein ligand interaction profile for potent compounds against Trypanothione reductase (a) and Synthetase (b). The black dots indicate the hydrogen bond.
The compounds under investigation also displayed significant binding affinity within the active sites of Leishmanolysin (GP63) and C24-sterol methyltransferase enzymes (SMT). SMT is required for the optimal mitochondrial function and virulence; similarly, GP63, which is the main protein found on the promastigote surface, helps in entry into the host cell and parasite survival68,69. While the inhibition of these enzymes presents a promising avenue for treating leishmaniasis, as inhibiting these enzymes is directly related to the cell death of the parasite.
The molecular docking study revealed key interactions between compounds and the active sites of GP63 and SMT enzymes. The crucial active site residues of GP63 are Glu220, Val223, Leu224, Ala225, Glu265, His268, Gly329, Pro34740 and that of SMT are Glu65, Trp68, Gln70, His71, Phe72, Arg140, His14470 respectively. Notably, Compound PG exhibited robust interactions with both enzyme active sites, forming bonds with essential residues. A detailed analysis of the interactions showed that Compound PG formed 3 hydrogen bonds with GP63 active site residues (Leu224, Ala225 and Glu265) and 2 hydrogen bonds with SMT active site residues (Phe72, and His 144). Compound T9, another potent inhibitor, formed three hydrogen bonds with crucial active site residues of Leishmanolysin (Glu220, Gly329 and Pro347). In contrast, Compounds T25 and PG showed strong interactions with SMT, with T25 establishing four hydrogen bonds (Phe72, His144 and Arg140) and PG showing two hydrogen bonds with residues (Phe72, His144). These interactions were facilitated by specific functional groups, such as thiadiazine, hydroxyl, and amino groups, which promoted binding with the active site amino acids. The docking scores and binding energies (Table 1) further supported the strong binding of these compounds to the enzyme active sites (Fig. 3a,b).

Represents the protein ligand interaction profile for potent compounds against Leishmanolysin (GP63) and C24-sterol methyl transferase. The black dots indicate the hydrogen bond.
The differences in binding profiles between the compounds and enzymes highlight the importance of structural specificity in enzyme inhibition. Compound PG’s ability to interact with both GP63 and SMT enzymes suggests a potential for dual-targeting, which could enhance its therapeutic efficacy. Further studies are warranted to explore the in-vitro and in-vivo efficacy of these compounds and to optimize their binding affinity and specificity for the target enzymes.
Docking study for anti-inflammatory potential
To study the binding interactions between the THTT derivatives and the active sites of target proteins COX1 and COX2, Molecular docking studies were performed using MOE software. Our test compounds have two types of side groups, one type of derivatives has bulky groups attached to thiadiazinane-2-thione core (compounds T24, T25, and T9), whereas the other type of derivatives have small side groups (PG and BG). The most active T24-compound docked (Fig. 4) in the active site of COX1 enzyme showed five hydrogen bonds with the active site residues Glu524, His513, His90, and Val116 showed Arene-hydrogen interaction. In contrast, PG exhibited strong binding with Leu117 and Arg120. Other derivatives of the series T25, T9 and BG also displayed strong interactions with the enzyme active sites.

3D representation of compounds T24 and PG (Cyan) interactions (black) with the active sites amino acids of COX1 (green) and COX2 (Sea green) enzymes. Docked pose of (a) compound T24 and (b) compound PG in the active site of COX1. Binding interactions of (c) compound T24 and (d) compound PG with COX2 active site residues.
Furthermore, these compounds also exhibited strong binding with the active site residues of the COX2 enzyme (Fig. 4). The results revealed good docking score, binding affinities, and important interactions of these compounds with the key residues of active sites of COX1 and COX2 enzymes, as shown in Table 2. Previously, a novel class of alkyl/aryl/aralkylamines, also known as amino acid-modified tetrahydro-2H-1,3,5-thiadiazine-2-thiones were synthesized and evaluated for anti-nociceptive and anti-inflammatory activities using an in-silico study. They have reported a high binding score of THTT derivatives with COX2/LOX5, respectively71. Thiadiazine derivatives were also reported to have inhibitory effects on the COX2 enzyme by a docking study, which emphasizes the importance of these compounds as potential anti-inflammatory agents72.
SAR analysis
The docking analysis suggests that the activity of these compounds is likely attributed to the presence of various functional groups. The compounds PG (2-(5-propyl-6-thioxo-1,3,5-thiadiazinan-3-yl) acetic acid) and BG (2-(5-butyl-6-thioxo-1,3,5-thiadiazinan-3-yl) acetic acid) exhibit comparable affinities towards COX’s enzymes and Leishmania specific enzymes despite differing by only a methyl group. Conversely, the bulkier compounds T24 [3,3′,3″-(nitrilotris(ethane-2,1-diyl))tris(5-(4-ethylphenyl)-1,3,5-thiadiazinane-2-thione)], T25 [3,3′,3″-(nitrilotris(ethane-2,1-diyl))tris(5-mesityl-1,3,5-thiadiazinane-2-thione)], and T9 [2,2′,2″-((nitrilotris(ethane-2,1-diyl))tris(6-thioxo-1,3,5-thiadiazinane-5,3-diyl))tris(3-phenylpropanoic acid)] demonstrate robust binding interactions with inflammatory and Leishmania specific enzymes, potentially due to their bulky moieties. All compounds share a common core structure, 1,3,5-thiadiazinane-2-thione, known for significant biological activity, which likely interacts crucially with target enzymes or receptors, impacting their anti-leishmanial and anti-inflammatory activities. Additionally, functional groups such as thiosemicarbazide moieties in T24, T25, and T9, aromatic rings with various substituents in T24, T25, and T9, and carboxylate groups in T9, PG, and BG contribute to the compounds’ activity through diverse interaction mechanisms including hydrogen bonding, hydrophobic interactions, and electronic properties. Structure–activity relationship (SAR) analyses indicate that substituents on aromatic rings influence electronic distribution and steric interactions, while alkyl chain length (PG and BG) affects flexibility and steric effects. Notably, the presence of bulky side groups enhances the binding interactions by increasing affinity toward active site residues of enzymes involved in inflammatory and leishmanial activities. Overall, the anti-leishmanial and anti-inflammatory activities of these compounds result from a complex interplay between their core structure, functional groups, and molecular interactions, emphasizing the importance of considering these factors in drug design and optimization efforts.
ADMET analysis
The pharmacokinetic and physiochemical properties were evaluated using an advanced bioinformatics tool, SwissADME predictor, which was utilized to analyze the ligand and corresponding metal complexes. The solubility of compounds in non-aqueous medium was assessed by determining the consensus logP based on their mean expected lipophilicity values, indicating that if a compound is more soluble, then the consensus logP will be more negative. The results indicated that our compounds are less soluble in non-aqueous medium. To estimate the aqueous solubility, the log S scale was employed. The assessment indicated that BG and PG were highly soluble, whereas the ligands were moderately soluble, and the complexes were poorly soluble. If log S < − 0 is highly soluble, < − 2 very soluble, < − 4 soluble, < − 6 moderately soluble, and < − 10 indicates poorly soluble. Moreover, the predicted topological polar surface areas (TPSAs) of compounds ranged from 101.17 to 230.85 Å2. Therefore, we conclude that the reference drug has significantly better intestinal absorption than the synthetic molecule.
The pharmacokinetic parameters, such as skin permeation, absorption, distribution, and metabolism, were predicted as given in Table 3. BG and PG demonstrated high GI absorption in contrast with T24, T25, and T9. All the compounds were found to be impermeable to the BBB, consequently, there is no probability of producing harmful toxicants in the blood and brain after being metabolized. The skin permeability values (log Kp) for the compounds ranged from − 6.55 to − 8.29 cms−1 indicating consistently low skin permeability73. The permeability glycoprotein (P-gp) is an important factor affecting the drug absorption and disposition; therefore, its assessment was conducted to identify the test compound’s role either as a substrate or an inhibitor of the p-glycoprotein. The results showed that BG and PG are not substrates of the P-gp except T24, T25, and T9. Drug ligands are predicted to exhibit non-inhibitor properties for every enzyme.
Drug likeness is a prediction that evaluates whether a specific compound has drug-like attributes and is recognized as an orally active drug or not. It is based on the bioavailability score and Lipinski rule of five (RO5). The results predicted that BG and PG, the synthesized compounds and the reference drug, adhere to the Lipinski rule of five without any violations, showing that these compounds have a drug-like molecular nature, except for the molecular weights of T24, T25, and T9. There are many FDA-approved drugs with a molecular weight greater than 500 Da that fail to conform to the Lipinski rule of five74. Many drugs among various important classes such as antibiotics (e.g., erythromycin, clarithromycin, and roxithromycin), anti-cancer (e.g., etoposide), Kinase inhibitors (e.g., lapatinib and nilotinib), HIV (atazanavir) and HCV protease inhibitors, immunosuppressant’s (e.g., cyclosporine), antifungal (e.g., amphotericin B), and other miscellaneous classes are approved by FDA or other drug regulatory authorities as clinical use lie beyond the rule of five75,76. Furthermore, Ro5 violation drugs, bromocriptine (an ergot alkaloid derivative), reserpine (a rauwolfia alkaloid), acarbose and everolimus (both microbial products), ivermectin (derived from a fungi product) and cyanocobalamin (vitamin B12) are natural products or derivatives, and are specifically excluded from the Ro5 criteria76. Currently, there are no FDA-approved THTT derivatives available. However, some research studies have explored mono-THTT derivatives with potential biological activities, which adhere to RO577; however, no prior study on tris-THTTs has been identified. This study reports the novel findings of tris derivatives. Hence, it can be concluded from the pharmacokinetic and physicochemical study of THTT derivatives that the compounds showed poor solubility in non-aqueous medium and varying aqueous solubility. BG and PG demonstrated high GI absorption, while T24, T25, and T9 showed lower absorption. None of the compounds were predicted to permeate the blood–brain barrier (BBB). The compounds showed low skin permeability and were predicted to be non-inhibitors of various enzymes. BG and PG adhered to RO5, while T24, T25, and T9 violated the rule due to high molecular weights.
In-vitro anti-leishmanial activity
Five THTT derivatives mentioned above (Fig. 1) were tested for their anti-leishmanial activity against L. tropica. All derivatives exhibited concentration-dependent inhibitory effects on the growth of promastigotes. In particular, PG showed the highest activity with an IC50 value of 82.79 µM among mono-THTT derivatives and T9 with an IC50 value of 20.01 µM among tris-THTT derivatives following 48 h of treatment, as shown in Table 4 below. Comparatively, Amphotericin B exhibited an IC50 value of 15.68 μM. The structure–activity relationship analysis revealed that the N-5 and N-3 substituents and the number of rings play crucial roles in leishmanicidal activity. For instance, tris-THTT derivatives (T9, T24, and T25) showed more activity as compared to mono-THTTs (BG and PG). The presence of a hydrophilic group, such as a carboxylic group at the N-3 position or a lipophilic group like alkyl/aryl at the N-5 position, can enhance the activity, as is evident from the activity of BG with butyl group and PG with propyl group at the N-5 position. Thiadiazine thiones are a biologically significant class of compounds as they produce the diathiocarbamate and isothiocyanate salts as metabolic byproducts and have a flexible structure with distinct substitution patterns at positions N-3 and N-5. The anti-parasitic bioactivity of thiadiazine derivatives is believed to be attributed to the interaction between protozoan cysteine proteinases and the isothiocyanates produced through the hydrolysis of the thiadiazine ring78. The synthesis and anti-leishmanial activity of THTT derivatives against L. major were previously reported, their research showed that this class of compounds has therapeutic value against the Leishmania parasite79. A number of 1,3,5-thiadiazine-2-thione derivatives were also synthesized, and their leishmanicidal effects against L. major were documented80,81. Furthermore, the effect of several alkyl-linked bis tetrahydro-(2H)-1,3,5-thiadiazine-2-thione (bis-THTT) on L. donovani has been reported, suggesting these compounds as potent trypanocidal agents60. To gain insight into the possible reasons for the activity, a molecular docking study was carried out with the parasite-specific critical enzymes Try-R, Try-S, GP63 and SMT. The results are aligned with our in-silico findings, where all of the tested compounds showed high affinity towards target enzymes. The high docking score of T series compounds (T9, T24, and T25) with the enzymes mentioned in (Figs. 2 and 3) may account for their high in-vitro anti-leishmanial potential (Table 4).
In-vitro cytotoxicity analysis (Hemolysis assay)
Cytotoxicity of THTT derivatives was evaluated by the Hemolysis assay. Hemolysis assay revealed that all compounds had significantly lower hemolytic effects, with percentages below 5% (p < 0.001), unlike Triton-X 100 (0.5%) (Positive control), which exhibited 100% hemolysis. As per ISO/TR 7405–1984(f) standards, samples will be classified as hemolytic if the hemolytic percentage exceeds 5%. Biosafety guidelines stipulate that substances (chemical or biological) are categorized as hemolytic if they induce over 5% hemolysis, slightly hemolytic if causing 2–5% hemolysis, and non-hemolytic if exhibiting less than 2% hemolysis activity. The findings of the hemolysis are expressed in terms of CC50 values (Table 5). Mono-THTTs (BG and PG) exhibited the highest CC50 values as compared to the tris-derivatives. Thus, it can be inferred that despite the low cytotoxicity of the two mono-THTT derivatives, less hydrophobic compounds exhibit lower cytotoxicity, possibly due to their limited interaction with the glycolipids and glycoproteins of cell membranes. Another study by Jácome Ortiz. and M. Poleth (2021)82 reported lower cytotoxicity and antigenicity of mono-THTT compounds compared to bis-THTT compounds. Our results are consistent with those reported by82,83, who also observed similar findings.
SI index
The ratio of the CC50 and IC50 values was used to calculate the selectivity index (SI) for each of the investigated drugs, as shown in Table 6. Higher SI values are indicative of more selective compounds84. Interestingly, none of the compounds showed toxicity, resulting in a high selectivity index for the parasite cells with the exception of T9 (SI value 0.19 µM), which is still greater than that of the reference drug Amp-B (SI value 0.18).
DNA laddering assay
Genotoxicity of the selected compounds was assessed through the DNA Laddering assay, which detects different bands indicative of double-stranded breaks, associated with genomic instability and chromosomal aberrations. THTT derivatives were assessed at concentrations ranging from 25 to 426 µM. Figure 5 of the data indicated that there was no discernible difference between the THTT’s treated groups and the negative controls. This suggests that even at the highest concentration tested, the THTT derivatives did not induce any damage. Similar findings were reported by Avuloğlu-Yılmaz E et al., 2017 on human lymphocytes in an in-vitro experiment, demonstrating that THTT derivatives do not pose any genetic risk32.

DNA damage induced by THTT derivatives (PG, BG, T9, T24, T25) was evaluated using DNA fragmentation assay. Lane M: DNA marker; Lane − C: Negative control (untreated cells); Lane EC: Untreated DNA control; Lane + C: Positive control (H2O2-treated DNA); Lanes PG, BG, T9, T24, and T25: DNA treated with respective THTT derivatives.
In-vitro evaluation of anti-inflammatory effect
HRBC’s membrane stabilization assay
Using the hypotonicity-induced Human red blood cells membrane stabilization approach, as previously reported54, the anti-inflammatory potential was evaluated for the test compounds. Hypotonic solutions have a hemolytic effect, whereas hemolysis occurs due to the accumulation of excessive fluid within cells, leading to the membrane lysis of RBCs. Cell membrane injury renders the cell more vulnerable to secondary damage, typically induced by lipid peroxidation85. Membrane stabilization serves to prevent the leakage of serum proteins and fluids into tissues, a process driven by inflammatory mediators that increase membrane permeability86. The findings of the current study demonstrated that the three derivatives, PG, BG, and T24, significantly inhibited hemolysis, with respective percentage inhibition values of 97.7 ± 2.11, 73.8 ± 2.01, and 73.6 ± 2.45 as indicated in Fig. 6. This suggests that these compounds may be involved in stabilizing the RBC membrane by inhibiting the release of lytic enzymes and active inflammatory mediators21. Our findings are consistent with another study on Thiadiazine thiones, which found that this class of compounds is potential anti-inflammatory drugs, and that the highest activity of compounds may be due to lipophilic character71.

Percent proctection of HRBCs by THTT derivatives at various concentrations. Values are Mean ± Standard Deviation, n = 3, values with * considered significant (p < 0.05) the remaining have no differences (KW-Test Sig).
Egg albumin protein denaturation assay.
The anti-inflammatory potential of THTT derivatives was also assessed by egg Albumin denaturation assay. According to the results, three compounds, T24, PG, and T25, exhibited notable concentration-dependent percentage inhibition of denaturation with respective values of 75.70 ± 2.1, 55.81 ± 1.1, and 49.81 ± 1.8. Moreover, the reference drug diclofenac sodium demonstrated significant inhibitory potential, as anticipated, with a value of 64.94 ± 0.94 (Fig. 7).

Percentage inhibitions of protein denaturation after heat shock by THTT derivatives at various concentrations, each bar shows the mean value of % inhibition, n = 3 and * are considered significant (p ˂ 0.05) while remaining has no differences (KW-Test Sig).
The heat-induced egg albumin protein denaturation bioassay is a widely utilized, validated, sensitive, and efficient in-vitro technique for investigating anti-inflammatory activity87. The rationale for employing this assay lies in the fact that the albumin denaturation leads to the generation of antigens, initiating a hypersensitive reaction, specifically type III, which triggers inflammation. Consequently, the inhibition of albumin denaturation by an agent signifies its anti-inflammatory properties, with a higher value of inhibition indicating greater anti-inflammatory potential88. HRBC membrane stabilization and egg albumin denaturation assays assess different mediators for inhibition of inflammation, while COX inhibition (which was assessed computationally) targets a specific enzyme-mediated pathway. Combining these approaches provides a more complete picture of the compounds’ anti-inflammatory potential, covering multiple aspects of inflammation. The results from both anti-inflammatory assays demonstrate that compound PG (mono-THTT derivative) and T24 (tris-THTT derivative) exhibit promising anti-inflammatory properties, highlighting their potential as therapeutic agents.
Conclusion
The current study revealed that THTT derivatives are pharmacologically active substances. Among both groups of compounds, tris-THTTs displayed significant leishmanicidal potential. Whereas, based on their high anti-leishmanial, low toxicity, PG among the mono derivatives and T9 among the tris derivatives were found to be more active. However, PG (mono-THTT) and T24 (tris-THTT) displayed profound anti-inflammatory activity. The Thiadiazine thione derivatives exhibit a multifaceted mechanism of action, further characterized by the in-silico inhibition of key Leishmania specific enzymes, including Try-R, Try-S, SMT, and GP63, as well as the anti-inflammatory enzymes COX1 and COX2, thereby suggesting a potential therapeutic strategy that concurrently targets both the parasitic infection and associated inflammatory responses.
Study limitations
This study provides valuable insights into the molecular interactions of compounds with different enzymes but it has several limitations. Notably, the lack of testing of the inhibitory effect on COX1 and COX2 enzymes in an in-vitro system. Additionally, the high molecular weight of some compounds may raise concerns about their bioavailability, which could impact their efficacy in-vivo. Future studies should prioritize in-vivo validation and investigate strategies to enhance the bioavailability of these compounds. Furthermore, mechanistic studies and the efficacy of THTT derivatives in in-vitro and in-vivo systems are in the pipeline.
Data availability
No data available to be deposited in online repository whereas the existing data is already presented in the form of regular tables and figures in this manuscript.
References
Ismail, N. E., Jimam, N. S., Goh, K. W., Tan, C. S. & Ming, L. C. Economic burdens of uncomplicated malaria in primary health care (PHC) facilities of Plateau State, Nigeria: patients’ perspectives. Int. J. Environ. Res. Public Health 20, 1093 (2023).
Li, Y., Geng, J., Liu, Y., Yu, S. & Zhao, G. Thiadiazole—a promising structure in medicinal chemistry. ChemMedChem 8, 27–41 (2013).
Plăcintă, G. et al. Cutaneous leishmaniasis. Mold. Med. J. 61, 38–42 (2018).
Sarwar, S. et al. Targeting cutaneous leishmaniasis with thiadiazine thione derivatives: An in vivo study of its anti-inflammatory, anti-pyretic, anti-nociceptive, and anti-sedative properties. Biomedicines 13, 93 (2025).
Kong, A.S.-Y. et al. Anti-and pro-oxidant properties of essential oils against antimicrobial resistance. Antioxidants 11, 1819 (2022).
Alvar, J. et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 7, e35671 (2012).
Wamai, R. G., Kahn, J., McGloin, J. & Ziaggi, G. Visceral leishmaniasis: A global overview. J. Global Health Sci. 2, e3 (2020).
Bailey, F. et al. A new perspective on cutaneous leishmaniasis—implications for global prevalence and burden of disease estimates. PLoS Negl. Trop. Dis. 11, e0005739 (2017).
Goto, H. & Lindoso, J. A. L. Cutaneous and mucocutaneous leishmaniasis. Infect. Dis. Clin. 26, 293–307 (2012).
Murray, H. W., Berman, J. D., Davies, C. R. & Saravia, N. G. Advances in leishmaniasis. The Lancet 366, 1561–1577 (2005).
Tiuman, T. S., Santos, A. O., Ueda-Nakamura, T., Dias Filho, B. P. & Nakamura, C. V. Recent advances in leishmaniasis treatment. Int. J. Infect. Dis. 15, e525–e532 (2011).
Rodrigues, J. C. F., Godinho, J. L. P. & de Souza, W. Biology of human pathogenic trypanosomatids: Epidemiology, lifecycle and ultrastructure. Proteins Proteom. Leishmania Trypanos. 1–42 (2013).
Sundar, S., Singh, A., Rai, M. & Chakravarty, J. Single-dose indigenous liposomal amphotericin B in the treatment of Indian visceral leishmaniasis: A phase 2 study. Am. J. Trop. Med. Hyg. 92, 513 (2015).
Dorlo, T. P., Balasegaram, M., Beijnen, J. H. & de Vries, P. J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 67, 2576–2597 (2012).
de Menezes, J. P. B., Guedes, C. E. S., Petersen, A. L. D. O. A., Fraga, D. B. M. & Veras, P. S. T. Advances in development of new treatment for leishmaniasis. BioMed Res. Int. 2015, 815023 (2015).
Hussain, H., Al-Harrasi, A., Al-Rawahi, A., Green, I. R. & Gibbons, S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 114, 10369–10428 (2014).
Trouiller, P. et al. Drug development for neglected diseases: A deficient market and a public-health policy failure. The Lancet 359, 2188–2194 (2002).
Lainson, R. et al. The dermal leishmaniases of Brazil, with special reference to the eco-epidemiology of the disease in Amazonia. Mem. Inst. Oswaldo Cruz 89, 435–443 (1994).
Coelho, A. C., Trinconi, C. T., Costa, C. H. & Uliana, S. R. In vitro and in vivo miltefosine susceptibility of a Leishmania amazonensis isolate from a patient with diffuse cutaneous leishmaniasis. PLoS Negl. Trop. Dis. 8, e2999 (2014).
Maspi, N., Abdoli, A. & Ghaffarifar, F. Pro-and anti-inflammatory cytokines in cutaneous leishmaniasis: A review. Pathog. Glob. Health 110, 247–260 (2016).
Yesmin, S. et al. Membrane stabilization as a mechanism of the anti-inflammatory activity of ethanolic root extract of Choi (Piper chaba). Clin. Phytosci. 6, 1–10 (2020).
Waqas, M. et al. Multi-fold computational analysis to discover novel putative inhibitors of isethionate sulfite-lyase (isla) from bilophila wadsworthia: Combating colorectal cancer and inflammatory bowel diseases. Cancers 15, 901 (2023).
Halim, S. A. et al. Discovering novel inhibitors of P2Y12 receptor using structure-based virtual screening, molecular dynamics simulation and MMPBSA approaches. Comput. Biol. Med. 147, 105743 (2022).
Khan, A. A., Iadarola, M., Yang, H.-Y.T. & Dionne, R. A. Expression of COX-1 and COX-2 in a clinical model of acute inflammation. J. Pain 8, 349–354 (2007).
Peitsch, M. C., Schwede, T. & Guex, N. Automated protein modelling-the proteome in 3D. Pharmacogenomics 1, 257–266 (2000).
Khan, H. et al. Identification of novel antileishmanial chemotypes by high-throughput virtual and in vitro screening. Acta Parasitol. 69, 1439–1457 (2024).
Ghosh, M. et al. Leishmania donovani infection enhances lateral mobility of macrophage membrane protein which is reversed by liposomal cholesterol. PLoS Negl. Trop. Dis. 8, e3367 (2014).
Yao, C. & Wilson, M. E. Dynamics of sterol synthesis during development of Leishmania spp. parasites to their virulent form. Parasites Vectors 9, 1–12 (2016).
Vakili, B. et al. Immunoinformatics-aided design of a potential multi-epitope peptide vaccine against Leishmania infantum. Int. J. Biol. Macromol. 120, 1127–1139 (2018).
El Shehry, M., Abu-Hashem, A. & El-Telbani, E. Synthesis of 3-((2, 4-dichlorophenoxy) methyl)-1, 2, 4-triazolo (thiadiazoles and thiadiazines) as anti-inflammatory and molluscicidal agents. Eur. J. Med. Chem. 45, 1906–1911 (2010).
Kang, L. et al. Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors. Mol. Divers. 22, 893–906 (2018).
Avuloğlu-Yılmaz, E., Yüzbaşıoğlu, D., Özçelik, A. B., Ersan, S. & Ünal, F. Evaluation of genotoxic effects of 3-methyl-5-(4-carboxycyclohexylmethyl)-tetrahydro-2H-1, 3, 5-thiadiazine-2-thione on human peripheral lymphocytes. Pharm. Biol. 55, 1228–1233 (2017).
Bermello, J. C., Piñeiro, R. P., Fidalgo, L. M., Cabrera, H. R. & Navarro, M. S. Thiadiazine derivatives as antiprotozoal new drugs. Open Med. Chem. J. 5, 51 (2011).
Muhammed, M. T. & Aki-Yalcin, E. Molecular docking: principles, advances, and its applications in drug discovery. Lett. Drug Des. Discov. 21, 480–495 (2024).
Liu, Y. et al. Recent advances in computer-aided virtual screening and docking optimization for aptamer. Curr. Top. Med. Chem. 23, 1985–2000 (2023).
Wadood, A. et al. Structure-based development of new and potent inhibitors of PIM kinases: A computational study. J. Chem. Soc. Pak. 39, 132 (2017).
Riaz, M. et al. Predicting multi-interfacial binding mechanisms of NLRP3 and ASC pyrin domains in inflammasome activation. ACS Chem. Neurosci. 12, 603–612 (2021).
Baiocco, P., Colotti, G., Franceschini, S. & Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 52, 2603–2612 (2009).
Fyfe, P. K., Oza, S. L., Fairlamb, A. H. & Hunter, W. N. Leishmania trypanothione synthetase-amidase structure reveals a basis for regulation of conflicting synthetic and hydrolytic activities. J. Biol. Chem. 283, 17672–17680 (2008).
Schlagenhauf, E., Etges, R. & Metcalf, P. The crystal structure of the Leishmania major surface proteinase leishmanolysin (gp63). Structure 6, 1035–1046 (1998).
Khan, H. et al. Investigating the role of Sterol C24-Methyl transferase mutation on drug resistance in leishmaniasis and identifying potential inhibitors. J. Biomol. Struct. Dyn. 1–14 (2023).
Miciaccia, M. et al. Three-dimensional structure of human cyclooxygenase (h COX)-1. Sci. Rep. 11, 4312 (2021).
Orlando, B. J. & Malkowski, M. G. Substrate-selective inhibition of cyclooxygeanse-2 by fenamic acid derivatives is dependent on peroxide tone. J. Biol. Chem. 291, 15069–15081 (2016).
Ranjith, D. & Ravikumar, C. SwissADME predictions of pharmacokinetics and drug-likeness properties of small molecules present in Ipomoea mauritiana Jacq. J. Pharmacogn. Phytochem. 8, 2063–2073 (2019).
Li, R., Luo, P., Guo, Y., He, Y. & Wang, C. Clinical features, treatment, and prognosis of SGLT2 inhibitors induced acute pancreatitis. Expert Opin. Drug Saf. 1–5 (2024).
Ahmad, B. et al. Comprehensive investigations on anti-leishmanial potentials of Euphorbia wallichii root extract and its effects on membrane permeability and apoptosis. Comp. Immunol. Microbiol. Infect. Dis. 64, 138–145 (2019).
Hayat, O. et al. A broad spectrum antiparasitic activity of organotin (IV) derivatives and its untargeted proteomic profiling using Leishmania donovani. Pathogens 11, 1424 (2022).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63 (1983).
Cheng, Y. et al. The investigation of Nfκb inhibitors to block cell proliferation in OSCC cells lines. Curr. Med. Chem. (2024).
Nadhman, A. et al. Visible-light-responsive ZnCuO nanoparticles: Benign photodynamic killers of infectious protozoans. Int. J. Nanomed. 6891–6903 (2015).
Sardar, A. et al. Spinigerin induces apoptotic like cell death in a caspase independent manner in Leishmania donovani. Exp. Parasitol. 135, 715–725 (2013).
Mehwish, S. et al. Bioflavonoid-induced apoptosis and DNA damage in amastigotes and promastigotes of Leishmania donovani: Deciphering the mode of action. Molecules 26, 5843 (2021).
Gandhidasan, R., Thamaraichelvan, A. & Baburaj, S. Anti inflammatory action of Lannea coromandelica by HRBC membrane stabilization. Fitoterapia 62, 81–83 (1991).
Shinde, U. et al. Membrane stabilizing activity—a possible mechanism of action for the anti-inflammatory activity of Cedrus deodara wood oil. Fitoterapia 70, 251–257 (1999).
Heendeniya, S., Ratnasooriya, W. & Pathirana, R. In vitro investigation of anti-inflammatory activity and evaluation of phytochemical profile of Syzygium caryophyllatum. J. Pharmacogn. Phytochem. 7, 1759–1763 (2018).
El-Shorbagi, A.-N. et al. Bis-(5-substituted-2-thiono-1, 3, 5-thiadiazinan-3-yl) butane as a scaffold of anti-proliferative activity, blended by a multicomponent process. Med. Chem. Res. 27, 1103–1110 (2018).
El-Shorbagi, A. N. New Tetrahydro-2H-1, 3, 5-thiadiazine-2-thione derivatives as potential antimicrobial agents. Archiv der Pharm. Int. J. Pharm. Med. Chem. 333, 281–286 (2000).
Semreen, M. H., El-Shorbagi, A.-N., Al-Tel, T. H. & Alsalahat, I. M. Targeting γ-aminobutyric acid (GABA) carriers to the brain: Potential relevance as antiepileptic pro-drugs. Med. Chem. 6, 144–149 (2010).
Vicentini, C. B., Forlani, G., Manfrini, M., Romagnoli, C. & Mares, D. Development of new fungicides against Magnaporthe grisea: Synthesis and biological activity of pyrazolo [3, 4-d][1, 3] thiazine, pyrazolo [1, 5-c][1, 3, 5] thiadiazine, and pyrazolo [3, 4-d] pyrimidine derivatives. J. Agric. Food Chem. 50, 4839–4845 (2002).
Coro, J. et al. Alkyl-linked bis-THTT derivatives as potent in vitro trypanocidal agents. Bioorg. Med. Chem. Lett. 16, 1312–1315 (2006).
Katiyar, D. et al. Synthesis and antimycobacterial activity of 3, 5-disubstituted thiadiazine thiones. Bioorg. Med. Chem. 11, 4369–4375 (2003).
Coro, J. et al. Synthesis and antiprotozoan evaluation of new alkyl-linked bis (2-thioxo-[1, 3, 5] thiadiazinan-3-yl) carboxylic acids. Bioorg. Med. Chem. 13, 3413–3421 (2005).
Ji, X. et al. Preparation of 1, 3, 5-thiadiazine-2-thione derivatives of chitosan and their potential antioxidant activity in vitro. Bioorg. Med. Chem. Lett. 17, 4275–4279 (2007).
Vicentini, C. B. et al. Pyrazole derivatives as photosynthetic electron transport inhibitors: New leads and structure− activity relationship. J. Agric. Food Chem. 53, 3848–3855 (2005).
Ettmayer, P., Amidon, G. L., Clement, B. & Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 47, 2393–2404 (2004).
Ali, H. et al. Synthesis, characterization, anticancer, anti-inflammatory activities, and docking studies of 3, 5-disubstituted thiadiazine-2-thiones. Green Process. Synth. 12, 20228136 (2023).
Mehwish, S. et al. Natural compounds from plants controlling leishmanial growth via DNA damage and inhibiting trypanothione reductase and trypanothione synthetase: An in vitro and in silico approach. 3 Biotech. 9, 1–14 (2019).
Ramu, D. & Singh, S. Potential molecular targets of Leishmania pathways in developing novel antileishmanials. Fut. Microbiol. 17, 41–57 (2022).
Schirmann, J. G. et al. In-vitro biological evaluation of 3, 3′, 5, 5′-tetramethoxy-biphenyl-4, 4′-diol and molecular docking studies on trypanothione reductase and Gp63 from Leishmania amazonensis demonstrated anti-leishmania potential. Sci. Rep. 13, 6928 (2023).
Khan, H. et al. Investigating the role of Sterol C24-Methyl transferase mutation on drug resistance in leishmaniasis and identifying potential inhibitors. J. Biomol. Struct. Dyn. 42, 10374–10387 (2024).
Arshad, N. et al. Highly potent anti-inflammatory, analgesic and antioxidant activities of 3, 5-disubstituted tetrahydro-2H-1, 3, 5-thiadiazine thiones. Bioorg. Med. Chem. Lett. 79, 129068 (2023).
Sever, B. et al. Indomethacin based new triazolothiadiazine derivatives: Synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur. J. Med. Chem. 113, 179–186 (2016).
Eswaramoorthy, R., Hailekiros, H., Kedir, F. & Endale, M. In silico molecular docking, DFT analysis and ADMET studies of carbazole alkaloid and coumarins from roots of Clausena anisata: A potent inhibitor for quorum sensing. Adv. Appl. Bioinf. Chem. 13–24 (2021).
Roskoski, R. Jr. Rule of five violations among the FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 191, 106774 (2023).
Lohit, N. et al. Description and in silico ADME studies of US-FDA approved drugs or drugs under clinical trial which violate the Lipinski’s rule of 5. Lett. Drug Des. Discov. 21, 1334–1358 (2024).
Benet, L. Z., Hosey, C. M., Ursu, O. & Oprea, T. I. BDDCS, the rule of 5 and drugability. Adv. Drug Deliv. Rev. 101, 89–98 (2016).
Shtaiwi, A. Thiadiazine-thiones as inhibitors of leishmania pteridine reductase (PTR1) target: Investigations and in silico approach. J. Biomol. Struct. Dyn. 42, 8588–8597 (2024).
Rodríguez, H., Suárez, M. & Albericio, F. Thiadiazines, N, N-heterocycles of biological relevance. Molecules 17, 7612–7628 (2012).
Arshad, N. et al. New series of 3, 5-disubstituted tetrahydro-2H-1, 3, 5-thiadiazine thione (THTT) derivatives: Synthesis and potent antileishmanial activity. Bioorg. Med. Chem. Lett. 28, 3251–3254 (2018).
Yan, J. et al. Design, synthesis and antimicrobial activities of novel 1, 3, 5-thiadiazine-2-thione derivatives containing a 1, 3, 4-thiadiazole group. PeerJ 7, e7581 (2019).
Tamanna, et al. Thiadiazine thione derivatives as anti-leishmanial agents: Synthesis, biological evaluation, structure activity relationship, ADMET, molecular docking and molecular dynamics simulation studies. J. Biomol. Struct. Dyn. 42, 7758–7772 (2024).
Jácome, O. & Poleth, M. Chemical Characterization of Six Thiadiazine Compounds with Antimalarial Activity and their Cross-Reactivity in the Humoral Response. Universidad de Investigación de Tecnología Experimental Yachay, (2021).
Arshad, N. et al. Antiproliferative activity of 3, 5-disubstituted tetrahydro-2H-1, 3, 5-thiadiazine thione (THTT) derivatives and evaluation as potential prodrug. Pak. J. Pharm. Sci. 34, 773 (2021).
Cherdtrakulkiat, R. et al. Derivatives (halogen, nitro and amino) of 8-hydroxyquinoline with highly potent antimicrobial and antioxidant activities. Biochem. Biophys. Rep. 6, 135–141 (2016).
Bukhari, I. A., Alhumayyd, M., Mahesar, A. & Gilani, A. The analgesic and anticonvulsant effects of piperine in mice. J. Physiol. Pharmacol. 64, 789 (2013).
Chaitanya, R., Sandhya, S., David, B., Vinod, K. & Murali, S. HRBC membrane stabilizing property of roor, stem and leaf of glochidion velutinum. Int. J. Res. Pharm. Biomed. Sci. 2, 256–259 (2011).
Chandra, S., Chatterjee, P., Dey, P. & Bhattacharya, S. Evaluation of in vitro anti-inflammatory activity of coffee against the denaturation of protein. Asian Pac. J. Trop. Biomed. 2, S178–S180 (2012).
Paridhavi, M. & Agrawal, S. Safety evaluation of a polyherbal formulation, Zuroor-E–Qula. (2007).
Acknowledgements
The authors would like to extend their sincere appreciation to the Ongoing Research Funding Program, (ORF-2025-350), King Saud University, Riyadh, Saudi Arabia.
Funding
Ongoing Research Funding Program, (ORF-2025-350), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Sarah Sarwar, Nazif Ullah and Rehana Masood contributed equally to this work; Huma khan has done the computational study; Haleema Ali and Rasool Khan has synthesized and provided the Test compounds; Data analysis, Said Hassan; Funding acquisition, Ajaz Ahmad; language improvement by Syed Sikandar Shah; Reviewing of manuscript, Naveed khan and Nadeem Ullah; Editing, Ho Soomin; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
This study was reviewed and approved by the research ethics committee of the Department of Biotechnology, Abdul Wali Khan University Mardan, under the protocol number (AWKUM/ BIOTECH/2022/2930).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sarwar, S., Masood, R., Khan, H. et al. Establishment of THTT derivatives as potential antileishmanial and anti-inflammatory agents through in vitro and in silico investigations. Sci Rep 15, 28246 (2025). https://doi.org/10.1038/s41598-025-12084-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12084-6
