
Abstract
Ovarian cancer (OC) is a highly fatal gynecological malignancy primarily attributable to late-stage detection and restricted treatment options. Aberrant glycolysis, exemplified by the Warburg effect, facilitates tumor development, immunological evasion, and alteration of the microenvironment. Identifying glycolysis-related biomarkers could provide novel insights into prognosis and potential therapeutic targets for OC.The transcriptomic and clinical information of OC patients were obtained from The Cancer Genome Atlas (TCGA), Genotype-Tissue Expression (GTEx), and Gene Expression Omnibus (GEO) databases. Differentially expressed glycolysis-related genes (GRGs) were identified and analyzed for their prognostic significance. Consensus clustering was employed to identify glycolysis subtypes, followed by pathway enrichment and immune infiltration analyses. A ten-gene GRG signature was developed with LASSO-Cox regression and verified in various cohorts. Single-cell RNA sequence and drug susceptibility analysis were performed to explore tumor microenvironment heterogeneity and potential therapeutic agents.A total of 457 differentially expressed GRGs were discovered, of which 30 were substantially linked with OC prognosis. Three molecular subtypes were characterized, with cluster C exhibiting the worst prognosis and activation of tumor-associated pathways. A prognostic model comprising ten genes (LMCD1, L1CAM, MYCN, GALT, IDO1, RPL18, XBP1, LPAR3, RUNX3, PLCG1) was developed and validated, demonstrating robust predictive efficacy across various cohorts. Immune analysis revealed substantial disparities in immune infiltration among risk groups, whereas single-cell analysis identified several critical genes essential for metabolism, proliferation, and interactions within the tumor microenvironment.This work highlights the prognostic and therapeutic significance of GRGs in OC. The ten-gene GRG signature serves as a reliable framework for risk assessment and the formulation of individualized treatment regimens. Nonetheless, further experimental validation and extensive clinical research are necessary to enable the application of these findings in clinical practice. These results highlight the potential of targeting glycolytic pathways as a promising approach to improve the management and treatment outcomes of OC.
Similar content being viewed by others


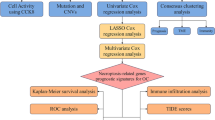
Introduction
Ovarian cancer (OC) denotes malignant tumors that originate in the ovary, with 90–95% classified as primary ovarian cancers and the remaining 5–10% resulting from metastatic lesions from other sites1. It demonstrates considerable histological heterogeneity, with the primary subtypes being epithelial tumors, germ cell tumors, and sex cord-stromal tumors, with epithelial tumors accounting for nearly 90% of cases2. As the third most prevalent gynecological malignancy, following cervical and endometrial cancers, OC remains difficult to diagnose due to the lack of early symptoms, often progressing to advanced stages before detection3,4. Consequently, 60–70% of patients present with late-stage disease, making OC the second leading cause of gynecological cancer-related mortality after cervical cancer5. Annually, approximately 240,000 women are diagnosed worldwide, with a five-year survival rate below 45%6. However, the biological mechanisms underlying the development of OC and efficient prognostic evaluation approaches remain ambiguous.
Previous studies have demonstrated that proliferating cancer cells increase glucose uptake and produce lactate even under aerobic conditions7. This atypical metabolic phenotype, known as aerobic glycolysis or the Warburg effect, is acknowledged as a characteristic of cancer, promoting tumor invasiveness and therapeutic resistance8. In recent years, increasing attention has been given to the role of glycolysis in OC progression. This metabolic reprogramming enhanced fatty acid synthesis, angiogenesis, and metastasis in OC, ultimately contributing to poor clinical outcomes9,10. Genes such as ESM1, PRMT5, and ZEB1 have been implicated in regulating glycolysis in OC9,10,11. However, the specific genes regulating the Warburg effect that are closely associated with prognosis in OC remain unclear. Therefore, it is imperative to examine the pivotal genetic modifications and therapeutic targets of aerobic glycolysis, together with the importance of metabolic coupling between cancer cells and the tumor microenvironment, to devise innovative treatments for this highly fatal illness.
Precision oncology, which tailors treatment based on a patient’s genomic profile and disease trajectory, is rapidly becoming a cornerstone of cancer management12. With the widespread adoption of next-generation sequencing technologies, an increasing number of molecularly defined cancer subtypes have been identified. However, these subtypes correspond to rare patient populations, posing challenges for the recruitment and execution of traditional large-scale clinical trials13,14. Computational oncology primarily encompasses the use of computational models to investigate tumor biology and cancer therapeutics, including the pharmacokinetic and pharmacodynamic relationships of anticancer agents15. By constructing gene co-expression networks and applying systems biology approaches, critical gene modules and prognostic biomarkers that conventional methods might overlook can be revealed16,17. Therefore, applying computational oncology tools to systematically and data-drivenly elucidate glycolytic reprogramming will advance the development of precision oncology.
This study aims to comprehensively explore the relationship between glycolysis-related genes (GRGs) and the development and progression of OC to identify critical therapeutic targets. By integrating data from The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) and employing machine learning techniques, we identified key GRGs associated with prognosis and conducted drug sensitivity screening. Additionally, we established a predictive model utilizing these genes, indicating significant potential for clinical application. This research can provide valuable insights into the Warburg effect and lays a foundation for advancing precision medicine in OC management.
Results
Characterizing GRGs in OC progression dynamics
A total of 457 GRGs were identified as significantly differentially expressed when comparing the transcriptomes of ovarian cancer samples from the TCGA-OC dataset with those of normal ovarian tissues from the GTEx database (Table S1). These differentially expressed genes (DEGs) were subsequently analyzed within the TCGA-OC-GSE26193 cohort to evaluate their association with patient survival outcomes. Univariate Cox regression analysis identified 30 GRGs that were significantly correlated with survival prognosis (Fig. 1 A). Among these, 18 genes were positively associated with poor prognosis in ovarian cancer patients, while 12 genes showed negative associations. Furthermore, a network diagram of the GRGs was constructed to elucidate their interactions and prognostic relevance, demonstrating that the majority of these genes exhibited positive regulatory effects, with a minority functioning as favorable factors (Fig. 1B). Considering the frequent chromosomal segment gains and losses observed in ovarian cancer, copy number variation (CNV) data were extracted from the TCGA database for further analysis. Significant copy number gains were observed in genes such as MECOM, KLF2, PDIA4, UCP2, GFPT2, L1CAM, DYRK1B, HMGB3, and IDO1, while genes such as HSPB7, RUNX3, and RPL18 exhibited notable copy number losses (Fig. 1 C). A circular plot of copy number alterations revealed that gains were predominantly located on chromosomes 6, 7, 8, 9, 11, and 12, whereas losses were primarily observed on chromosomes 1, 4, and 20 (Fig. 1D). Overall, this part suggested a potential correlation between the variations in GRGs and the prognosis of OC.

Differentially expressed GRGs in OC and their correlation with prognosis. (A) A forest plot illustrating the most significant 30 GRGs (P < 0.05) identified through univariate Cox regression analysis in TCGA-OC-GSE26193 cohort. (B) A network diagram depicting the interrelationships among the GRGs. (C) Analysis of CNVs in the 30 GRGs within the TCGA-OC-GSE26193 cohort. (D) Examination of chromosomal locations and alterations in the GRGs.
Molecular and immune landscape characterization of OC subtypes and their prognostic implications
To further delineate the heterogeneity among OC subtypes, the TCGA-OC-GSE26193 cohort was classified into three molecular subtypes based on the expression profiles of 30 prognosis-related GRGs. This classification achieved optimal separation when k = 3, with minimal overlap between the subtypes (Fig. 2 A). Survival analysis revealed significant differences among the subtypes, with GRG cluster C associated with the poorest prognosis, while cluster A exhibited the most favorable survival outcomes (Fig. 2B). The distinct spatial distributions of these subtypes were visualized using principal component analysis (PCA) and Uniform Manifold Approximation and Projection (UMAP), which highlighted clear separation among the clusters (Fig. 2C-D). Analysis of GRG expression patterns across the three subtypes indicated that GFPT2, SUN2, LMCD1, TPM1, HSPB7, CBX7, L1CAM, ADH1B, FHL2, KLF2 and NR4A1 were predominantly expressed in GRG cluster C, aligning with its association with the worst prognosis. In contrast, genes such as IDO1, UCP2 and XBP1 were more highly expressed in GRG cluster A, corresponding to a more favorable prognosis (Fig. 2E).
To further characterize the biological features of the three subtypes, immune infiltration analysis revealed that subtype B exhibited the lowest level of immune cell infiltration, while no significant differences were observed between subtypes A and C in terms of immune cell composition (Fig. 3 A). The heatmap analysis showed no notable differences in age distribution; however, it revealed elevated expression of genes such as ADH1B, GFPT2, HSPB7, TPM1, FHL2, LMCD1, KLF2 and NR4A1 in subtype C, while CYBA, UCP2, IDO1 and TREM2 were highly expressed in subtype A (Fig. 3B). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis identified several pathways significantly enriched in subtype C, which is associated with a poorer prognosis. These pathways included the focal adhesion, TGF-β signaling pathway, WNT signaling pathway, ECM-receptor interaction, glycosaminoglycan biosynthesis, calcium signaling pathway, GnRH signaling pathway, MAPK signaling pathway, pathways in cancer, and gap junctions (Fig. 3 C, Fig. S1). These pathways are frequently linked to distant metastasis and adverse cancer outcomes. Additionally, gene set enrichment analysis (GSEA) highlighted the enrichment of the calcium signaling pathway and other key pathways in subtype C (Fig. 3D). In summary, the findings in this section demonstrated the distinct biological characteristics of the three subtypes classified based on GRGs, particularly the tumor-associated signaling pathways activated in the low-prognosis C subtype.

GRGs-based subgroup categorization in OC. (A) Utilizing consensus clustering, a consensus matrix was established for k=3. (B) The analysis of overall survival revealed significant differences between A, B and C subgroups (P < 0.001). (C, D) Employing PCA and UMAP techniques, these three subtypes were discerned based on GRG expression patterns. (E) Analysis of GRGs expression across these three different subtype clusters.
A prognostic model was constructed for OC based on GRGs
Using the 30 differentially expressed GRGs previously identified, we conducted a Lasso-penalized Cox regression analysis. The lambda (λ) value that minimized cross-validation error was found to be 15. Through this approach, we selected ten genes—LMCD1, L1CAM, MYCN, GALT, IDO1, RPL18, XBP1, LPAR3, RUNX3 and PLCG1—as key contributors to the construction of the prognostic model (Fig. 4 A). Based on the corresponding weight coefficients for each gene, we calculated an individual “GRG score” using a formula in Table S2. Based on the expression of these ten genes, individuals were stratified into high-risk and low-risk groups (Table S3). To enhance the model’s validation, we randomly partitioned the TCGA-OC-GSE26193 cohort into training and testing subsets. Survival analysis curves demonstrated that the prognosis of the high-risk group was significantly worse than that of the low-risk group in both the training and testing cohorts (Fig. 4B-D). The receiver operating characteristic (ROC) curves illustrated the model’s performance in predicting 1-year, 3-year, and 5-year survival rates, with the area under the curve (AUC) for the training set exceeding 0.685, and the AUC for the testing set above 0.583, further confirming the excellent predictive ability of our model (Fig. 4E-G). The risk score for subtype C of OC was significantly elevated compared to subtypes A and B (Fig. 4H). A Sankey diagram was employed to illustrate the connections among GRG clustering, risk categories, and survival status (Fig. 4I). This part effectively developed a predictive diagnostic model utilizing GRGs, which showed a robust capacity to forecast the prognosis of OC patients.

GSEA and immune infiltration in distinct subtype clusters. (A) Examination of immune infiltration characteristics within these three subtype clusters. (B) A comprehensive heat map displayed the expression profiles of GRGs alongside the clinicopathological attributes of these three subtypes. (C) KEGG enrichment analysis of subtype A relative to subtype C. (D) Significant pathways enriched in the three molecular subtypes.
External validation of the model and nomogram characterization
To further validate the prognostic model, two independent datasets, GSE53963 (N = 174) and GSE140082 (N = 380), were utilized to classify samples into high-risk and low-risk groups based on their GRG scores (Fig. 5A-B). Cox survival analysis indicated that the low-risk group, as determined by the GRG score, showed markedly enhanced long-term survival relative to the high-risk group, with P-values of 0.014 for GSE53963 and 0.023 for GSE140082. The univariate Cox regression analysis further validated the predictive significance of the 10 modeling genes in the TCGA-OC-GSE26193 cohort (Fig. 5 C). A heatmap visualization illustrated the differential expression of these genes between the high-risk and low-risk groups (Fig. 5D). A nomogram was created to predict patient prognosis by integrating the GRG score and clinical data (Fig. 5E). The nomogram’s predicted accuracy was evaluated by a calibration plot, indicating that the model’s predictions sustained reliability throughout time (Fig. 5 F). The cumulative risk curve demonstrated notable survival disparities between the high- and low-risk groups (Fig. 5G). A pharmacological susceptibility analysis was conducted to investigate potential treatment implications, revealing 48 medicines with varying sensitivities across the high- and low-risk groups (Fig. 5H, Fig. S2).
To further validate the accuracy of our model, the expression and prognostic relevance of the modeled genes were assessed across different OC cohorts using the BEST platform. The expression patterns of GALT, L1CAM and LMCD1 were consistent with our initial findings, whereas XBP1 expression in the GSE26712 cohort showed an inverse trend. Additionally, no differential expression was observed for MYCN, PLCG1, RPL18 and RUNX3 in the GSE26712 cohort (Fig. 6 A). Further analysis confirmed the independent prognostic significance of these ten GRGs across various OC cohorts, with results closely aligning with our previous findings (Fig. 6B). GSEA of Hallmark pathways highlighted the significant involvement of these genes in glycolytic processes, which are known to promote tumor initiation and progression (Fig. 6 C and D).

Development of a GRG-based prognostic signature. (A) Identification of prognostic GRGs using LASSO regression with 10-fold cross-validation. (B-D) Kaplan-Meier survival curves showed the prognosis of high-risk and low-risk groups, with the training set on the left, the testing set in the middle, and all samples on the right. (E-G) Time-dependent ROC analysis for OS at 1, 3, and 5 years, with the training set on the left, the testing set in the middle, and all samples on the right. (H) Risk score evaluation across three established clusters. (I) Sankey diagram depicting transitions between subtypes and survival status.
Association analysis of GRGs revealed differences in immune activity between the high-risk and low-risk groups
The tumor microenvironment (TME) is crucial in tumor initiation and development, necessitating the examination of immune microenvironment disparities between high- and low-risk populations for prospective treatment approaches. The comparative analysis of immune cell types across samples involved normalizing each sample’s immune cell components to a total of 1. The variation in immune cell proportions between the low-risk and high-risk groups is shown in Fig. 7A. Subsequent examination of immune cell correlations yielded an enhanced understanding of the immunological microenvironment in ovarian cancer (Fig. 7B). The high-risk group had a greater number of resting dendritic cells and M0 macrophages relative to the low-risk group, while activated dendritic cells, M1 macrophages, activated CD4 + memory T cells, and follicular helper T cells were markedly diminished (Fig. 7 C). The expression patterns of the ten genes in the GRG scoring model differed between high- and low-risk groups, exhibiting a robust connection with immune cell infiltration. L1CAM, MYCN, IDO1, RPL18, LPAR3, RUNX3 and PLCG1 exhibited a strong correlation with the patterns of immune cell infiltration observed (Fig. 7D). Spearman correlation analysis indicated that the proportions of resting and activated dendritic cells, activated CD4 + memory T cells, and follicular helper T cells significantly altered with increasing risk scores (Fig. 7E). Notably, the proportion of M1 macrophages decreased significantly with increasing risk score (R = −0.29, P = 1.7 × 10⁻⁶) (Fig. 7 F). To evaluate the total TME, stromal and immunological scores for both high-risk and low-risk groups were computed, revealing that the high-risk group exhibited elevated stromal scores and diminished immune scores relative to the low-risk group (Fig. 7G).
To further investigate the role of the ten GRGs in TME at the single-cell level, their expression across various stromal cell types was analyzed using the OV_GSE154600 dataset from the TISCH database. A total of 26 cell clusters were identified, comprising 11 distinct cell types (Fig. 8 A). Notably, RPL18 was significantly overexpressed in all 11 cell types, while L1CAM showed higher expression specifically in malignant cells (Fig. 8B, S3). Hallmark analysis revealed that malignant cells, epithelial cells, and fibroblasts exhibited elevated levels of glycolysis (Fig. 8 C). To explore the relationship between gene expression and functional scores, we utilized the OC_GSE150864 dataset in the LnCeCell 2.0 platform. Further analysis of the relationship between gene expression and functional scores revealed a significant correlation between the expression of L1CAM, MYCN, PLCG1 and XBP1 with both proliferation and metastasis functional scores. In contrast, the expression of RPL18 and RUNX3 showed a negative correlation with these functional scores (Fig. 8D and E). Overall, the results in this section demonstrated a close relationship between specific GRGs and immune cell infiltration in different risk groups of OC.

Validation of external datasets and independent prognostic role of GRG. (A, B) The ten-gene prognostic model also achieved significant results in the two datasets GSE53963 and GSE140082. (C) Independent prognostic role of LMCD1, L1CAM, MYCN, GALT, IDO1, RPL18, XBP1, LPAR3, RUNX3 and PLCG1 in cohort TCGA-OC-GSE26193. (D) A heatmap presenting the expression patterns of ten central GRGs. (E) Construction of a nomogram integrating GRG-score with various clinicopathological parameters. (F) A calibration curve is employed to verify the accuracy of the nomogram. (G) A cumulative hazard curve depicting survival probabilities over time for patients. (H) Drug sensitivity analysis of high-risk and low-risk groups showed that the high-risk group was more sensitive to dasatinib and foretinib. The vertical axis is IC50, and the smaller the value, the more sensitive it is.
Validation of model gene expression by qRT-PCR
To experimentally validate the expression patterns of the ten model genes identified from our bioinformatic analysis, we performed quantitative real-time PCR (qRT-PCR) in normal human ovarian epithelial cells (IOSE-80) and ovarian cancer cells (OVCAR-3). As shown in Fig. 9, the expression levels of L1CAM, LMCD1, PLCG1 and RUNX3 were significantly upregulated in cancer cells, while GALT, MYCN and XBP1 were markedly downregulated. No statistical difference was found in the expression of IDO1, LPAR3, and RPL18. These findings are basically consistent with the differential expression trends predicted by our computational analysis, supporting the relevance of the identified genes in ovarian cancer progression.

Validation of the reliability of GRGs in different cohorts. (A) Comparison of GRGs expression in both normal and pathological tissues. (B) Prognostic implications of GRGs for patients categorized into high and low-risk groups in different cohorts. (C) GSEA-Hallmark analysis was performed on these ten GRGs. (D) GSEA was obviously enriched in the glycolysis pathway, indicating that these ten GRGs were closely related to glycolysis.
Discussion
This study thoroughly examined GRGs in OC, identifying 457 differently expressed GRGs, with 30 showing a significant correlation with patient prognosis. Molecular and immunological profiling identified three different OC subtypes, with GRG cluster C exhibiting the worse prognosis and activation of tumor-associated signaling pathways. A prognostic model utilizing ten essential GRGs was developed and validated, exhibiting robust prediction accuracy across various cohorts. External validation further substantiated the model’s strong applicability, including the discovery of GRGs associated with glycolysis and tumor development. Furthermore, the study of the immunological milieu revealed notable disparities in immune cell infiltration between high- and low-risk groups, with particular GRGs closely associated with immune activity. These findings enhance our understanding of GRG-related mechanisms in OC and provide a basis for developing tailored therapeutic options.
We identified 30 GRGs significantly associated with survival prognosis. Among these, GFPT2, SUN2, TPM1, L1CAM, DYRK1B, KLF2, NR4A1, and TREM2 exhibited notable copy number gains, which were positively correlated with OC risk. GFPT2 and KLF2 demonstrated the most direct roles in glycolysis, influencing glycolytic activity through the regulation of metabolic pathways and signal transduction18,19. Other genes likely impacted glycolysis indirectly by modulating cellular structures and gene expression19,20. Additionally, GFPT2, TPM1 and L1CAM were closely linked to OC chemoresistance21,22,23. Tpm1.8/9 isoforms have been reported to be specifically expressed in ascites-derived OC cells, where they promote invasion through epithelial-mesenchymal transition (EMT), Wnt signaling, and inflammatory pathways23. Similarly, L1CAM has been shown to enhance stemness-related traits in OC cells, indicating its potential as a druggable target22. Furthermore, KLF2 acts as a downstream effector of epigenetic regulation in OC24while targeting TREM2 has been shown to improve the efficacy of cancer immunotherapy25. The present study further supports these findings, underscoring the potential application of GRGs in OC prognosis and as targets for therapeutic intervention.
Among the three molecular subtypes of OC, the C subtype was found to be closely associated with the poorest prognosis. This subtype was significantly associated with the aberrant overexpression of GRGs, including GFPT2, SUN2, and TPM1. These molecular markers may serve as potential therapeutic targets. Tumor cells resistant to phagocytosis have been shown to inhibit macrophage mitochondrial fission by overexpressing GFPT2, thereby negatively impacting treatment outcomes26. Recent research has also highlighted the interaction between SUN2 and lamins, particularly Lamin A, which is closely related to immune evasion in tumors27. Furthermore, SUN2 was involved in the formation of spliceosome complexes and was highly correlated with various tumor proliferation markers20. Although no studies have yet directly elucidated the underlying mechanisms of these GRGs in OC, based on the findings of the present study and the aforementioned related research, these genes are likely to serve as potential therapeutic targets for OC with poor prognosis.
The pivotal role of TME in the onset and advancement of ovarian cancer, along with its influence on resistance to anti-tumor therapies, has garnered heightened scrutiny28,29. The metabolically adverse tumor microenvironment creates obstacles for tumor-infiltrating immune cells, impeding sustained clinical remission after immunotherapy29. The metabolic interaction between cancer cells and adjacent immune cells may influence the magnitude and kind of immunological responses, underscoring the possible role of metabolic crosstalk in immune surveillance and evasion. Acidosis resulting from aerobic glycolysis might hinder the function of anti-tumor immune cells and diminish the phagocytic capabilities of tumor-associated macrophages, whereas nutritional scarcity fosters the emergence of regulatory T cells and M2-like macrophages30. The current work indicates that the minimal immune cell infiltration in Subtype B may signify immune evasion, resulting in a worse prognosis relative to Subtype A, and is marked by reduced expression of IDO1, TREM2, and RUNX3. Nonetheless, Subtype C, exhibiting the worse prognosis, possesses immune infiltration features akin to those of Subtype A, which demonstrates the most favorable prognosis. The variability in immune cell infiltration may not entirely account for the clinical prognosis of malignancies; other mechanisms, including metabolic reprogramming and functional alterations in the immune milieu, may be more significant in explaining prognostic discrepancies.
The study also identified a negative correlation between the infiltration of activated dendritic cells (DCs), CD4 memory-activated T cells, follicular helper T cells, and M1 macrophages, and high tumor risk scores, with significant associations between these favorable factors in the immune microenvironment and IDO1. Although DCs constitute a rare immune cell population in tumors and lymphoid organs, they play a pivotal role in initiating antigen-specific immunity and tolerance. Manipulating DCs holds excellent potential for inducing effective anti-tumor immunity31,32. DCs promote immune responses or tolerance by sampling and presenting antigens to T cells, as well as providing immune regulatory signals through intercellular contact and cytokines33. Tumor-associated macrophages can exert their role in the tumor immune response through metabolic reprogramming, with significant metabolic differences existing between different macrophage subtypes34. In specific contexts, M1 macrophages can promote immune cell recruitment, especially T cells and natural killer cells, by secreting large amounts of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6) and chemokines (e.g., CXCL9, CXCL10), thereby inhibiting tumor growth35,36. This finding is consistent with the results of our study. Furthermore, the role of IDO1 in tumor therapy is primarily reflected in its ability to suppress anti-tumor immune responses and promote tumor immune escape through the metabolism of tryptophan37,38. Therefore, IDO1 holds the potential to become a target for future immunotherapy in OC.
The glycolysis-related model constructed in this study comprises ten genes (LMCD1, L1CAM, MYCN, GALT, IDO1, RPL18, XBP1, LPAR3, RUNX3, and PLCG1), which were validated across ten different OC datasets. Other studies have also attempted to predict the prognosis of ovarian cancer based on various biological processes. For instance, anoikis-related features were used to predict ovarian cancer prognosis and characterize the immune landscape39. Pyroptosis-related genes were identified as correlating with clinical staging of OC40. Molecular subtype identification and prognosis estimation of OC based on cuproptosis yielded promising results41. In comparison, the glycolysis-related nomogram we generated demonstrates favorable predictive performance and stability across multiple datasets relative to earlier models. Moreover, we examined the expression patterns of these ten genes using single‐cell RNA sequence data, which suggested a potential clinical utility. Furthermore, this study also demonstrated that high-risk groups based on GRGs exhibited reduced sensitivity to some conventional anticancer drugs (e.g., Dasatinib and Foretinib), suggesting that GRGs may be involved in tumor cell drug resistance mechanisms to some extent.
Notably, beyond modulating the Warburg effect and glycolysis, model-derived GRGs are implicated in additional oncogenic processes. GSEA analysis revealed EMT as the top-ranked pathway. EMT facilitates invasion, metastasis, and therapy resistance; enhanced glycolysis supports its energy and redox demands42,43,44. Signature genes LMCD1, L1CAM, and IDO1 have been linked to both glycolysis and EMT45,46,47,48,49. Interferon‐related pathways were also enriched in the high‐risk cohort. Interferons orchestrate the tumor immune microenvironment, exhibiting context-dependent antitumor or immunosuppressive effects50,51. Glycolysis can modulate interferon signaling via altered cytokine secretion, antigen presentation, and epigenetic mechanisms52. Thus, GRGs may both drive EMT and reshape immunity, potentially influencing immunotherapy outcomes. To validate these hypotheses, future work should perturb LMCD1, L1CAM or IDO1 in ovarian cancer stem cells and assess EMT markers (E-cadherin, N-cadherin, vimentin) alongside interferon signaling components (PD-L1, MHC-I, cytokine profiles). Complementary in vivo studies would further substantiate these mechanistic links and therapeutic potential.
Notwithstanding the exemplary performance of our model in both the training and validation cohorts, certain limitations persist. The patients were recruited retrospectively, which may obviously introduce some degree of bias. Secondly, the biological roles of certain GRGs in our model have not been investigated in ovarian cancer cells, which is essential for subsequent experimental investigation. Consequently, additional in vitro and in vivo research are required to substantiate the GRGs found in this work, alongside subsequent high-quality, large-sample, and rigorously monitored multi-center randomized controlled trials for additional confirmation.

Exploring the immune landscape in TME across varying risk scores. (A) Analysis of the composition of immune cell infiltration at different risk levels. (B) Examination of interrelations among different immune cells. (C) Comparison of immune cell profiles in high-risk and low-risk groups. (D) Study of the associations between immune cells and these ten pivotal GRGs. (E) Correlation analysis between immune cells and risk scores. (F) Macrophage M1 type and risk score showed a significant negative correlation (G) Comparative analysis of estimate scores based on expression profiles in high-risk versus low-risk groups.
Methods
Data collection and processing
Transcriptomic and clinical data for 429 OC patients were obtained from TCGA (https://www.cancer.gov/ccg/research/genome-sequencing/tcga), along with transcriptomic data from 88 normal ovarian tissues from the GTEx portal (http://www.gtexportal.org/home/) as a control group. Differentially expressed genes were identified by comparing TCGA-OC samples with normal ovarian tissues. Additionally, the GSE26193 dataset, containing transcriptomic and clinical data for 107 OC patients, was retrieved from GEO (https://www.ncbi.nlm.nih.gov/geo/). To reduce batch effects, the TCGA-OC and GSE26193 datasets were combined into a new cohort, designated as TCGA-OC-GSE26193. For further validation, two external datasets, GSE53963 (N = 174) and GSE140082 (N = 380), were downloaded from GEO. Transcriptomic and clinical data were extracted using custom Perl scripts, with log2 transformations applied to the transcriptomic data, and gene expression levels averaged for repeated measures. All analyses were performed using R (version 4.4.1) and Bioconductor (version 3.18) for data preprocessing, statistical analysis, and visualization.

Analysis of GRGs in OC’s TME cells using scRNA-seq database. (A) Cell type identification and quantification in dataset OV_GSE154600. (B) Analysis of the percentages and expression levels of key GRGs. (C) Glycolysis in different cell types. (D, E) The OC_GSE150864 dataset was used to evaluate the correlation between the expression of six genes, including L1CAM, MYCN, RPL18, RUNX3, PLCG1, and XBP1, and the proliferation function score and metastasis function score.
Identification of GRGs
A total of 4,110 GRGs were identified by searching for the keyword “glycolysis” in the GeneCards database (https://www.genecards.org/), as referenced in relevant publications (Table S4). A detailed analysis was then conducted using the ‘limma’ package (version 3.58.1) from Bioconductor (version 3.18) on the TCGA-OC cohort. By comparing these data with normal ovarian tissue samples from the GTEx database. Additionally, these GRGs were subsequently analyzed for their association with survival outcomes in the merged cohort, TCGA-OC-GSE26193. Univariate Cox regression analysis, performed using the ‘Survival’ package (version 3.5-7), was used to evaluate hazard ratios (HRs), 95% confidence intervals (CIs), and p-values for each gene. The intricate interrelationships and prognostic implications of these GRGs were then visualized in a network diagram, illustrating their complex interactions and underscoring their prognostic significance in ovarian cancer.
Advanced clustering and pathway analysis of glycolysis subtypes in OC
Consensus clustering was performed using the ‘ConsensusClusterPlus’ package (version 1.66.0) within the R environment, enabling the identification and visualization of distinct GRG expression patterns in OC samples. To assess the robustness and stability of the identified clusters, advanced dimensionality reduction techniques, including UMAP and PCA, were applied, with the ‘ggplot2’ package used for visualization. A heatmap was subsequently constructed to visualize the clinical characteristics associated with each subtype.
To further explore the biological relevance of these subtypes, functional and pathway enrichment analyses were conducted53,54,55. Pathway enrichment was performed using the ‘GSEABase’ (version 1.64.0) and ‘GSVA’ (version 1.50.0) packages, utilizing ‘c2.cp.kegg.symbol’ and ‘c5.go. symbols’ gene sets derived from the GSEA database. Additionally, single-sample gene set enrichment analysis (ssGSEA) was applied to quantify TME cell scores across individual OC samples. To depict immune infiltration patterns across the different glycolysis subtypes, a box plot was created, highlighting variations in TME cell composition.

Validation of model gene expression by qRT-PCR in normal and ovarian cancer cell lines. The qRT-PCR was performed to assess the mRNA expression levels of the ten prognostic model genes in normal human ovarian epithelial cells (IOSE-80) and ovarian cancer cells (OVCAR-3). Relative expression levels were normalized to ACTB and calculated using the 2−ΔΔCt method. *P < 0.05, **P < 0.01,***P < 0.001, ns: not significant (Student’s t-test).
Developing a prognostic model for glycolysis in OC
The merged TCGA-OC-GSE26193 cohort was randomly partitioned into training and testing sets to ensure balanced representation. The training set was utilized for model development, while the testing set was reserved for internal validation. To further validate the model externally, two additional GSE datasets were incorporated. A multivariate Cox regression analysis, combined with LASSO, was applied to identify the most prognostically significant GRGs. The RiskScore was calculated as the cumulative product of each gene’s risk coefficient and expression level (\(\:{\sum\:}_{i}^{n}Coefi*Expi\)). The glycolysis score was calculated by summing weighted gene expression levels, where the weights were determined by their corresponding coefficients. OC samples were categorized into high-risk and low-risk groups based on a median score of 0.983. Survival analysis was conducted using the ‘survminer’ package (version 0.4.9), and predictive accuracy was assessed via ROC curves from the ‘timeROC’ package (version 0.4), with AUC calculation. A regression nomogram integrating the glycolysis score and clinical factors was constructed using ‘regplot’ (version 1.1), and model calibration was evaluated with ‘rms’ (version 6.7-1). To assess the potential therapeutic implications of the risk model, drug response analysis was performed using the “oncoPredict” R package. This tool utilizes gene expression profiles to predict the half-maximal inhibitory concentration (IC50) values for a panel of common chemotherapeutic and targeted agents based on the Genomics of Drug Sensitivity in Cancer (GDSC) database. Predicted IC50 values were compared between high- and low-risk groups to identify drugs that may be more effective in specific subpopulations. A p-value < 0.05 was considered statistically significant.
Analyzing tumor microenvironment and prognostic indicators
The ESTIMATE algorithm, implemented via the ‘estimate’ package (version 1.0.13), was used to quantify stromal and immune cell fractions, generating “ESTIMATEScore”, “StromalScore”, and “ImmuneScore” for each tumor sample. Immune cell composition was further analyzed using CIBERSORT, a method that accurately characterizes tumor microenvironment composition based on gene expression data. This analysis enabled the identification of correlations between immune infiltration patterns and the expression of survival-related GRGs (e.g., LMCD1, L1CAM, MYCN, GALT, IDO1, RPL18, XBP1, LPAR3, RUNX3, PLCG1). Spearman rank correlation analysis was performed to evaluate the relationship between risk scores and immune cell infiltration. Differences in “ESTIMATEScores”, including “StromalScore” and “ImmuneScore”, between high- and low-glycolysis score groups were visualized using violin plots.
TME analysis via single-cell RNA sequence
The Tumor Immune Single-Cell Hub (TISCH, http://tisch.comp-genomics.org), an online repository for single-cell RNA sequence data on TME, was used to explore TME diversity across multiple datasets and cell types56. Additionally, the updated LnCeCell 2.0 resource (http://bio-bigdata.hrbmu.edu.cn/LnCeCell/), focusing on lncRNA-associated ceRNA networks, was utilized for further analysis of TME interactions, leveraging single-cell and spatial transcriptomics data57. These tools facilitated the examination of correlations between GRGs and functional scores linked to ovarian cancer cell proliferation and metastasis, with detailed analysis conducted through the CeStateTalk feature of the LnCeCell platform.
Validating the accuracy of prognostic indicators using the BEST data platform
The Biomarker Exploration for Solid Tumors (BEST) platform (https://rookieutopia.hiplot.com.cn/) provides a robust tool for multi-cohort, multi-level biomarker analysis, encompassing 27 solid tumor types and a total of 363 high-quality cohorts, with nearly 50,000 samples. Designed to identify biomarkers with consistent performance across independent cohorts, the platform is ideal for validating the reliability of the GRGs identified in this study across various ovarian cancer cohorts58.
Cell culture
The human ovarian surface epithelial cell line IOSE-80 and ovarian cancer cell line OVCAR-3 (SUNNCELL Biotechnology Co., Ltd., Wuhan, China) were used. IOSE-80 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (P/S), whereas OVCAR-3 cells were cultured in RPMI-1640 medium supplemented with 20% FBS, 10 µg/mL insulin, and 1% P/S. All cultures were kept at 37 °C with 5% CO₂ and passaged every 2–3 days upon reaching ~ 80–90% confluence. Briefly, cells were washed with phosphate-buffered saline (PBS), detached using 0.25% trypsin-EDTA, neutralized with complete medium, and centrifuged at 900 rpm for 3–5 min. The resulting pellet was resuspended in fresh medium and reseeded at a 1:2–1:4 split ratio. Experiments utilized cells in the logarithmic growth phase.
mRNA extraction and qRT-PCR
Total RNA was extracted using TransZol™ Up reagent (TransGen Biotech, Beijing, China) according to the manufacturer’s instructions. RNA concentration and purity were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). Subsequently, 1 µg of total RNA was reverse transcribed into cDNA using the TransScript™ All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) (TransGen Biotech, Beijing, China). Quantitative real-time PCR was performed using a Bio-Rad CFX96 Real-Time PCR Detection System with PerfectStart™ Green qPCR SuperMix (TransGen Biotech, Beijing, China). The qPCR reaction was conducted using a standard three-step amplification protocol for 42 cycles. Each reaction was run in triplicate. ACTB was used as the internal control. Relative gene expression levels were calculated using the 2−ΔΔCt method. Primer sequences are listed in Table 1.
Comprehensive statistical analysis
All statistical analyses were conducted using R software (Version 4.4.1). Data were analyzed using appropriate statistical tests, including t-tests and ANOVA. A significance threshold of P < 0.05 was applied, and the false discovery rate (FDR) was controlled with a cutoff value of q < 0.05.
Data availability
The datasets generated and analyzed during the current study are publicly accessible via the GTEx Portal (http://www.gtexportal.org/home/), the GEO database (https://www.ncbi.nlm.nih.gov/geo/), and The Cancer Genome Atlas (TCGA) repository (http://cancergenome.nih.gov/). All raw data, together with detailed data‐processing workflows, can be downloaded from the following link: https://www.jianguoyun.com/p/DTayFkQQvvidDBi8x-QFIAA. The scripts and code used to design this study are available in the GitHub repository: https://github.com/mingwei3516/OC.git.
References
Cho, K. R. & Shih Ie, M. Ovarian cancer. Annu. Rev. Pathol. 4, 287–313. https://doi.org/10.1146/annurev.pathol.4.110807.092246 (2009).
Davidson, B. & Tropé, C. G. Ovarian cancer: Diagnostic, biological and prognostic aspects. Womens Health (Lond)10, 519–533. https://doi.org/10.2217/whe.14.37 (2014).
Kuroki, L. & Guntupalli, S. R. Treatment of epithelial ovarian cancer. BMJ 371, m3773. https://doi.org/10.1136/bmj.m3773 (2020).
Chornokur, G., Amankwah, E. K., Schildkraut, J. M. & Phelan, C. M. Global ovarian cancer health disparities. Gynecol. Oncol.129, 258–264. https://doi.org/10.1016/j.ygyno.2012.12.016 (2013).
Lheureux, S., Braunstein, M. & Oza, A. M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin.69, 280–304. https://doi.org/10.3322/caac.21559 (2019).
Webb, P. M. & Jordan, S. J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 41, 3–14. https://doi.org/10.1016/j.bpobgyn.2016.08.006 (2017).
Warburg, O. On the origin of cancer cells. Science123, 309–314 (1956).
Icard, P. et al. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updat. 38, 1–11. https://doi.org/10.1016/j.drup.2018.03.001 (2018).
Zhang, J. et al. ESM1 enhances fatty acid synthesis and vascular mimicry in ovarian cancer by utilizing the PKM2-dependent Warburg effect within the hypoxic tumor microenvironment. Mol. Cancer23, 94. https://doi.org/10.1186/s12943-024-02009-8 (2024).
Lin, F. et al. GLUT3 transcriptional activation by ZEB1 fuels the Warburg effect and promotes ovarian cancer progression. Biochimica et Biophysica Acta (BBA)1871, 119715. https://doi.org/10.1016/j.bbamcr.2024.119715 (2024).
Xie, F. et al. PRMT5 promotes ovarian cancer growth through enhancing Warburg effect by methylating ENO1. MedComm 4, e245. https://doi.org/10.1002/mco2.245 (2023).
Malone, E. R., Oliva, M., Sabatini, P. J. B., Stockley, T. L. & Siu, L. L. Molecular profiling for precision cancer therapies. Genome Med.12, 8. https://doi.org/10.1186/s13073-019-0703-1 (2020).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature575, 210–216. https://doi.org/10.1038/s41586-019-1689-y (2019).
Dienstmann, R. et al. Evolving landscape of molecular Prescreening strategies for oncology early clinical trials. JCO Precision Oncol. 4 https://doi.org/10.1200/PO.19.00398 (2020).
Barbolosi, D., Ciccolini, J., Lacarelle, B., Barlési, F. & André, N. Computational oncology–mathematical modelling of drug regimens for precision medicine. Nat. Rev. Clin. Oncol.13, 242–254. https://doi.org/10.1038/nrclinonc.2015.204 (2016).
Yu, Y. et al. Multi-omics identification of a polyamine metabolism related signature for hepatocellular carcinoma and revealing tumor microenvironment characteristics. Front. Immunol. 16, 1570378. https://doi.org/10.3389/fimmu.2025.1570378 (2025).
Jeong, S. H. et al. Nomogram for predicting gastric cancer recurrence using biomarker gene expression. Eur. J. Surg. Oncol.46, 195–201. https://doi.org/10.1016/j.ejso.2019.09.143 (2020).
Kim, D. et al. Nutlin-3a induces KRAS mutant/p53 wild type lung cancer specific methuosis-like cell death that is dependent on GFPT2. J. Experimental Clin. Cancer Research: CR. 42, 338. https://doi.org/10.1186/s13046-023-02922-8 (2023).
Li, L. et al. Calenduloside E modulates macrophage polarization via KLF2-regulated glycolysis, contributing to attenuates atherosclerosis. Int. Immunopharmacol.117, 109730. https://doi.org/10.1016/j.intimp.2023.109730 (2023).
Koedoot, E. et al. Splicing factors control triple-negative breast cancer cell mitosis through SUN2 interaction and Sororin intron retention. J. Experimental Clin. Cancer Research: CR. 40, 82. https://doi.org/10.1186/s13046-021-01863-4 (2021).
Leung, D. et al. Platinum-resistance in epithelial ovarian cancer: an interplay of epithelial-mesenchymal transition interlinked with reprogrammed metabolism. J. Translational Med. 20, 556. https://doi.org/10.1186/s12967-022-03776-y (2022).
Giordano, M. et al. L1CAM promotes ovarian cancer stemness and tumor initiation via FGFR1/SRC/STAT3 signaling. J. Exp. Clin. Cancer Res.40, 319. https://doi.org/10.1186/s13046-021-02117-z (2021).
Xu, T. et al. Tropomyosin1 isoforms underlie epithelial to mesenchymal plasticity, metastatic dissemination, and resistance to chemotherapy in high-grade serous ovarian cancer. Cell Death Differ. 31, 360–377. https://doi.org/10.1038/s41418-024-01267-9 (2024).
Bai, Z. et al. Long non-coding RNA SNGH7 is activated by SP1 and exerts oncogenic properties by interacting with EZH2 in ovarian cancer. J. Cell. Mol. Med.24, 7479–7489. https://doi.org/10.1111/jcmm.15373 (2020).
Binnewies, M. et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell. Rep. 37, 109844. https://doi.org/10.1016/j.celrep.2021.109844 (2021).
Li, J. et al. Macrophage mitochondrial fission improves cancer cell phagocytosis induced by therapeutic antibodies and is impaired by glutamine competition. Nat. Cancer3, 453–470. https://doi.org/10.1038/s43018-022-00354-5 (2022).
Li, Y. et al. Tumor cells impair immunological synapse formation via central nervous system-enriched metabolite. Cancer Cell. 42 https://doi.org/10.1016/j.ccell.2024.05.006 (2024).
Hanahan, D. & Coussens, L. M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell.21, 309–322. https://doi.org/10.1016/j.ccr.2012.02.022 (2012).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med.19, 1423–1437. https://doi.org/10.1038/nm.3394 (2013).
Nicolini, A. & Ferrari, P. Involvement of tumor immune microenvironment metabolic reprogramming in colorectal cancer progression, immune escape, and response to immunotherapy. Front. Immunol. 15, 1353787. https://doi.org/10.3389/fimmu.2024.1353787 (2024).
Steinman, R. M. Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 30 https://doi.org/10.1146/annurev-immunol-100311-102839 (2012).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20 https://doi.org/10.1038/s41577-019-0210-z (2020).
Collin, M. & Bigley, V. Human dendritic cell subsets: an update. Immunology 154 https://doi.org/10.1111/imm.12888 (2018).
Li, M. et al. Metabolism, metabolites, and macrophages in cancer. J. Hematol. Oncol. 16 https://doi.org/10.1186/s13045-023-01478-6 (2023).
Xia, Y. et al. Engineering macrophages for Cancer immunotherapy and drug delivery. Adv. Mater. (Deerfield Beach Fla). 32, e2002054. https://doi.org/10.1002/adma.202002054 (2020).
Locati, M., Curtale, G., Mantovani, A. & Diversity Mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 15, 123–147. https://doi.org/10.1146/annurev-pathmechdis-012418-012718 (2020).
Sadik, A. et al. IL4I1 is a metabolic immune checkpoint that activates the AHR and promotes tumor progression. Cell 182 https://doi.org/10.1016/j.cell.2020.07.038 (2020).
Shi, D. et al. USP14 promotes tryptophan metabolism and immune suppression by stabilizing IDO1 in colorectal cancer. Nat. Commun.13, 5644. https://doi.org/10.1038/s41467-022-33285-x (2022).
Yang, J. et al. Anoikis-related signature predicts prognosis and characterizes immune landscape of ovarian cancer. Cancer Cell Int. 24 https://doi.org/10.1186/s12935-023-03170-8 (2024).
Zhang, B. et al. Exploration of pyroptosis-associated prognostic gene signature and lncRNA regulatory network in ovarian cancer. Comput. Biol. Med.164, 107343. https://doi.org/10.1016/j.compbiomed.2023.107343 (2023).
Zhang, J., Lu, M., Xu, H., Ren, F. & Zhu, L. Molecular subtypes based on cuproptosis-related genes and tumor microenvironment infiltration characterization in ovarian cancer. Cancer Cell Int. 22, 328. https://doi.org/10.1186/s12935-022-02756-y (2022).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol.20, 69–84. https://doi.org/10.1038/s41580-018-0080-4 (2018).
Marcucci, F. & Rumio, C. Tumor cell Glycolysis—At the crossroad of Epithelial–Mesenchymal transition and autophagy. Cells 11 https://doi.org/10.3390/cells11061041 (2022).
Zhao, H. et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine‐resistant pancreatic cancer cells. J. Cell. Mol. Med.21, 2055–2067. https://doi.org/10.1111/jcmm.13126 (2017).
Ye, Z. et al. LMCD1 facilitates the induction of pluripotency via cell proliferation, metabolism, and epithelial-mesenchymal transition. Cell. Biol. Int. 46, 1409–1422. https://doi.org/10.1002/cbin.11858 (2022).
Kiefel, H. et al. EMT-associated up-regulation of L1CAM provides insights into L1CAM-mediated integrin signalling and NF-kappaB activation. Carcinogenesis 33, 1919–1929. https://doi.org/10.1093/carcin/bgs220 (2012).
Chien, M. H. et al. Cyclic increase in the ADAMTS1-L1CAM-EGFR axis promotes the EMT and cervical lymph node metastasis of oral squamous cell carcinoma. Cell Death Dis.15, 82. https://doi.org/10.1038/s41419-024-06452-9 (2024).
Kolijn, K. et al. Epithelial–mesenchymal transition in human prostate cancer demonstrates enhanced immune evasion marked by IDO1 expression. Cancer Res.78, 4671–4679. https://doi.org/10.1158/0008-5472.Can-17-3752 (2018).
Liang, H. et al. Tryptophan deficiency induced by indoleamine 2,3-dioxygenase 1 results in glucose transporter 1-dependent promotion of aerobic Glycolysis in pancreatic cancer. MedComm (2020). 5, e555. https://doi.org/10.1002/mco2.555 (2024).
Chang, C. H. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell162, 1229–1241. https://doi.org/10.1016/j.cell.2015.08.016 (2015).
Shen, H. et al. HIF1alpha-regulated Glycolysis promotes activation-induced cell death and IFN-gamma induction in hypoxic T cells. Nat. Commun. 15, 9394. https://doi.org/10.1038/s41467-024-53593-8 (2024).
Peng, M. et al. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science354, 481–484 (2016).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res.53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci.28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Sun, D. et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res.49(D1), D1420–D1430. https://doi.org/10.1093/nar/gkaa1020 (2021).
Guo, Q. et al. Lncecell 2.0: An updated resource for lncRNA-associated ceRNA networks and web tools based on single-cell and spatial transcriptomics sequencing data. Nucleic Acids Res.https://doi.org/10.1093/nar/gkae947 (2024).
Liu, Z. et al. BEST: a web application for comprehensive biomarker exploration on large-scale data in solid tumors. J. Big Data. 10 https://doi.org/10.1186/s40537-023-00844-y (2023).
Funding
This work was supported by CAMS Innovation Fund for Medical Sciences, grant number: 2021-I2M-1-060.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. The conceptualization of the research was jointly undertaken by M.W and Q.Y. The initial draft of the manuscript was prepared collaboratively by M.W, Q.Y, Y.X, S.D and J.W. The analysis and visualization of the data were carried out by M.W and Q.Y. Z.L provided critical revisions to the manuscript and offered support for the study. The final version of the manuscript received a thorough review and endorsement from all contributing authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This is an observational study. The Institute of Blood Transfusion, Chinese Academy of Medical Sciences Ethics Committee has confirmed that no ethical approval is required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Ying, Q., Xing, Y. et al. Metabolic reprogramming and prognostic insights in molecular landscapes driven by glycolysis in ovarian cancer. Sci Rep 15, 26956 (2025). https://doi.org/10.1038/s41598-025-12350-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-12350-7
