
Chronic obstructive pulmonary disease (COPD) is a heterogeneous lung condition characterized by chronic respiratory symptoms due to abnormalities of the airways and/or alveoli that cause airflow obstruction. It is a leading cause of death worldwide. While alterations in airway bacterial community have been linked to exacerbation frequency, the underlying mechanisms remain unclear. We aimed to characterize associations between airway bacterial community structure and exacerbation history in stable COPD patients and to identify candidate microbial markers that could assist in risk assessment for the clinical management of COPD patients. Sixty-two stable COPD patients were enrolled and categorized into two groups based on their exacerbation history: low risk (LR) and high risk (HR) of exacerbation. Sputum samples were collected and analyzed using both bacterial extended culture and 16S rRNA gene sequencing. The combination of these approaches provided complementary insights, enabling a more comprehensive characterization of the bacterial community. To our knowledge, this is the first study to combine these two approaches in this context and to evaluate their relative performance in detecting microbiological markers associated with exacerbation risk. Microbial composition analysis revealed a loss of α-diversity in HR patients based on extended culture data, a finding not corroborated by sequencing. This discrepancy suggests that the observed impoverishment of diversity may primarily affect the viable fraction of the airway microbial community. The HR group also exhibited increased relative abundances of Pseudomonadota and Bacteroidota, alongside a marked decrease in relative abundances of Lactobacillus and Streptococcus. Notably, significant reductions in the proportion of positive samples were observed at the species level for Streptococcus salivarius and Streptococcus mutans. A comparison of the two methods underlined that 16S rRNA gene sequencing identified five additional phyla and 84 genera not detected by culture, notably strict anaerobes. However, extended culture demonstrated robust sensitivity in detecting Enterobacterales and the pathogenic Moraxella and Pseudomonas. This study revealed microbiological features linked to exacerbation history in stable COPD patients, highlighting the need for future functional and longitudinal research to validate these airway bacterial community signatures and develop targeted preventive strategies.
Similar content being viewed by others

Introduction
Sequencing-based approaches have allowed for an exhaustive description of the airway microbiota1,2,3,4. They have revealed a lower bacterial density compared to the gut microbiota5, a high α-diversity6, and a predominance of strict anaerobes, especially Bacteroidota, along with Bacillota, Pseudomonadota, and Actinomycetota7,8,9.
The airway microbiota has been proposed to maintain lung architecture, enhance antibacterial defenses, and modulate immune system functions9,10,11,12. Its importance is particularly evident in chronic respiratory diseases such as chronic obstructive pulmonary disease (COPD), characterized by lung dysbiosis with alterations in the composition and distribution of the microbiota13,14.
Patients with COPD may experience acute exacerbations (AE-COPD), which are critical and pejorative events in the course of the disease15,16. Understanding the mechanisms that lead to exacerbations has become a primary focus, particularly in steady-state patients, intending to improve prevention strategies, which are a critical aspect of COPD management17. AE-COPD are frequently triggered by viral and/or bacterial airway proliferation, including pathogens such as Haemophilus influenzae, Streptococcus pneumoniae, Moraxella catarrhalis, Pseudomonas aeruginosa, and Staphylococcus aureus. These bacterial species are commonly classified as potentially pathogenic microorganisms (PPMs) in the context of COPD18,19. AE-COPD are associated with significant compositional and functional remodeling of the airway bacterial community20, notably an increase abundance of the phylum Pseudomonadota21,22,23. Currently, the primary practical method for predicting exacerbation risk relies on the history of exacerbations in the previous year16. Nevertheless, the airway bacterial community may also offer measurable parameters, such as microbial signatures and diversity metrics which could serve as potential diagnostic, therapeutic, and prognostic biomarkers.
Since the sequencing-based approaches provide valuable insights into the entire microbial community22, conventional culture-based techniques are frequently disregarded due to their perceived limitations. However, they provide distinct advantages, as they represent standard operating procedures for sputum analysis and focus on viable and culturable microorganisms24.
In this study, we employed both extended culture- and 16S rRNA gene sequencing-based methods to describe the airway bacterial community in sputum samples from stable COPD patients with low risk (LR) and high risk (HR) of exacerbation based on exacerbation history in the previous year (Fig. 1).

Overview of the extended bacterial culture and 16S rRNA gene sequencing protocols. COPD: Chronic Obstructive Pulmonary Disease; DNA: DesoxyriboNucleic Acid; FEV1: Forced Expiratory Volume in 1 s; FVC: Forced Vital Capacity; MALDI-TOF: Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight; PCR: Polymerase Chain Reaction; rRNA: ribosomal RiboNucleic Acid. Created with BioRender®.
We aimed to investigate the associations between airway bacterial community composition in stable COPD and exacerbation risk and to identify novel microbiological markers linked to this risk. Additionally, we assessed whether 16S rRNA gene sequencing provides superior predictive value over extended culture methods in this context. A deeper understanding of the relationships between COPD exacerbations and lung bacterial community—regarded as a potentially modifiable factor—will reveal new opportunities for therapeutic strategies in COPD.
Methods
Study population
Patients with COPD were prospectively included in the Recherche et INNOvation en PAthologie Respiratoire Inflammatoire (RINNOPARI) cohort (University Hospital of Reims, France; NCT02924818; registered on October 4, 2016). The study was approved by the regional ethics committee (Comité de Protection des Personnes—Dijon EST I, no. 2016-A00242-49) and all patients provided informed consent. Exclusion criteria were patients with asthma, cystic fibrosis (CF), bronchiectasis, or pulmonary fibrosis. Enrollment occurred during stable state periods, defined as at least four weeks after the last exacerbation25. Baseline data collection encompassed demographic data, smoking history, treatment, respiratory symptoms [modified Medical Research Council dyspnea scale (mMRC), chronic bronchitis, COPD assessment test (CAT score) assessing the global impact of COPD on health status, exacerbation history in the previous year], arterial blood gas analysis, 6-min walking distance, and pulmonary function test results. COPD diagnosis was defined by postbronchodilator FEV1/FVC < 70% and GOLD (Global initiative for chronic Obstructive Lung Disease) grades were defined by the severity of airflow obstruction measured by spirometry (GOLD 1 : FEV1 ≥ 80% ; GOLD 2 : 50% ≤ FEV1 < 80% ; GOLD 3 : 30% ≤ FEV1 < 50% ; GOLD 4 : FEV1 < 30%)15. Emphysema presence and severity were assessed through computed tomography (CT) scan images by two independent investigators (SD, GD) with a final consensus interpretation26,27. Patients were stratified into two groups based on their exacerbation history over the preceding year: Low Risk of exacerbation (LR) characterized by ≤ 1 exacerbation with no exacerbation-related hospitalization and High Risk of exacerbation (HR), defined by ≥ 2 exacerbations or ≥ 1 exacerbation-related hospitalization(s)15.
Extended culture of sputum samples and bacterial identification
For each patient, induced or non-induced sputum was collected at inclusion, and processed by an extended microbiological culture as previously described25. Compared with conventional sputum culture used in laboratory routine, extended culture included additional media (notably selective media), multiple atmospheres (including anaerobic culture), and multiple dilutions to detect low-abundance bacteria. Serial dilutions (1/1,000, 1/10,000, and 1/100,000) of the liquefied sputum, processed with N-acetylcysteine, were cultured on Columbia blood agar, chocolate agar, Schaedler agar, and Pseudomonas-selective cetrimide agar (Thermo Fisher Scientific, USA) at 37 °C for 48 h for aerobic and 5% CO2 cultures, and for five days for anaerobic cultures. All morphologically distinct colonies—without any predefined limit on the number analyzed—were quantified as colony-forming units (CFU) per milliliter (ranging from 10² to 10¹⁰ CFU/mL) and identified using MALDI-TOF mass spectrometry (MALDI Biotyper®, Bruker Daltonics, Bremen, Germany) (Fig. 1). The α-diversity of the viable and culturable respiratory microbiota was assessed using the Shannon, Simpson, and Chao1 indices. α-diversity represents a measure of species diversity within a specific location and is composed of richness and evenness21.
DNA extraction and 16S rRNA sequencing of sputum samples
Sputum samples were stored in cryotubes at − 80 °C, for further processing using 16S rRNA sequencing (Fig. 1). For each sputum sample, 150 µL was sonicated for 5 min, and bacterial DNA was extracted using the QIAamp® DNA Mini Kit (Qiagen). Environmental DNA contamination was monitored by processing a negative control for each extraction series. PCR amplification of the V3-V4 regions of the 16S rRNA bacterial gene was performed with a mix of 5 µL of extracted DNA, 25 µL of KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cape Town, South Africa), 17 µL of water, 1.5 µL of 10 µM 341 F forward primer, and 1.5 µL of 10 µM 785R reverse primer. The PCR protocol included an initial denaturation step at 95 °C for 3 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 59 °C for 30 s, and extension at 72 °C for 30 s, with a final extension at 72 °C for 5 min.
Amplicon libraries were normalized and sequenced on an Illumina MiSeq (Illumina, San Diego, California, USA), generating 300 bp paired-end reads using PE300, 600-cycle kits (Genomer platform, Roscoff, France). Extraction negative controls and a positive control of known microbial composition (ZYMO D6305, ZymoBIOMICS) were processed and sequenced in parallel with each pool of study samples.
Figure S1 provides an overview of the steps involved in data acquisition and quality control analysis. Sequence data were demultiplexed and separated into forward and reverse FASTQ files. The quality of the demultiplexed raw sequence reads was assessed using the FastQC and MultiQC tools. Primers were removed and sequence quality scores consistently higher than 20 were maintained using CutAdapt and BBDuk. DADA2 was used to infer amplicon sequence variants (ASVs) and assign taxonomy28. Sequencing reads were dereplicated, pooled, and ASVs were inferred for each sample using the DADA2 sample inference algorithm and the estimated error model. Denoised sequences were generated by merging forward and reverse reads. Chimeric sequences were identified by reconstructing them from the left and right segments of more abundant sequences and then removed from the ASVs table.
ASVs were first clustered into operational taxonomic units (OTUs) using dbOTU prior to taxonomic assignment. Taxonomy was then assigned using the SILVA version 138 species classifier implementation for DADA2 (assignTaxonomy() and assignSpecies()). Following taxonomic assignment, additional clustering was performed using the TreeCluster algorithm at 98% similarity on a SATé-enabled phylogenetic placement (SEPP) tree. Non-bacterial and spurious ASVs (i.e., those with fewer than 5 reads across all sequenced biological specimens and no-template controls) were removed.
Nucleic acid extraction and sequencing efficiency were assessed by comparing the mock bacterial community extraction and sequencing controls to the manufacturer’s profiles. Sequence data from biological specimens and extraction-negative controls were used to identify potential contaminants by applying the microDecon package.
Following data processing, two samples were excluded due to an insufficient number of reads (< 2000) (Fig. 1).
Statistical analysis
Data are presented as mean values ± standard deviation, median (interquartile range), or numbers and percentages, as appropriate. Comparisons were made using Fisher’s exact test for qualitative variables, and either Student’s t-test or the Mann–Whitney U test for quantitative variables, as appropriate. A p-value (p) < 0.05 was considered statistically significant.
To summarize and visualize the dissimilarities in bacterial communities between groups in a low-dimensional Euclidean space, an unsupervised principal component analysis (PCA) was performed and plotted along the first two principal components which explain most of the variance.
Results
Patients
Sixty-two patients were enrolled in the study between November 2016 and April 2022, with 28 (45.2%) assigned to the LR group and 34 (54.8%) to the HR group. Detailed patient characteristics are provided in Table 1. They were predominantly male (58.1%) with a mean age of 61.5 ± 9.4 years. Most were former smokers (66.1%) and had cardiovascular comorbidities (59.7%). Fifty-one patients (82.2%) received inhaled treatment, including bronchodilators (61.3% long-acting beta-agonists and 17.7% long-acting muscarinic antagonists) and/or inhaled corticosteroids (33.9%). Sixteen patients (25.8%) were on triple inhaled therapy, with no significant differences between groups. Most patients experienced at least one exacerbation in the previous year (66.1%), with a mean of 2.5 exacerbations, and 56.5% had received antibiotics in the past six months. Of note, 2 patients (3.2%) received antibiotics for a reason other than a COPD exacerbation in the past month. COPD was classified as severe or very severe (GOLD 3 or 4) in 58.1% of the patients.
Compared with the LR group, the HR group had a lower proportion of males (41.2% vs. 78.6%, p = 0.003), a younger age (59.4 vs. 64.2, p = 0.02), and, as expected, more frequent symptoms of chronic bronchitis, as well as a higher CAT score. The HR group was also characterized by more impaired lung function, more severe airway obstruction, and more frequent use of antibiotics and oral corticosteroids in the last six months. Notably, no significant differences were observed between groups in terms of COPD maintenance treatment, CT emphysema severity, comorbidities, smoking history and antibiotic use in the month prior to inclusion.
Viable and culturable airway microbiota of COPD patients using an extended culture-based approach
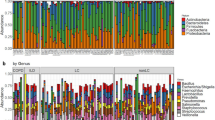
The viable and culturable airway microbiota of the 62 sputum samples (one per patient) was analyzed. A total of 410 bacterial isolates were identified across all samples, representing 71 distinct species, distributed among 34 genera and four phyla (Table S1). The mean number of species per sample was 6.6 with no significant difference between groups (LR group: 6.5 vs. HR group: 6.7; Fig. 2A). The total bacterial counts per sample ranged from 2.1 × 10⁴ CFU/mL to 1.82 × 10¹⁰ CFU/mL, with a median of 3.2 × 10⁷ CFU/mL. Similarly, no difference was observed in overall bacterial load between the LR and the HR groups (median of 3.5 × 10⁷ vs. 3.2 × 10⁷ CFU/mL respectively). The Shannon index was significantly lower in the HR group compared to the LR group (0.9 vs. 1.2 respectively; p = 0.015; Fig. 2B). No significant differences were observed between groups for the Simpson (Fig. 2C) or Chao1 indexes (Fig. 2D). In both groups, the distribution of bacterial phyla was predominantly Bacillota, followed by Actinomycetota, Pseudomonadota, and a much smaller proportion of Bacteroidota29. Notably, the HR group exhibited a significantly higher relative abundance of Pseudomonadota compared to the LR group (26.9% vs. 15.3% respectively; p = 0.005; Fig. 2E). Within this phylum, the Gammaproteobacteria class was also present at a significantly higher relative abundance in the HR group (HR: 15.9% vs. LR: 8.7% of total bacteria; p = 0.036), whereas no significant differences were observed for the Αlphaproteobacteria and Betaproteobacteria classes.
The distribution of bacterial genera was found to be similar between the two groups, with Streptococcus, Rothia, Veillonella, Neisseria, and Actinomyces as the most prevalent, collectively accounting for 72.9% of the identified bacteria (Fig. 2F). Although it was not statistically significant, a lower relative abundance of Streptococcus in the HR group was observed (HR: 32.2% vs. LR: 36.6%; p = 0.35). Strict anaerobes were identified in 74.2% of samples (46/62), accounting for 18.3% of the total isolates, with no significant difference observed between groups (LR: 19.7% vs. HR: 17.2%; Fig. 2G). Focusing on Enterobacterales, 22.6% of samples were positive (14/62), including the genus Citrobacter, Enterobacter, Escherichia, Hafnia, Klebsiella, Morganella, Proteus, and Raoultella (Table S1). The relative abundance of Enterobacterales represented only 4.1% of the total isolates, with no statistically significant difference between the HR and LR groups (HR: 5.3% vs. LR: 2.7%; p = 0.22; Fig. 2G).
At the species level, the most frequently isolated bacteria were Streptococcus oralis/mitis/pneumoniae, followed by Veillonella parvula/dispar/atypica and Streptococcus salivarius, detected in 90.3%, 54.8%, and 48.4% of samples, respectively. Analysis of the species data revealed a significant difference between groups, with a lower prevalence of Streptococcus mutans in the HR sputa (HR: 0% vs. LR: 14.3% of positive samples; p = 0.037; Fig. 2H). Bacterial load of S. mutans was also significantly lower in the HR group (p = 0.037; Table S1). Excluding S. pneumoniae, which could not yet be reliably distinguished from S. mitis and S. oralis using MALDI-TOF at the start of the study30, several PPMs were identified: S. aureus (n = 9 isolates; 14.5% of positive samples), H. influenzae (n = 6; 9.7%), M. catarrhalis (n = 6; 9.7%), and P. aeruginosa (n = 4; 6.5%), collectively representing only 6.1% of the total bacterial isolates (Table S1). The total prevalence of these PPMs did not significantly differ between the LR and HR groups (6.6% vs. 5.7%, respectively).
Differences in airway bacterial community composition between HR vs. LR patients, as determined by culture results, are summarized on the left side (blue) of Figs. 3 and 4.
Finally, a principal component analysis (PCA) was conducted to assess the similarities in viable and culturable airway microbiota between COPD patients. This analysis revealed no significant differences in the overall microbial composition between the two groups, and no distinct clusters or “pulmotypes” could be identified (Fig. 2I).

Extended culture-based approach results. (A) Number of species per sample. (B) Shannon Index. (C) Simpson Index. (D) Chao1 index. (E) Phyla distribution (relative abundance). (F) Genera distribution (relative abundance). (G) Strict anaerobes and Enterobacterales relative abundance. (H) Species prevalence. Species with a prevalence lower than 4% are not listed. – (I) Principal Component Analysis (PCA). The two principal components (PC1 and PC2) explaining 11% and 9% of the variance, respectively, were used for visualization. Statistical significance is indicated as follows: * for p < 0.05, ** for p < 0.01 and NS for Not Significant. Statistical analyses were performed using Student’s t-test, the Mann–Whitney U test, or Fisher’s exact test, as appropriate.
Airway microbiota of COPD patients using 16S rRNA gene sequencing
Among the 62 sputum samples, four did not meet the required volume for microbiota analyses, and following quality filtering, 2 did not pass quality control. Consequently, the airway microbiota was investigated using a 16 S rRNA gene sequencing-based approach on 56 samples (LR: 27 (48.2%) vs. HR: 29 (51.8%); one sample per patient). The rarefaction curves reached a plateau, indicating that the sequencing depth was sufficient to capture most of the bacterial diversity present in the samples (Fig. S2). A total of 1,631,976 high-quality reads were retained, enabling the identification of 3,307 OTUs (364 distinct), distributed across 111 genera and 9 phyla. On average, each sample contained 59 OTUs, ranging from 14 to 119 OTUs. There was no significant difference in the average number of OTUs per sample between the LR and HR groups (mean of 56.3 vs. 61.6 OTUs, respectively; Fig. 3A).
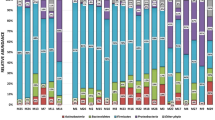
Unlike the culture results, the 16 S rRNA gene sequencing data revealed no differences in the α-diversity between the overall microbiota of the LR and HR groups, as none of the Shannon, Chao1, and Simpson indices exhibited significant variation (Fig. 3B-D). The relative abundance of bacterial phyla was evaluated using the percentage of reads for each sample, providing a quantitative overview of phyla distribution across patients. The most abundant phyla were Bacillota, Pseudomonadota, Bacteroidota, and Actinomycetota, with global mean relative abundances of 54.2%, 16.7%, 14.2%, and 10.2%, respectively. Despite considerable inter-sample variability, this pattern was consistent in both the LR and HR groups (Fig. 3E).
Phylum distribution was further analyzed as a percentage of total OTUs (Fig. 3F), to facilitate comparison with bacterial culture data. Overall, the most abundant phyla were Bacillota (35.6%), Bacteroidota (27.8%), and Pseudomonadota (12.7%), with the same hierarchy observed in both the LR and HR groups. Interestingly, we found a significantly higher relative abundance of Bacteroidota in the HR group compared to the LR group (29.3% vs. 25.9%, respectively; p = 0.032) and a non-significant lower relative abundance of Bacillota in the HR group (LR: 37.1% vs. HR: 34.3%). In contrast to the results observed with the culture method, no significant difference was observed between the two groups in the relative abundance of Pseudomonadota (LR: 12.4% vs. HR: 13.0%; p = 0.60).
We next examined the genus-level taxonomy distribution, focusing on the percentage of OTUs (Fig. 3G). Prevotella (13.8%) emerged as the most dominant genus overall, followed by Leptotrichia (6.0%) and Capnocytophaga (4.5%). This ranking was maintained in the LR group; however, in the HR group, Streptococcus surpassed Capnocytophaga and was the third most abundant genus. We observed significantly lower relative abundance of Streptococcus (LR: 5.3% vs. HR: 3.8%; p = 0.042) and Lactobacillus (LR: 3.3% vs. HR: 1.7%; p = 0.005) in the HR group. Strict anaerobes, encompassing 48 distinct bacterial genera, were identified in all samples (56/56; Fig. 5C). They accounted for more than half of the total OTUs (55.3%), with no significant difference in relative abundance observed between the groups (LR: 54.1% vs. HR: 56.3%; data not shown). Enterobacterales were detected in only 23.2% of the samples (13/56; Fig. 5D), including the genera Citrobacter, Enterobacter, Escherichia, Hafnia, Klebsiella, Morganella, and Proteus. The relative abundance of Enterobacterales was notably low, representing only 0.5% of the total OTUs, with no significant difference between the groups (LR: 0.6% vs. HR: 0.3%; data not shown).
At the species level, the five groups of species with the highest prevalence of positive samples were S. mitis/oralis/pneumoniae/parasanguinis (96.4%), Gemella morbillorum/parahaemo-lysans/sanguinis (92.9%), V. atypica/dispar/parvula/rogosae/tobetsuensis (92.9%), Rothia mucilaginosa (91.1%), and Capnocytophaga gingivalis/granulosa (85.7%) (Fig. 3H). Interestingly, we observed significant differences in the prevalence of positive samples between HR and LR groups for eight species. Six species showed a higher prevalence of positive samples in the HR group: Prevotella oris (LR: 51.9% vs. HR: 79.3%; p = 0.048) (Fig. 3H), Prevotella conceptionensis (LR: 18.5% vs. HR: 44.8%; p = 0.047), Alloprevotella_otu7057 (LR: 25.9% vs. HR: 65.5%; p = 0.004), Eikenella corrodens (LR: 25.9% vs. HR: 62.1%; p = 0.008), Selenomonas artemidis (LR: 11.1% vs. HR: 41.4%; p = 0.015), and Leptotrichia_otu12783 (LR: 11.1% vs. HR: 37.9%; p = 0.030) (data not shown). Two Streptococcus species had a lower prevalence of positive samples in the HR group: S. salivarius (LR: 77.8% vs. HR: 48.3%; p = 0.029) (Fig. 3H) and, consistent with the bacterial culture results, S. mutans (LR: 44.4% vs. HR: 13.8%; p = 0.017) (data not shown). Excluding S. pneumoniae, which could not be distinguished from Streptococcus mitis, S. oralis, and S. parasanguinis in this study, and considering PPMs only at the genus level, Staphylococcus (no positive samples), Haemophilus (78.6% of positive samples), Moraxella (3.6% of positive samples), and Pseudomonas (5.4% of positive samples) collectively accounted for just 1.8% of the total OTUs. No significant difference in the relative abundance of PPMs was detected between the two groups (LR: 1.7% vs. HR: 1.9% of total OTUs) (data not shown).
Differences in airway bacterial community composition between HR vs. LR patients, as determined by 16S rRNA gene sequencing, are summarized on the right side (red) of Fig. 4.
Consistent with bacterial culture results, PCA based on 16S rRNA sequencing data revealed no significant difference in overall microbial composition between the LR and HR groups, and no distinct clusters or “pulmotypes” were identified (Fig. 3I).
To assess microbial co-occurrence patterns, we inferred an interaction network from global 16 S rRNA gene sequencing data. Eight distinct modules were identified, each comprising taxa with significant co-occurrence relationships (Fig. S3-A). These modules varied in connectivity: some, such as M1 and M5, formed densely connected networks indicative of tightly interacting microbial consortia, whereas others, notably M6 and M8, were sparsely connected, potentially reflecting specialized or peripheral taxa. This network structure provides insight into community organization and highlights potential keystone taxa involved in ecosystem-level interactions. As we observed a decreased relative abundance of the Lactobacillus genus and a lower prevalence of S. mutans in high-risk patients, it is noteworthy that S. mutans clustered within the same module (M2) as two Lactobacillus species (Lactobacillus_otu13571 and Lactobacillus salivarius).
Several microbial modules showed significant associations with clinical parameters (Fig. S3-B). Module M4 was positively associated with female gender, while M6 correlated with higher epithelial cell counts in sputum. In contrast, M8 was negatively associated with both epithelial cell and leukocyte counts. Notably, no module showed a significant association with exacerbation frequency or antibiotic use in the month preceding sampling.

16 S rRNA gene sequencing results. (A) Number of OTUs per sample. (B) Shannon Index. (C) Simpson Index. (D) Chao1 index. (E) Phyla relative abundance per patient. (F) Phyla distribution (relative abundance). (G) Genera distribution (relative abundance). (H) Species prevalence. Only the 30 most prevalent bacterial species are represented. – (I) Principal Component Analysis (PCA). The two principal components (PC1 and PC2) explaining 59% and 15% of the variance, respectively, were used for visualization. Statistical significance is indicated as follows: * for p < 0.05, ** for p < 0.01 and NS for Not Significant. Statistical analyses were performed using Student’s t-test, the Mann–Whitney U test, or Fisher’s exact test, as appropriate.

Airway bacterial community differences between HR vs. LR AE-COPD patients detected by extended bacterial culture and 16 S rRNA gene sequencing.
This figure summarizes the main differences in airway bacterial community composition between patients at high risk (HR) vs. low risk (LR) of AE-COPD. Taxa are indicated as increased (↑) or decreased (↓) in the HR group. Results from extended culture-based analysis are presented on the left (blue), and those from 16S rRNA gene sequencing on the right (red). Created with BioRender®.
Comparison of extended bacterial culture and 16S rRNA gene sequencing for analyzing the airway COPD bacterial community
Although we characterized differences in airway bacterial community composition between HR vs. LR COPD patients (Fig. 4), we further explored the effectiveness of bacterial culture compared to 16S rRNA gene sequencing for analyzing the airway bacterial community in COPD. We conducted a comparative analysis of their efficiency in detecting various bacterial genera from 56 sputa.
As anticipated, the 16S rRNA gene sequencing-based approach detected bacteria from five additional phyla and 84 additional genera compared to extended bacterial culture (Fig. 5A and B). Surprisingly, the extended bacterial culture identified seven genera (Staphylococcus, Enterococcus, Lactococcus, Paracoccus, Cutibacterium, Raoultella, and Rhizobium) that were not detected by 16S rRNA gene sequencing.

Venn diagrams comparing bacterial detection by culture and 16S rRNA gene sequencing. (A) Phyla detection. (B) Genera detection. The values represent the number of different phyla and genera detected from the 56 sputa analyzed using both bacterial culture and 16S rRNA gene sequencing. The area of each circle is proportional to the count of phyla/genera. –(C) Strict anaerobes detection (prevalence). (D) Enterobacterales detection (prevalence). (E–H) Detection of bacterial genera associated with COPD exacerbations (prevalence). The values represent the count of positive samples from a total of 56 sputa analyzed using both bacterial culture and 16S rRNA gene sequencing. The area of each circle is proportional to the count of positive samples. –Yellow circles indicate culture data, while green circles represent 16 S rRNA gene sequencing data.
Our analysis revealed significant differences in the effectiveness of the 16S rRNA gene sequencing analysis compared to the extended culture-based approach for detecting major bacterial genera in the 56 sputum samples. Of the 1,727 bacterial detections depicted in Figs. 6 and 84.4% were uniquely identified by 16S rRNA gene sequencing, 2.9% were exclusive to culture, and 12.8% were detected by both methods. It is noteworthy that only Streptococcus and Prevotella were detected in all samples, either by metagenomics alone or by both metagenomics and culture. Among the genera detected in a high proportion of samples (> 70%), Streptococcus, Rothia, Neisseria, and Veillonella achieved concordance of detection rates exceeding 50% between the two methods (100%, 71%, 65%, and 54%, respectively; Fig. 6).
Semi-quantitative comparison based on the percentage of positive samples underscored the prominent contribution of the 16 S rRNA gene sequencing-based analysis for bacterial genera identification (Fig. 7). As expected, these included genera that are not routinely culturable, such as Treponema, Mycoplasma, and Solobacterium, as well as those that are fastidious, such as strict anaerobes, HACEK bacteria (Haemophilus spp. excluding H. influenzae species, Aggregatibacter actinomycetemcomitans, Capnocytophaga spp., Cardiobacterium hominis, Eikenella corrodens, Kingella kingae), and Nutritionally Variant Streptococci (NVS) species (Abiotrophia spp. and Granulicatella spp.). It is worth noting that 16 S rRNA gene sequencing allowed better detection of strict anaerobes. While the culture-based method identified anaerobes in 40 out of 56 samples (71.4%), the 16S rRNA gene sequencing approach detected them in all the 56 samples (Fig. 5C). Genera typically regarded as easy to cultivate, such as Corynebacterium and Haemophilus, also exhibited enhanced detection rates with 16S rRNA gene sequencing. Specifically, Haemophilus was detected in only 17 of 56 samples (30.4%) using culture, compared to 44 samples (78.6%) with sequencing (Fig. 5E).
For several genera the percentage of positive samples was nonetheless equivalent between the two detection approaches (Fig. 7). Excluding genera detected in less than 5% of samples by either method, we found the two detection approaches equivalent for Streptococcus, Neisseria, Micrococcaceae, Pseudomonas, Moraxella, Citrobacter, Escherichia, and the entire Enterobacterales order (Fig. 7). Moraxella and Pseudomonas, two major PPMs, were better detected —although not significant— by culture (6/56 (10.7%) and 5/56 (8.9%), respectively) compared with 16S rRNA gene sequencing (2/56 (3.6%) and 3/56 (5.4%), respectively) (Fig. 5F and H). Additionally, Enterobacterales were detected in 13 samples (23.2%) by both methods, with overlapping detection in 10 samples (17.9%) (Fig. 5D).
Finally, Staphylococcus was the only genus detected significantly more frequently by culture (Fig. 7), being found in 11 samples (19.6%), whereas it was detected in none of the samples by 16 S rRNA sequencing (Fig. 5G).

Qualitative comparison of genus-level bacterial detection by extended culture and 16S rRNA gene sequencing.
Heatmap displaying qualitative detection patterns (prevalence) for the 68 most frequently identified genera across bacterial culture and/or 16S rRNA gene sequencing. Blue boxes represent genera detected exclusively by 16S rRNA gene sequencing, red boxes indicate those identified only by culture, and purple boxes denote genera detected by both methods.

Semi-quantitative comparison of genus-level bacterial detection by extended culture and 16S rRNA gene sequencing.
Scatter plot depicting semi-quantitative detection patterns (prevalence) relative to each detection method for the 68 most frequent bacterial genera identified. Each point represents a bacterial genus, clearly labeled for reference. Points near the diagonal line, within the purple area, indicate genera with similar detection rates between culture and 16S rRNA gene sequencing methods. Points in the green zone indicate genera detected in significantly more samples by 16S rRNA gene sequencing, while points in the red zone indicate genera detected in significantly more samples by extended bacterial culture. Statistical significance (Fisher’s exact test) is indicated as follows: ** for p < 0.01, *** for p < 0.001, and ns for not significant.
Discussion
Our results delineate distinct features of the airway microbiota associated with an increased risk of COPD exacerbation. HR patients exhibited reduced viable bacterial α-diversity and a marked enrichment of Pseudomonadota in culture-based analyses. Sequencing further revealed an increased relative abundance of Bacteroidota, coupled with a depletion of the Streptococcus and Lactobacillus genera, and a lower prevalence of S. salivarius. Notably, S. mutans was the only microbial marker to show a consistent pattern across both methodologies, with a reduced prevalence in HR patients. These results are summarized in Fig. 4.
In this study, we presented a comprehensive characterization of the airway bacterial community in stable COPD patients by simultaneously integrating results from extended culture- and 16S rRNA gene sequencing-based approaches. To our knowledge, this is the first study to combine these methods in this context and to assess their relative capabilities in detecting microbiological markers associated with the risk of COPD exacerbation. While 16S rRNA gene sequencing and metagenomics have emerged as leading methods in microbiota research22 due to their ability to detect bacterial communities not identifiable by conventional culturing methods, their clinical application is often constrained by factors such as cost, time, and complexity31,32. It neither allows taxonomic resolution at the species level for all taxa22,33,34 nor distinguishes between viable and non-viable bacteria, which can limit its diagnostic effectiveness31. This latter point has been underlined by demonstrating that microbiota sputum composition identified by 16S rRNA sequencing did not correlate with viable microorganisms, as revealed by RNA-based metatranscriptomic analysis22,35. By integrating these two approaches, our study aimed to deepen the understanding of the complex airway bacterial community and enhance the identification of readily assessable microbial markers associated with exacerbation history.
The analysis of extended culture data revealed a significant loss in the α-diversity among HR patients. To our knowledge, no previous study has substantially assessed microbial diversity using extended culture-based methods regarding exacerbation risk in stable COPD patients. This decline suggests a less stable and less robust viable and culturable airway microbiota, with relative dysbiosis persisting even under stable conditions for this patient group. Such microbial imbalance could promote the colonization and/or proliferation of PPMs and contribute to an increased risk of exacerbation36. Although HR patients in the RINNOPARI cohort received significantly more antibiotics over the past six months, which may have adversely affected viable bacterial α-diversity37, they were included only if they were stable without exacerbations for at least four weeks prior to sampling. Furthermore, a detailed review of clinical data revealed that only two patients (one per group) had received antibiotics within the month preceding inclusion (for a reason other than a COPD exacerbation), minimizing the likelihood of a substantial impact on overall bacterial diversity. While antibiotic-induced perturbations alone are unlikely to fully explain the observed reduction in α-diversity, this possibility cannot be entirely excluded. Whether this perturbation reflects intrinsic disease pathophysiology or antibiotic effects remains uncertain and warrants further investigation through longitudinal and functional studies to disentangle their respective contributions. Notably, our 16S rRNA sequencing analysis revealed no significant difference in α-diversity between patient groups, suggesting that dysbiosis may primarily affect viable and/or non-fastidious cultivable microbiota. This discrepancy highlights the added value of culture-based approaches in capturing relevant shifts in microbial diversity that may be overlooked by sequencing alone. Of note, most studies using sequencing approaches have reported decreased α-diversity in the sputum microbiota of frequent compared to infrequent exacerbators during stable periods38,39,40,41. However, this finding has not been consistently replicated42. These discrepancies likely reflect differences in study design, such as inclusion and stratification criteria or variations in sampling methodology.
Next, we compared whether the patients in the HR and LR groups could be differentiated using distinct airway bacterial community features during stable periods. PCAs comparing the overall bacterial community composition between the two patient groups revealed strikingly similar profiles, with no distinct clusters or “pulmotypes” identified. These findings align with several recent studies using 16S rRNA sequencing, which reported similar overall sputum microbiota structures in frequent versus infrequent exacerbators23,38,39,40. Collectively, these findings support the hypothesis that the global bacterial community structure in COPD patients undergoes a “homeostatic shift” between exacerbations, reflecting a significant capacity for recovery. Consequently, identifying precise microbiological markers for exacerbation risk may require more detailed analyses at various taxonomic levels.
At the phyla level, both approaches used to analyze the airway bacterial community in COPD showed a phylum distribution predominantly composed of Bacillota, which notably includes the genera Streptococcus, Lactobacillus, Staphylococcus, Veillonella, and Gemella. Our findings align with previous studies on stable COPD patients either conducted using extended culture25,43 or 16S rRNA gene sequencing40,44. With culture-based analysis, we found, firstly, an increased relative abundance of Pseudomonadota in the HR group, including a higher abundance of Gammaproteobacteria. The class Gammaproteobacteria encompasses several major human pathogens, including the genera Pseudomonas and Haemophilus, as well as the order Enterobacterales. This observed elevation in the HR group appeared to result from a global enrichment of various members within this class, as no significant increase was observed in any individual genus or species within the Gammaproteobacteria class. Secondly, the 16 S rRNA gene sequencing approach revealed an increased relative abundance in the phylum Bacteroidota, which encompasses a substantial proportion of anaerobes. It may explain the significant increase detected exclusively through sequencing.
At the genus level, while the culture data indicated only a non-significant trend for Streptococcus, the 16S rRNA gene sequencing identified a statistically significant lower relative abundance in the HR group. These results were in line with a previous study on COPD patients with a high risk of exacerbation40. We also evidenced, through sequencing, a lower relative abundance of the genus Lactobacillus in the HR group. Lactobacillales, an order that includes both Lactobacillus and Streptococcus genera, has previously been associated with a low risk of AE-COPD40. Altogether, these findings suggested a potential protective role for the Streptococcus and Lactobacillus genera in the airway microbiota. It is established that a reduction in commensal microflora increases the risk of subsequent exacerbations and that the sputum of AE-COPD patients are poor in the Streptococcus genus36. Our findings on Lactobacillus spp. may have significant implications for future interventions. Indeed, studies have shown that the administration of probiotics containing Lactobacillus species, such as L. rhamnosus and L. gasseri, may be beneficial in COPD, primarily due to their anti-inflammatory and immunomodulatory effects22,45,46. In addition, a recent multicenter randomized controlled trial reported that long-term oral administration of L. rhamnosus significantly delayed the onset of moderate-to-severe AE-COPD47.
At the species level, 16S rRNA gene sequencing analyses showed that S. mutans and S. salivarius were significantly less prevalent in the HR group compared to the LR group. This finding was confirmed by culture analysis for S. mutans. Such findings were consistent with our observation at the genus level and support the hypothesis of a potential protective effect against dysbiosis and exacerbation. Interestingly, this is sustained by three pathophysiological reports showing that S. salivarius (i) produced bacteriocins that inhibited S. pneumoniae growth and reduced its adhesion to airway epithelial cells, and (ii) lowered the burden of P. aeruginosa in a rat infection model, and (iii) inhibited the growth of M. catarrhalis and S. aureus in vitro48,49,50,51,52. Five anaerobic species more prevalent in the HR group were identified only by the 16S rRNA gene sequencing analysis, including three species of Prevotella/Alloprevotella. Both Prevotella and Alloprevotella belong to the Bacteroidota phylum and are part of the core airway anaerobiome of patients with CF53,54 and COPD44,55,56,57. Despite their prevalence, the role of Prevotella in COPD remains ambiguous, due to conflicting evidence regarding their pathogenic versus protective effects, warranting further research to elucidate their precise role58. For instance, Prevotella melaninogenica has been associated with anti-inflammatory effects in AE-COPD59, whereas Prevotella nigrescens strains have been implicated in tissue-destructive activities via protease production60. The anaerobic species Selenomonas artemidis and Leptotrichia_otu12783 were also more prevalent in the HR group samples. This finding aligns with previous studies reporting an increased relative abundance of Selenomonas and Leptotrichia, along with Pseudomonas, in the sputum of stable COPD patients who experienced frequent exacerbations, and severe COPD patients, respectively42,61. Despite these results regarding specific anaerobes, it should be noted that (i) the overall anaerobes accounted for 18% of the total isolates in extended-culture and more than 55% of the total OTUs in our 16S rRNA gene sequencing analysis, confirming they represent an important group within the airway bacterial community7, and (ii) we found no significant differences in terms of global relative abundance or prevalence of positive samples between HR and LR patients. We also assessed the distribution of Enterobacterales across LR and HR groups, based on findings by Muggeo et al., who identified a COPD patient cluster with sputum enriched in this bacterial order using an extended bacterial culture approach. This cluster was associated with reduced bacterial diversity, predominant cough, and negative impact on mental health25. However, our study did not reveal an association between the HR group and the relative abundance or prevalence of Enterobacterales, regardless of whether the analysis was performed by culture or sequencing. It is noteworthy that Enterobacterales constituted only 4.1% of the culture isolates and 0.45% of the total detected OTUs. Finally, culture analysis identified several PPMs, including S. aureus, H. influenzae, M. catarrhalis, and P. aeruginosa in proportions fairly comparable to those previously observed in stable COPD patients25,62,63,64. PPMs collectively accounted for only 6.1% of the total isolates and showed equal relative abundance or prevalence in both groups.
Interestingly, network analysis based on 16S rRNA sequencing data revealed that S. mutans clustered within the same module as two Lactobacillus species. This co-clustering may reflect a shared ecological pattern or functional association between these taxa. However, this module did not exhibit significant associations with the clinical parameters analyzed.
Our study confirmed the superior sensitivity of the 16S rRNA gene sequencing approach, particularly for detecting non-culturable or fastidious bacteria. It undeniably identified a greater number of potential airway microbiota features associated with exacerbation risk at both the genus and species level compared to the culture-based approach. However, our results highlighted the complementary value of extended bacterial culture. Specifically, culture revealed an increased relative abundance of Pseudomonadota in the HR group—a finding not captured by 16S rRNA sequencing—suggesting a higher abundance of viable and cultivable bacteria within this phylum. In addition, several pathogenic species, particularly respiratory PPMs, as well as members of Enterobacterales, were detected with equal performance regarding the number of positive samples using culture and 16S rRNA sequencing. These results underscore the effectiveness of commercial culture media—Columbia blood, chocolate, Schaedler, and cetrimide agar—in isolating clinically relevant pathogens65. An unexpected outcome of our comparative analysis was that the Staphylococcus genus was detected solely by culture-based methods, with no corresponding signal in 16S rRNA gene sequencing. We hypothesize that this discrepancy may result from a combination of factors, including the low relative abundance of Staphylococcus among cultured isolates (2.93%), its modest bacterial load (median: 5.50 × 10⁵ CFU/ml), and the selective nature of culture-based methods, which are particularly favorable for the growth of PPMs65. Primer and extraction biases appear unlikely, as multiple Staphylococcus OTUs were successfully identified using the same DNA extraction and 16S rRNA sequencing protocol in other concurrent studies of the lung microbiota conducted by our group. Moreover, no specific limitations of 16S rRNA sequencing for this genus have been reported in the literature.
The primary strength and uniqueness of our study lay in the dual methodological approach employed. Furthermore, our findings emphasize the relevance of bacterial culture, which can be extended for exhaustivity as described here, as a standard operational procedure to analyze the airway bacterial community of COPD patients.
This study has several limitations. First, its monocentric, cross-sectional design, based on single time-point sampling, and the relatively modest sample size compared with that of some previous studies may have limited the statistical power to detect subtle associations. The absence of longitudinal follow-up precluded assessment of the temporal stability and variability of microbial markers associated with exacerbation risk. We employed 16S rRNA gene sequencing rather than whole-metagenome sequencing, which introduces several constraints. Most notably, the limited taxonomic resolution of 16S rRNA sequencing, due to the short length of the amplified region, often prevents confident species-level identification33. Primer selection can also bias detection toward specific bacterial taxa, and the compositional nature of sequencing data complicates interpretation of relative abundance changes66. The use of sputum samples, rather than more invasive approaches such as bronchoalveolar lavage (BAL), presents both advantages and limitations. While sputum collection carries a risk of contamination from the upper airway and may not accurately represent the microbiota of the lower respiratory tract as precisely as BAL22, it is non-invasive, widely accessible, and routinely used in clinical microbiology laboratories, thereby enhancing the translational potential of our findings. Although primarily descriptive and taxonomic in nature, this study was designed to serve as a foundation for future investigations that incorporate functional and longitudinal analyses, advancing our understanding of microbial community dynamics and their contribution to COPD pathogenesis.
Conclusion
In conclusion, we analyzed the airway bacterial community of stable COPD patients using a dual approach that combined extended bacterial culture and 16S rRNA gene sequencing.
While culture-based analysis revealed a loss of viable α-diversity in high-risk patients, overall diversity appeared similar across groups when assessed by sequencing. In addition, sequencing revealed a decrease in the relative abundance of the genera Streptococcus and Lactobacillus in high-risk individuals. Such findings may confirm other studies suggesting a potential protective role for commensal bacteria and probiotics for preventing exacerbations. Importantly, reductions in S. mutans prevalence, as well as in α-diversity, were readily detectable through bacterial culture. Future research should incorporate functional and longitudinal studies to further validate the identified bacterial community features and support the development of targeted preventive strategies for COPD exacerbations.
Data availability
The datasets of 16S rRNA sequencing generated and/or analyzed during the current study have been deposited in the European Nucleotide Archive (ENA) under the project reference PRJEB85758. Supplementary figures and tables are provided in Supplementary File 1. Raw data from extended cultures supporting the findings are available in this published article (Supplementary File 2), along with control data (Supplementary File 3) and the completed STORMS checklist (Supplementary File 4). The analysis code is accessible at: https://github.com/lvelosuarez/amplicon.
Abbreviations
- AE:
-
Acute exacerbation
- BMI:
-
Body Mass Index
- CAT:
-
COPD assessment test
- CF:
-
Cystic fibrosis
- COPD:
-
Chronic obstructive pulmonary disease
- CT:
-
Computed tomography
- CFU:
-
Colony-forming units
- DNA:
-
DeoxyriboNucleic acid
- DLCO:
-
Diffusing capacity of the Lung for Carbon Monoxide
- FEV1 :
-
Forced Expiratory Volume in 1 second
- FVC:
-
Forced Vital Capacity
- GOLD:
-
Global Initiative for Chronic Obstructive Lung Disease
- HACEK:
-
Haemophilus spp., Aggregatibacter actinomycetemcomitans, Capnocytophaga spp., Cardiobacterium hominis, Eikenella corrodens, Kingella kingae
- HR:
-
High risk
- LR:
-
Low risk
- MALDI-TOF:
-
Matrix-Assisted Laser Desorption/Ionization - Time-Of-Flight
- mMRC:
-
modified Medical Research Council
- NVS:
-
Nutritionally Variant Streptococci
- OTU:
-
Operational taxonomic unit
- PCA:
-
Principal component analysis
- PCR:
-
Polymerase Chain Reaction
- PPM:
-
Potentially oathogenic microorganisms
- RNA:
-
Ribonucleic Acid
- rRNA:
-
ribosomal RiboNucleic Acid
- RV:
-
Residual volume
- SATé:
-
Simultaneous alignment and tree estimation
- SD:
-
Standard deviation
- SEPP:
-
SATé-Enabled Phylogenetic Placement
- TLC:
-
Total Lung Capacity
References
Erb-Downward, J. R. et al. Analysis of the lung Microbiome in the « healthy » smoker and in COPD. PloS One . 6 (2), e16384 (2011).
Charlson, E. S. et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir Crit. Care Med. 15 (8), 957–963 (2011).
Dickson, R. P. et al. Spatial variation in the healthy human lung Microbiome and the adapted Island model of lung biogeography. Ann. Am. Thorac. Soc. . 12 (6), 821–830 (2015).
Morris, A. et al. Comparison of the Respiratory Microbiome in Healthy Nonsmokers and Smokers. Am. J. Respir. Crit. Care Med. 187(10), 1067–75. (2013).
Sze, M. A. et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 185(10), 1073–80. (2012).
Kim, H. J. et al. The microbiome of the lung and its extracellular vesicles in nonsmokers, healthy smokers and COPD patients. Exp Mol Med. 49(4):e316. (2017).
Lamoureux, C., Guilloux, C. A., Beauruelle, C., Jolivet-Gougeon, A. & Héry-Arnaud, G. Anaerobes in cystic fibrosis patients’ airways. Crit. Rev. Microbiol. . 45 (1), 103–117 (2019).
Dickson, R. P. et al. Bacterial topography of the healthy human lower respiratory tract. mBio . 8 (1). https://doi.org/10.1128/mbio.02287-16 (2017).
Héry-Arnaud, G. & Société Française de, M. [cité 27 août 2024]. Le microbiote pulmonaire, un enjeu récent en microbiologie médicale. Disponible sur: https://www.sfm-microbiologie.org/2020/06/10/9870( 2020).
Karmarkar, D. & Rock, K. L. Microbiota signalling through MyD88 is necessary for a systemic neutrophilic inflammatory response. Immunol.. 140 (4), 483–492 (2013).
Yun, Y. et al. Environmentally determined differences in the murine lung microbiota and their relation to alveolar architecture. PloS One. 9 (12), e113466 (2014).
Brown, R. L., Sequeira, R. P. & Clarke, T. B. The microbiota protects against respiratory infection via GM-CSF signaling. Nat. Commun.. 8 (1), 1512 (2017).
Budden, K. F. et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 7 (10), 907–920 (2019).
Liu, J. et al. Role of pulmonary microorganisms in the development of chronic obstructive pulmonary disease. Crit. Rev. Microbiol. 2 Janv. 47 (1), 1–12 (2021).
Global Initiative for Chronic Obstructive Lung Disease [Internet]. (2024 Gold reports). Disponible sur: https://goldcopd.org/2024-gold-report/(2024).
Anzueto, A. Impact of exacerbations on COPD. Eur. Respir Rev.. 19 (116), 113–118 (2010).
Wedzicha, J. A. et al. Prevention of COPD exacerbations: a European respiratory society/american thoracic society guideline. Eur. Respir J.. 50 (3), 1602265 (2017).
Sapey, E. & Stockley, R. A. COPD exacerbations. 2: aetiology. Thorax Mars. 61 (3), 250–258 (2006).
Ko, F. W. et al. Acute exacerbation of COPD. Respirology 21 (7), 1152–1165 (2016).
Molyneaux, P. L. et al. Outgrowth of the bacterial airway Microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 15 (10), 1224–1231 (2013).
Sin, D. D. Chronic obstructive pulmonary disease and the airway microbiome: what respirologists need to know. Tuberc Respir Dis. . 86 (3), 166–175 (2023).
Yu, S. et al. The association between the respiratory tract Microbiome and clinical outcomes in patients with COPD. Microbiol. Res.. 266, 127244 (2023).
Dicker, A. J. et al. The sputum microbiome, airway inflammation, and mortality in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol.. 147 (1), 158–167 (2021).
Dingle, T. C. & Butler-Wu, S. M. Maldi-tof mass spectrometry for microorganism identification. Clin. Lab. Med.. 33 (3), 589–609 (2013).
Muggeo, A. et al. Extended bacteria Culture-Based clustering identifies a phenotype associating increased cough and enterobacterales in stable chronic obstructive pulmonary disease. Front. Microbiol.. 12, 781797 (2021).
Washko, G. R. et al. Computed Tomographic-Based quantification of emphysema and correlation to pulmonary function and mechanics. COPD J. Chronic Obstr. Pulm Dis. . 5 (3), 177–186 (2008).
Perotin, J. M. et al. Delay of airway epithelial wound repair in COPD is associated with airflow obstruction severity. Respir Res. . 15 (1), 151 (2014).
Callahan, B. J. et al. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods . 13 (7), 581–583 (2016).
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C. & Göker, M. List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70 (11), 5607–5612 (2020).
Farfour, E. et al. Accurate identification of S. pneumoniae using MALDI-TOF mass spectrometry, still a challenge for clinical laboratories? Eur. J. Clin. Microbiol. Infect. Dis. . 39 (1), 209–211 (2020).
Ditz, B. et al. Sputum Microbiome profiling in COPD: beyond singular pathogen detection. Thorax . 75 (4), 338–344 (2020).
Han, M. K. et al. Significance of the Microbiome in obstructive lung disease. Thorax . 67 (5), 456–463 (2012).
Watson, R. L., De Koff, E. M. & Bogaert, D. Characterising the respiratory Microbiome. Eur. Respir J. . 53 (2), 1801711 (2019).
Shima, K., Coopmeiners, J., Graspeuntner, S., Dalhoff, K. & Rupp, J. Impact of micro-environmental changes on respiratory tract infections with intracellular bacteria. FEBS Lett. . 590 (21), 3887–3904 (2016).
Lee, S. W., Kuan, C. S., Wu, L. S. H. & Weng, J. T. Y. Metagenome and metatranscriptome profiling of moderate and severe COPD sputum in Taiwanese Han males. Tunney M, éditeur. PLOS ONE . 11 (7), e0159066 (2016).
Sun, Z., Zhu, Q., Shen, Y., Yan, T. & Zhou, X. Dynamic changes of gut and lung microorganisms during chronic obstructive pulmonary disease exacerbations. Kaohsiung J. Med. Sci. . 36 (2), 107–113 (2020).
Rogers, G. B., Bruce, K. D., Martin, M. L., Burr, L. D. & Serisier, D. J. The effect of long-term macrolide treatment on respiratory microbiota composition in non-cystic fibrosis bronchiectasis: an analysis from the randomised, double-blind, placebo-controlled BLESS trial. Lancet Respir Med.. 2 (12), 988–996 (2014).
Dang, X. et al. Frequent exacerbators of chronic obstructive pulmonary disease have distinguishable sputum Microbiome signatures during clinical stability. Front. Microbiol. . 13, 1037037 (2022).
Li, W. et al. Analysis of sputum microbial metagenome in COPD based on exacerbation frequency and lung function: a case control study. Respir Res. . 23 (1), 321 (2022).
Yang, C. Y. et al. Association of exacerbation phenotype with the sputum microbiome in chronic obstructive pulmonary disease patients during the clinically stable state. J. Transl. Med. 19(1), 121. (2021).
Pragman, A. A. et al. Sputum Microbiome α-diversity is a key feature of the COPD frequent exacerbator phenotype. ERJ Open. Res.. 10 (1), 00595–2023 (2024).
Millares, L. et al. Relationship between the respiratory Microbiome and the severity of airflow limitation, history of exacerbations and Circulating eosinophils in COPD patients. BMC Pulm Med. . 19 (1), 112 (2019).
Perotin, J. M. et al. High blood eosinophil count at stable state is not associated with airway microbiota distinct profile in COPD. Int. J. Chron. Obstruct Pulmon Dis. 19:765–771 (2024).
Tangedal, S. et al. Sputum microbiota and inflammation at stable state and during exacerbations in a cohort of chronic obstructive pulmonary disease (COPD) patients. Singanayagam A, éditeur. PLOS ONE. 17 (9), e0222449 (2019).
Carvalho, J. L. et al. Oral feeding with probiotic Lactobacillus rhamnosus attenuates cigarette smoke-induced COPD in C57Bl/6 mice: relevance to inflammatory markers in human bronchial epithelial cells. Chu HW, éditeur. PLOS ONE. 15 (4), e0225560 (2020).
Salva, S., Villena, J. & Alvarez, S. Immunomodulatory activity of Lactobacillus rhamnosus strains isolated from goat milk: impact on intestinal and respiratory infections. Int. J. Food Microbiol. . 141 (1–2), 82–89 (2010).
Hua, J. et al. feng, tu, ran, Prevention of exacerbation in patients with moderate-to-very severe COPD with the intent to modulate respiratory microbiome: a pilot prospective, multi-center, randomized controlled trial. Front Med. 10, 1265544. (2024).
Drigot, Z. G. & Clark, S. E. Insights into the role of the respiratory tract Microbiome in defense against bacterial pneumonia. Curr. Opin. Microbiol. Févr. 77, 102428 (2024).
Manning, J. et al. Investigation of Streptococcus salivarius-mediated Inhibition of Pneumococcal adherence to pharyngeal epithelial cells. BMC Microbiol. . 16 (1), 225 (2016).
Santagati, M., Scillato, M., Patanè, F., Aiello, C. & Stefani, S. Bacteriocin-producing oral Streptococci and Inhibition of respiratory pathogens. FEMS Immunol. Med. Microbiol. . 65 (1), 23–31 (2012).
Stoner, S. N., Baty, J. J., Novak, L. & Scoffield, J. A. Commensal colonization reduces Pseudomonas aeruginosa burden and subsequent airway damage. Front. Cell. Infect. Microbiol.. 13, 1144157 (2023).
Jörissen, J. et al. Case-Control Microbiome Study of Chronic Otitis Media with Effusion in Children Points at Streptococcus salivarius as a Pathobiont-Inhibiting Species. mSystems 6(2), 10–1128 (2021).
Harris, J. K. et al. Molecular identification of bacteria in Bronchoalveolar lavage fluid from children with cystic fibrosis. Proc. Natl. Acad. Sci.. 104 (51), 20529–20533 (2007).
Thornton, C. S. & Surette, M. G. Potential contributions of anaerobes in cystic fibrosis airways. Kraft CS, éditeur. J. Clin. Microbiol. . 59 (3), e01813–e01819 (2021).
Wang, Z. et al. Sputum Microbiome Temporal variability and dysbiosis in chronic obstructive pulmonary disease exacerbations: an analysis of the COPDMAP study. Thorax . 73 (4), 331–338 (2018).
Leiten, E. O. et al. The airway microbiota and exacerbations of COPD. ERJ Open. Res.. 6 (3), 00168–2020 (2020).
Wang, Z. et al. Airway host-microbiome interactions in chronic obstructive pulmonary disease. Respir Res.. 20 (1), 113 (2019).
Webb, K. A. et al. Genomic diversity and antimicrobial resistance of prevotella species isolated from chronic lung disease airways. Microb. Genomics. 8 (2), 000754 (2022).
Mao, X. et al. Analysis of sputum microbial flora in chronic obstructive pulmonary disease patients with different phenotypes during acute exacerbations. Microb. Pathog . 184, 106335 (2023).
Yanagisawa, M., Kuriyama, T., Williams, D. W., Nakagawa, K. & Karasawa, T. Proteinase activity of prevotella species associated with oral purulent infection. Curr. Microbiol.. 52 (5), 375–378 (2006).
Hazra, D. et al. The altered sputum Microbiome profile in patients with moderate and severe COPD exacerbations, compared to the healthy group in the Indian population. F1000Research 12, 528 (2023).
Leung, J. M. et al. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology. 22 (4), 634–650 (2017).
Einarsson, G. G. et al. Community dynamics and the lower airway microbiota in stable chronic obstructive pulmonary disease, smokers and healthy non-smokers. Thorax. 71 (9), 795–803 (2016).
Garcia-Nuñez, M. et al. Severity-Related changes of bronchial Microbiome in chronic obstructive pulmonary disease. Munson E, éditeur. J. Clin. Microbiol.. 52 (12), 4217–4223 (2014).
Sibley, C. D. et al. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. Planet PJ, éditeur. PLoS ONE . 6 (7), e22702 (2011).
Abellan-Schneyder, I. et al. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere. 6(1):e01202-20 (2021).
Acknowledgements
The authors are deeply grateful to all healthcare professionals who contributed to the RINNOPARI cohort.
Funding
This work was funded by a grant from the University Hospital of Reims and the University of Reims Champagne-Ardenne (Hospital University Project RINNOPARI).
Author information
Authors and Affiliations
Consortia
Contributions
The study was designed by AM, JMP, GD and TG. The microbiological data acquisition was performed by QLT, AC and AM. The patients were included, and their clinical data were acquired by JMP, SD and GD. NGS experiments were designed by GHA and performed by SG. Bioinformatics analyses were conducted by LVS, AB, and QLT. The original draft was written by QLT and AM. Editing of the manuscript was performed by JMP, GD, GHA and TG. All the authors contributed to the final data interpretation and manuscript writing. All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.M. Perotin reports lecture honoraria from AstraZeneca, and support for attending meetings from AstraZeneca and Chiesi, outside the submitted work. G. Deslée reports lecture honoraria from Chiesi, AstraZeneca and GlaxoSmithKline; outside the submitted work. S. Dury reports fees from Boehringer-Ingelheim and Sanofi-Adventis, outside the submitted work. Rest of the authors have no conflict of interest.
Consent for publication
Not applicable.
Ethics approval and informed consent
This research was conducted in accordance with the Declaration of Helsinki, followed the rules applicable to medical research in France, and received the authorization needed. The study was approved by the regional ethics committee (Comité de Protection des Personnes—Dijon EST I, no. 2016-A00242-49). Informed consent was obtained from all the patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lecomte-Thenot, Q., Perotin, JM., Héry-Arnaud, G. et al. Association between exacerbation history and airway bacterial community assessed by extended bacterial culture and sequencing approaches in stable COPD. Sci Rep 15, 32767 (2025). https://doi.org/10.1038/s41598-025-14994-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-14994-x
