
Abstract
Genetically modified (GM) soybean (Glycine max) expressing a human thioredoxin (trx) gene under the control of a seed-specific promoter has been developed for cosmetic applications, but its ecological effects remain poorly understood. We examined the rhizosphere microbiomes of GM soybean, wild soybean (Glycine soja), and F3 interspecific hybrids segregating for the transgene under low-input field conditions. Rhizosphere soil samples were collected at the vegetative and flowering stages, and microbial communities were analyzed via high-throughput sequencing of 16S rRNA and ITS regions. This study represents the first application of amplicon sequence variant (ASV)-level niche breadth analysis to evaluate the rhizosphere effects of a trx-expressing GM soybean. ASV-level analysis and ecological niche breadth classification revealed that genotype-specific microbial shifts were not apparent at relatively high taxonomic levels. Notably, several bacterial ASVs from the class Bacilli were more abundant in GM and homozygous plants at flowering. A fungal ASV from Tausonia also showed increased abundance in the GM lines. These findings highlight that genotype-driven microbial shifts can occur in a stage-specific manner and underscore the importance of fine-resolution microbial analyses in environmental risk assessment.
Similar content being viewed by others


Introduction
Genetically modified (GM) crops are increasingly cultivated worldwide because of their enhanced traits, such as herbicide tolerance, pest resistance, and improved nutritional profiles1. Despite these advantages, concerns remain regarding their potential impacts on surrounding ecosystems, particularly on soil microbial communities, which play vital roles in nutrient cycling, plant health, and overall ecosystem functioning2,3. Among these, the rhizosphere microbiome—microorganisms directly influenced by plant root exudates—is especially responsive to plant genotype and physiological traits4.
Although recent studies suggest that GM traits generally do not induce major shifts in rhizosphere microbial communities at the broader community level (e.g., alpha or beta diversity), subtle or transient changes in the abundance of specific microbial taxa can occur, depending on factors such as plant developmental stage, genotype, and soil conditions5,6,7,8,9. Such fine-scale changes, while not altering overall diversity metrics, may still impact key microbial functions (e.g., nitrogen cycling, disease suppression) or disrupt microbe–plant interactions, thereby influencing plant health or ecosystem services4,5,6,7,9. Capturing and analyzing these shifts is therefore critical for a more ecologically nuanced and functionally relevant assessment of GM crop impacts. Interspecific hybridization has been proposed as a strategy to reintroduce adaptive traits from wild progenitors into modern cultivars8. Such traits include tolerance to abiotic stress and improved microbial symbiosis, which can enhance crop resilience and ecological compatibility10. These complexities underscore the need to investigate how GM traits and interspecific hybridization influence root–microbe interactions, as even subtle shifts may have ecologically significant implications.
Soybean is one of the most widely grown GM crops globally, and its interactions with the soil microbiota have been a key focus of numerous environmental risk assessment studies. Several investigations have employed advanced analytical approaches–including niche partitioning, network analysis, and functional prediction–to evaluate how transgenic traits influence rhizosphere microbial communities11,12,13. While these studies have significantly advanced our understanding of GM crop–microbiome interactions, many rely on broader taxonomic resolution or are conducted under high-input agricultural conditions. In contrast, our study utilized amplicon sequence variant (ASV)-level resolution and ecological niche breadth analysis to examine fine-scale microbial shifts associated with the interspecific hybrid background and transgene zygosity in a GM soybean under low-input field conditions.
The GM soybean used in this study expresses a human thioredoxin (trx) gene under the control of a seed-specific promoter. Thioredoxins are involved in a wide range of plant physiological processes, including redox homeostasis, carbon metabolism, and embryo development14. However, the potential indirect effects of thioredoxin expression on the rhizosphere microbiome–mediated through changes in plant physiology or root traits–remain poorly understood. To our knowledge, this is the first study to investigate how the expression of human thioredoxin in a transgenic crop may affect root-associated microbial communities at the ASV-level. Although the trx gene in the GM soybean is driven by a seed-specific promoter, pleiotropic effects on plant physiology, such as altered root development or metabolic shifts, may indirectly influence rhizosphere microbial communities. Therefore, even seed-specific transgenes warrant evaluation of their potential belowground ecological impacts.
We analyzed the rhizosphere microbiome of GM soybean expressing the trx gene, wild soybean and F3 interspecific hybrid progenies segregating for the transgene (homozygous, hemizygous, and nullizygous lines). Rhizosphere soil samples were collected at both the vegetative and flowering stages, and the bacterial and fungal communities were characterized via high-throughput sequencing of the 16S rRNA and ITS regions, respectively. The objective of this study was to determine whether the introduction of the trx transgene and interspecific hybridization with wild soybean influences the rhizosphere microbial community and, if so, whether these effects are detectable at the ASV-level and vary according to plant developmental stage or transgene zygosity. In addition, because non-GM hybrid controls from the same G. max × G. soja background were not available, it is challenging to fully disentangle the effects of trx expression from those of interspecific hybridization. Thus, the observed microbial responses may represent combined outcomes of both factors. However, by integrating fine-scale genotype resolution and microbial niche breadth analysis, this study offers novel insights into the nuanced and context-dependent effects of GM traits on soil microbial ecology.
Results
Plant growth parameters
To evaluate whether the expression of the trx genes in GM soybean or their segregation in F3 progenies affects plant growth, shoot and root dry weights were measured at both the vegetative (July) and flowering (August) stages (Fig. 1). During the vegetative stage, shoot dry weight did not differ significantly among the GM, wild soybean, and F3 genotypes. However, by the flowering stage, shoot dry weight was significantly greater in homozygous F3 plants than in GM plants (p < 0.05). GM plants presented significantly greater root dry weight than did wild soybean and all F3 progenies during the vegetative stage (p < 0.05). By the flowering stage, the root dry weight of the F3 lines—regardless of zygosity—had increased to levels comparable to those of the GM plants, whereas the root biomass of the wild soybean plants continued to decrease. These findings indicate that both shoot and root biomass are influenced by genotype and developmental stage.

Shoot and root dry weights of genetically modified (GM) soybean, wild soybean, and F3 progenies with different transgene zygosities–homozygous (Homo), hemizygous (Hemi), and nullizygous (Null)–measured at the vegetative and flowering stages (n = 15 per genotype). Each boxplot shows the median (line within the box), the 25th and 75th percentiles (lower and upper box boundaries), and the 10th and the 90th percentiles (whiskers). Dots represent outliers. Different letters above the boxplots indicate statistically significant differences among genotypes (Tukey’s HSD test, p < 0.05).
Microbial community composition and alpha diversity
Amplicon sequencing of the rhizosphere samples yielded 6,809 bacterial and 599 fungal ASVs. On average, each sample contained 1,360 ± 273 bacterial ASVs and 105 ± 26 fungal ASVs. The community structure was visualized via NMDS based on the basis of Bray–Curtis dissimilarity (Fig. 2C). Across genotype groups—including, GM, wild soybean, and F3 segregants—neither bacterial nor fungal communities formed distinct clusters by genotype at either developmental stage. The NMDS stress values were below 0.1, indicating a reliable representation of community dissimilarities. Both bacterial and fungal communities in the rhizosphere were significantly influenced by plant growth stage (PERMANOVA p = 0.001; ANOSIM p = 0.001), whereas plant genotype had no significant effect (PERMANOVA p > 0.3; ANOSIM p > 0.6) (Table 1). No significant interaction between genotype and growth stage was observed.At the class level, the bacterial communities were dominated by Gammaproteobacteria, Alphaproteobacteria, Bacilli, and Actinobacteria (Fig. 2A). The fungal communities were mainly composed of Dothideomycetes, Agaricomycetes, and Mortierellomycetes (Fig. 2B). At the ASV-level, Bacillus (BASV0001) was the most dominant bacterial taxon, detected in all the samples, with a mean relative abundance of 9.2 ± 5.5%. For fungi, FASV0001 (Geastrales) was present in 93% of the samples, with a mean relative abundance of 8.4 ± 12.6%.

Relative abundance of A bacterial and B fungal community compositions and nonmetric multidimensional scaling (NMDS) plots based on Bray–Curtis dissimilarity for C bacterial and fungal communities associated with the rhizosphere of soybean.
Alpha diversity, assessed via the Shannon index, Chao1 richness, and Pielou’s evenness (Fig. 3), revealed no significant differences among the genotypes for either the bacterial or fungal communities at either developmental stage. One wild soybean sample collected at the vegetative stage presented unusually low fungal diversity across all indices, slightly reducing the group mean. Nevertheless, alpha diversity was largely consistent across genotypes and time points.

Bacterial and fungal diversity indices across soybean genotypes. A Shannon diversity index, B Pielou’s evenness index, and C Chao 1 richness index. Different letters above the boxplots indicate statistically significant differences among genotypes (p < 0.05).
Niche breadth and taxonomic composition
Ecological niche breadth analysis classified ASVs into generalists, common taxa, and specialists on the basis of their distribution across samples (Fig. 4A–D). Among the bacterial ASVs, 78% were assigned to one of these categories: 58% were common taxa, 3.3% were generalists, and 17.1% were specialists. For fungal ASVs, 91% were classified, with 69% being common taxa, 13.8% being generalists, and 8.8% being specialists. Compared with bacterial communities, fungal communities contained a greater proportion of generalists. These findings suggest that, in both bacterial and fungal rhizosphere communities, the majority of ASVs are moderately distributed across genotypes and samples, with relatively few taxa exhibiting highly restricted (specialist) or broadly ubiquitous (generalist) distributions.

Niche breadth analysis of bacterial and fungal ASVs. A, B Average relative log-abundances of individual ASVs classified as generalists, common taxa, or specialists; each dot represents one ASV. Specialists are defined as ASVs with niche breadths below the 5th percentile, and generalists are defined as those above the 95th percentile. C, D Proportions of generalists, common taxa, and specialists within the bacterial and fungal communities. E, F Taxonomic composition of generalists, common taxa, and specialists at the order level.
Taxonomic analysis of niche groups revealed that bacterial generalists were dominated by Rhizobiales (mainly Devosia sp. and Rhizobium sp.) and Alicyclobacillales (mainly Tumebacillus sp.), whereas common taxa included Micrococcales and Vicinamibacterales (Fig. 4E). Bacterial specialists were more often affiliated with Enterobacterales and other minor groups. In fungal communities, Alternaria sp. dominated the generalist group, whereas Cladosporium sp. was frequent among common taxa. Fungal specialists primarily consisted of taxonomically minor groups.
To further investigate their ecological roles, heatmaps and hierarchical clustering were generated separately for generalist and specialist ASVs (Fig. 5A–B). The generalist ASVs showed no clear clustering by genotype or developmental stage, indicating a broad distribution. In contrast, specialist ASVs showed distinct clustering by developmental stage, particularly between the vegetative and flowering stages. However, within each stage, the samples did not cluster by genotype.

Heatmap of niche partitioning based on the relative abundance of A generalist ASVs and B specialist ASVs. Relative abundances were arcsine-transformed, and the z scores were normalized prior to clustering.
ASV-level analysis reveals genotype-specific shifts
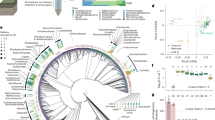
While the microbial community composition varied slightly with developmental stage, no distinct clustering by genotype was observed at the community level (Fig. 2). To uncover finer-scale differences, we conducted differential abundance analysis at the ASV level. ASVs showing > 3-fold differences in relative abundance between GM soybean and wild soybean or between homozygous F3 plants and wild soybean in at least 66% of the samples (n = 30) were selected, resulting in the identification of 25 bacterial and 20 fungal ASVs. Phylogenetic trees were constructed using 16S rRNA or ITS sequences to determine taxonomic affiliations (Fig. 6). Most of these ASVs have been classified as either common taxa or specialists on the basis of niche breadth.

Phylogenetic tree, GM/homozygous-to-wild soybean abundance ratios, and niche/taxonomic classifications of selected ASVs. A Bacterial ASVs and B fungal ASVs were selected on the basis of a relative abundance ratio greater than 3.0 or less than 0.33 when comparing GM or homozygous plants to wild soybean. In the ratio tables, the color highlights the ASVs meeting these selection thresholds. Labels were added to highlight key taxa discussed in the text. Phylogenetic trees were constructed via the neighbor‒joining method on the basis of partial 16S rRNA (bacteria) or ITS (fungi) sequences. Bootstrap values (%) are shown at nodes for values exceeding 50%. One wild soybean sample from the vegetative stage was excluded from the calculation because of abnormally low sequencing depth and diversity.
Among the bacterial ASVs, members of Acinetobacter and Chryseobacterium were enriched in the wild soybean samples at the vegetative stage. In contrast, all nine ASVs assigned to Bacilli were significantly more abundant in GM and homozygous plants at the flowering stage. At the class level, Bacilli exhibited contrasting temporal dynamics: in wild soybean, their relative abundance decreased ~ 4.6-fold from the vegetative stage to the flowering stage, whereas in GM plants, their relative abundance nearly doubled. The homozygous F3 lines presented a moderate (~ 2-fold) decline, whereas the hemizygous and nullizygous lines presented sharper (~ 3-fold) reductions. In fungal communities, ASVs from Pleosporales, Mucorales, and Spizellomycetales vary substantially by genotype. Notably, the FASV0058 (Tausonia) consistently exhibited 3.6- to 9.4-fold greater abundance in GM and homozygous plants than in wild soybean across both stages. These results demonstrate that although community-level differences were minimal, specific microbial taxa responded sensitively to plant genotype, particularly during the flowering stage.
Discussion
Thioredoxins (Trxs) play multifaceted roles throughout plant development, including embryo development, seed reserve mobilization, chloroplast biogenesis, carbon metabolism, and cellular redox regulation14. They also contribute to intercellular signaling, influencing a broad range of physiological processes across the plant life cycle. For example, the overexpression of an h-type thioredoxin gene (SlTrxh) in tobacco was shown to increase root length under nitrate stress, suggesting that Trx proteins may promote root development under specific environmental conditions15. In the present study, although the trx transgene encodes a human thioredoxin and is not root specific, we examined its potential impact on the rhizosphere microbial communities of GM soybean, F3 progenies segregating for the transgene, and wild soybean across two developmental stages. While shoot biomass did not differ significantly among the genotypes at the vegetative stage, the GM plants presented significantly greater root dry weights than did the wild soybean plants and all the F3 progenies (Fig. 1). This increase is likely attributable to inherent growth differences between cultivated and wild soybeans rather than to thioredoxin expression per se. Nonetheless, such variation in root traits—driven by genotype—may influence rhizosphere microbial structure and function.
The potential impact of GM crops on soil microbial communities has been extensively studied as part of environmental risk assessments5,8,16,17. Rhizosphere microorganisms play critical roles in nutrient cycling, plant health, and soil ecosystem services4making it essential to understand how transgenes may influence these interactions. Most studies, including those on Bt and herbicide-tolerant crops, have reported no consistent or significant effects on microbial community composition or diversity at broad taxonomic levels6,18. These findings suggest that the presence of a transgene alone does not inherently disrupt the overall structure of rhizosphere microbial communities. However, context-dependent and transient shifts in specific taxa have been observed, often mediated by environmental factors such as soil type, plant developmental stage, and cultivation practices7,19. For example, Li et al.7 reported that Bt rice altered the abundance of specific bacterial taxa without affecting overall diversity or network stability. Similarly, Ge et al.9 reported bacterial but not fungal diversity changes in GM rice, potentially linked to nutrient cycling indicators. Fazal et al.13 further demonstrated that GM soybean plants treated with glyphosate recruited beneficial microbial taxa associated with stress tolerance and nitrogen cycling. These findings underscore the need for case-by-case assessment, particularly at finer taxonomical scales, to capture subtle and ecologically relevant shifts.
High-resolution ASV-level analysis can reveal genotype-associated microbial differences that are undetectable at coarse taxonomic levels20,21. In our study, although NMDS analyses revealed limited separation among genotypes, ASV-level comparisons revealed distinct differences in microbial abundance patterns. Notably, several ASVs affiliated with the class Bacilli were significantly more abundant in GM and homozygous F3 plants than in wild soybean plants during the flowering stage (Fig. 6A). At the class level, Bacilli showed contrasting temporal trends: a more than fourfold decrease in wild soybean from the vegetative to the flowering stage, versus a twofold increase in GM plants. These patterns suggest that the presence of the trx transgene–or associated physiological changes–may create rhizosphere conditions favorable to Bacilli. Bacillus species are well-known plant growth-promoting rhizobacteria in soybean systems22. While some studies reported greater Bacillus abundance in wild soybeans than in cultivated soybeans under field conditions23 our findings indicate a marked enrichment of Bacilli in GM and homozygous lines during the flowering stage, where they constitute ~ 15% of the bacterial community. This suggested that their ecological role may vary on the basis of genotype and developmental timing. Among the fungal communities, Tausonia pullulans (FASV0058), classified as a common taxon, was consistently more abundant in the GM and homozygous lines across both stages (Fig. 6B). Given its reported capacity to delay mycorrhizal colonization in tomato roots24 this species could influence symbiotic interactions in the soybean rhizosphere. In addition, T. pullulans possesses a rich repertoire of carbohydrate-active enzymes (CAZymes), including glucoamylases and lignocellulose-degrading enzymes25 which may contribute to rhizosphere carbon turnover and nutrient availability, thereby indirectly shaping microbial community dynamics around soybean roots. Incorporating ecological niche characteristics, such as specialist, generalist, and common taxa, offers additional insight into which microbial groups are more likely to respond to genotype-related environmental changes. Such genotype-linked shifts in functional taxa, even if subtle, could influence key rhizosphere processes such as nutrient mineralization, disease suppression, and symbiotic interactions, thereby affecting plant fitness and broader soil ecosystem services.
Kim et al.26 reported that environmental conditions exerted a stronger influence on the nutrient and metabolite profiles of the GM soybean used in this study than did the genotype, which is consistent with our present results and shows that the rhizosphere microbial community structure is more strongly shaped by the plant developmental stage than by the genotype at the community level. Similarly, our previous environmental persistence study demonstrated that GM soybeans could germinate and produce seeds under unmanaged field conditions but did not persist beyond 1.5 years27 suggesting a low risk of long-term establishment in natural ecosystems and limited potential for gene flow to wild soybean populations. Collectively, these findings indicate that although genotype-specific effects–detectable at finer biological resolution–can influence root traits and associated microbial taxa, such effects are likely to be transient and ecologically constrained under field conditions, where environmental factors predominantly shape both plant and microbial phenotypes.
Our study also highlights the value of ASV-level resolution in detecting genotype-associated shifts masked at the community level. Importantly, these effects are strongly dependent on plant developmental stage, reinforcing the need to consider temporal dynamics in microbial assessments. We recommend that future environmental risk assessments of GM crops incorporate high-resolution taxonomic profiling, expanded temporal sampling, and sufficient biological replication to capture subtle but ecologically meaningful changes.
Despite the insights gained, several limitations should be acknowledged. First, soil heterogeneity can introduce significant spatial variation in microbial composition, even within the same field. Although our randomized block design included three replicates per genotype, greater replication would improve the statistical power and ecological generalizability. Second, the rhizosphere is not a uniform zone; microbial assemblages can vary depending on root architecture and proximity to the root surface. Future studies targeting more defined microhabitats (e.g., soil tightly bound to lateral roots) could improve spatial resolution. Third, our analysis focused exclusively on rhizosphere soil, whereas the plant microbiome extends to aboveground compartments such as the phyllosphere. Integrating rhizosphere and phyllosphere data under a plant holobiont framework may yield a more holistic view of genotype–microbiome interactions.
Additionally, the lack of a nontransgenic hybrid control limits our ability to fully disentangle the effects of the trx transgene from those of interspecific hybridization. Nevertheless, by comparing homozygous, hemizygous, and nullizygous F3 lines within the same hybrid background, our study provides a valuable approximation of transgene effects under ecologically relevant conditions, addressing a key question in environmental biosafety research.
In summary, while the overall structure of rhizosphere microbial communities in soybean was not significantly altered by the introduction of the trx transgene or interspecies hybridization, genotype-specific differences in the abundance of particular microbial taxa, especially Bacilli, were detected at fine taxonomic resolution. These effects were most pronounced during the flowering stage and were further clarified by ecological niche classification. However, our results also emphasize that plant developmental stage and environmental conditions exert stronger influences on microbial community structure than does genotype alone. These findings underscore the importance of incorporating ASV-level taxonomic resolution, developmental stage considerations, and spatial replication in future assessments. This research contributes to our understanding of the ecological consequences of interspecific hybridization involving GM crops and provides a theoretical basis for refining biosafety evaluations under field conditions.
Methods
Plant materials
We used GM soybean seeds (event CT-4025, hereafter referred to as “GM”), a wild soybean accession (IT241179, hereafter referred to as “wild soybean”), and F3 interspecific hybrid progenies derived from a cross between wild soybean (♀) and GM soybean (♂). The GM soybean harbors the human trx gene under the control of the ββ-conglycinin promoter (seed-specific expression) and the PinII terminator and contains the bar gene for phosphinothricin selection28. GM seeds were provided by the Research & Development Center of CELLTRION (Incheon, Korea), and the wild soybean seeds were obtained from the National Agrobiodiversity Center, Rural Development Administration, Republic of Korea.
F1 hybrids were generated through natural hybridization in a field experiment at the Korea Research Institute of Bioscience & Biotechnology (KRIBB), Cheongju, Republic of Korea, in 201828. The F2 and F3 generations were produced via self-pollination of the F1 plants.
Zygosity determination of F3 progeny
To evaluate the effects of transgene zygosity on the rhizosphere microbiome, F3 seeds from selfed F2 plants were sterilized with 10% NaOCl for 10 min and germinated in plastic trays filled with commercial potting soil (BioPlus, Hungnong, Korea) in a greenhouse on May 3, 2023. Leaf tissue was collected at the two- or three-leaf stage for genomic DNA extraction (100 mg) via the FastDNA Kit (MP Bio, USA). Transgene zygosity was determined via PCR targeting a 185-bp region of the trx gene via the primers TRX-RBF (5′-GCA AGA TGA TCA AGC CCT TCT T-3′) and TRX-LBR (5′-CTT CTC TTT GTT AGC ACC GGA G-3′)28.
The PCRs (20 µL total volume) contained 2 µL of 10 ng gDNA, 1 µL of each primer (10 pmol), and 16 µL distilled water in AccuPower PCR PreMix tubes (Bioneer, Korea). The thermal cycling conditions were as follows: initial denaturation at 94 °C for 3 min; 35 cycles of 94 °C for 30 s, 57 °C (for trx) or 64 °C (for zygosity) for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min. PCR products were separated on 2% agarose gels and visualized via ethidium bromide staining.
Field cultivation and rhizosphere sampling
After zygosity determination, the seedlings were transplanted into field plots at KRIBB on June 4, 2023. The experiment followed a randomized block design with five genotypes (GM, wild soybean, and F3 lines that were homozygous, hemizygous, or nullizygous for the transgene), each with three replicate plots. Ten plants per genotype were spaced 0.5 m × 1.0 m apart. No fertilizers or pesticides were applied.
The soil physicochemical properties were assessed via five composite samples collected around the plots on July 7, 2023. The soil was a neutral sandy loam (pH 7.2; 73.8% sand, 6.3% silt, and 19.9% clay) with 3.46% organic matter. Analyses were conducted by the National Instrumentation Center for Environmental Management (NICEM), Seoul National University, Korea. The total nitrogen content was 0.09%; the available phosphorus content was 210.8 mg kg−1; and the exchangeable calcium, magnesium, potassium, and sodium contents were 1795.1, 230.4, 43.0, and 12.4 mg kg−1 dry soil, respectively.
Rhizosphere sampling was performed on July 7 (vegetative stage) and August 14 (flowering stage). Five plants per plot were randomly selected, and their shoots and roots were separated. Roots were shaken to remove loosely attached soil, and fine roots (< 1 mm diameter) were placed in 50 ml tubes with distilled water and shaken to dislodge rhizosphere soil. The samples were freeze-dried (FreeZone 2.5, Labconco, USA). The shoots and roots were dried at 65 °C for seven days, after which the dry weights were recorded.
DNA extraction and sequencing
Genomic DNA was extracted from 0.5 g of rhizosphere soil using the FastDNA® Spin Kit for Soil (MP Biomedicals, Irvine, CA, USA). Amplicon sequencing of bacterial 16S rRNA and fungal ITS regions was conducted by CJ Bioscience (Seoul, Korea). The bacterial V4 region was amplified via the primers 515F and 806R29,30 and the fungal ITS2 region was amplified using the gITS7 and ITS4ngs primers31,32. The primers were fused with P5/P7 adaptors, indices, and sequencing adapters for Illumina MiSeq. The PCR conditions included initial denaturation at 95 °C for 3 min; 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min. The PCR products were confirmed by 1% agarose gel electrophoresis and visualized with a Gel Doc system (Bio-Rad, USA). Products were purified via magnetic beads and size-selected with the ProNex® system (Promega, UK). The product quality was verified with a PicoGreen assay (Invitrogen, USA), and sequencing was performed on the Illumina Miseq platform according to the manufacturer’s protocol.
Bioinformatics and statistical analysis
Amplicon sequence variants (ASVs) were inferred via the DADA2 pipeline (v1.8) in R33 following the tutorial for 16S and ITS data (https://benjjneb.github.io/dada2). Singleton, doubleton, and tripleton reads were excluded to minimize sequencing error. Taxonomic classification was performed via the SILVA database (v138.1) for 16S rRNA34 and the UNITE database (release 04.04.2024) for ITS35. Sequences assigned to chloroplasts, mitochondria, archaea, or nontarget eukaryotes were removed. The read counts were rarefied to the minimum observed depth via the ‘rrarefy’ function in the Vegan package36: 93,451 reads for bacteria and 4,155 for fungi. Rarefaction curves were generated with the ‘rarecurve’ function. Alpha diversity metrics (Shannon index, Pielou’s evenness, and Chao1) were calculated via the ‘diversity’ function. Microbial niche breadth was estimated via the ‘levins.Bn’ function in the MicroNiche package37. ASVs with niche breadths below the 5th percentile were defined as specialists, and those above the 95th percentile were defined as generalists37. Intermediate ASVs were considered common taxa.
The community structure was visualized via nonmetric multidimensional scaling (NMDS) via the ‘metaMDS’ function in the Vegan package. The effect of genotype on community structure was assessed via permutational multivariate analysis of variance (PERMANOVA) and analysis of similarities (ANOSIM), which were performed with the ‘adonis2’ and ‘anosim’ functions, respectively. Heatmaps were generated with the ‘heatmap.2’ function (gplots package), via arcsine-transformed and z-score normalized relative abundances. One-way ANOVA followed by Tukey’s HSD test (p < 0.05) was used to assess differences in alpha diversity among genotypes.
Data availability
The sequencing results generated for this study were deposited in K-BDS (Korea BioData Station, https://kbds.re.kr) with the accession ID KAP241515.
References
Kumar, K. et al. Genetically modified crops: current status and future prospects. Planta 251, 91 (2020).
Conner, A. J., Glare, T. R. & Nap, J. P. The release of genetically modified crops into the environment: part II. Overview of ecological risk assessment. Plant J. 33, 19–46 (2003).
Nap, J. P., Metz, P. L., Escaler, M. & Conner, A. J. The release of genetically modified crops into the environment: part I. Overview of current status and regulations. Plant J. 33, 1–18 (2003).
Mendes, R., Garbeva, P. & Raaijmakers, J. M. The rhizosphere microbiome: significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 37, 634–663 (2013).
Dunfield, K. E. & Germida, J. J. Impact of genetically modified crops on soil-and plant‐associated microbial communities. J. Environ. Qual. 33, 806–815 (2004).
Kapur, M. et al. A case study for assessment of microbial community dynamics in genetically modified Bt cotton crop fields. Curr. Microbiol. 61, 118–124 (2010).
Li, P. et al. Combined metagenomic and metabolomic analyses reveal that Bt rice planting alters soil CN metabolism. ISME Commun. 3, 4 (2023).
Oh, S. D. et al. Assessment of the effect on soil microbial communities of genetically modified soybean and a hybrid from crossing with wild soybean. Plant Biotechnol. Rep. 15, 855–862 (2021).
Ge, L. et al. Soil nutrient cycling and Microbiome responses to Bt rice cultivation. Plant Soil https://doi.org/10.1007/s11104-024-06856-8 (2024).
Warschefsky, E., Penmetsa, R. V., Cook, D. R. & Von Wettberg, E. J. Back to the wilds: tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 101, 1791–1800 (2014).
Xu, Y. et al. Bacterial communities in soybean rhizosphere in response to soil type, soybean genotype, and their growth stage. Soil. Biol. Biochem. 41, 919–925 (2009).
Tao, Z. et al. Rhizosphere soil bacterial community composition in soybean genotypes and feedback to soil P availability. J. Integr. Agric. 18, 2230–2241 (2019).
Fazal, A. et al. Triple-transgenic soybean in conjunction with glyphosate drive patterns in the rhizosphere microbial community assembly. Environ. Pollut. 335, 122337 (2023).
Rouhier, N. et al. Involvement of thiol-based mechanisms in plant development. Biochim. Biophys. Acta 1850, 1479–1496 (2015).
Zhai, J. et al. Overexpression of tomato thioredoxin h (SlTrxh) enhances excess nitrate stress tolerance in transgenic tobacco interacting with SlPrx protein. Plant Sci. 315, 111137 (2022).
Bruinsma, M., Kowalchuk, G. & Van Veen, J. Effects of genetically modified plants on microbial communities and processes in soil. Biol. Fertil. Soils. 37, 329–337 (2003).
Chun, Y. J. et al. Do Transgenic Chili pepper plants producing viral coat protein affect the structure of a soil microbial community? Appl. Soil. Ecol. 51, 130–138 (2011).
Wang, L. et al. Bt toxins alter bacterial communities and their potential functions in earthworm intestines. Environ. Pollut. 367, 125591 (2025).
Van Capelle, C., Schrader, S. & Arpaia, S. Selection of focal earthworm species as non-target soil organisms for environmental risk assessment of genetically modified plants. Sci. Total Environ. 548, 360–369 (2016).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643 (2017).
Joos, L. et al. Daring to be differential: metabarcoding analysis of soil and plant-related microbial communities using amplicon sequence variants and operational taxonomical units. BMC Genom. 21, 1–17 (2020).
Sugiyama, A. et al. Changes in the bacterial community of soybean rhizospheres during growth in the field. PLoS One 9, e100709 (2014).
Ma, L. et al. Different effects of wild and cultivated soybean on rhizosphere bacteria. Microbiology 88, 720–728 (2019).
Mestre, M. C., Navarrete, T., García Garrido, J. M. & M. I. & Exploring the yeast-mycorrhiza-plant interaction: Saccharomyces Eubayanus negative effects on arbuscular mycorrhizal formation in tomato plants. Plant Soil 479, 529–542 (2022).
Kim, Y. J. et al. Effects of genotype and environment on the nutrient and metabolic profiles of soybeans genetically modified with epidermal growth factor or thioredoxin compared with conventional soybeans. Ind. Crop Prod. 175, 114229 (2022).
Trochine, A. et al. Genomic and proteomic analysis of Tausonia pullulans reveals a key role for a GH15 glucoamylase in starch hydrolysis. Appl. Microbiol. Biotechnol. 106, 4655–4667 (2022).
Kim, D. Y. et al. Assessing invasiveness of genetically modified soybean expressing human epidermal growth factor gene. Weed Turf Sci. 9, 119–128 (2020).
Kim, D. Y. et al. Natural hybridization between Transgenic and wild soybean genotypes. Plant. Biotechnol. Rep. 15, 299–308 (2021).
Apprill, A., McNally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137 (2015).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Ihrmark, K. et al. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 82, 666–677 (2012).
Tedersoo, L. et al. Global diversity and geography of soil fungi. Science 346, 1256688 (2014).
Callahan, B. J. et al. DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Abarenkov, K. et al. UNITE general FASTA release for Fungi 2. UNITE Community https://doi.org/10.15156/BIO/2959333 (2024).
Oksanen, J. et al. Package ‘vegan’. Community ecology package, version 2(9), 1–295 (2013).
Finn, D. et al. MicroNiche: an R package for assessing microbial niche breadth and overlap from amplicon sequencing data. FEMS Microbiol. Ecol. 96, fiaa131 (2020).
Acknowledgements
We thank Dr. Ju Seok Seo at CELLTRION for providing the GM soybean seeds.
Funding
This research was supported by the KRIBB Research Initiative Program (KGM1121511) and the National Institute of Ecology (NIE) funded by the Ministry of the Environment (MOE) of the Republic of Korea (NIE-A-2025-04).
Author information
Authors and Affiliations
Contributions
S. J. C. analyzed the data and wrote the manuscript. I. S. P., D. Y. K., and J. H. H. performed the experiment. K. H. N. and C. G. K. designed the experiments, analyzed the data and wrote the manuscript. All the authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chun, SJ., Pack, I.S., Kim, D.Y. et al. Ecological impacts of human thioredoxin expression and interspecific hybridization on the soybean rhizosphere microbiome: insights from ASV-level niche analysis. Sci Rep 15, 29806 (2025). https://doi.org/10.1038/s41598-025-15799-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15799-8
