
Abstract
Mowat‒Wilson syndrome (MWS), a rare genetic disorder caused by heterozygous loss-of-function mutations in ZEB2, is characterised by significant growth retardation with unclear mechanisms. In this study, we developed a Zeb2 haploinsufficient (Zeb2+/−) mouse model that recapitulates key features of MWS, including reduced body weight, impaired intestinal development and skeletal hypoplasia. RNA sequencing revealed significant downregulation of nutrient digestion and absorption pathways in the duodenum of Zeb2+/− mice, which was associated with reduced body fat and bone mass loss. Additionally, Zeb2+/− mice presented severe gut microbiota dysbiosis, as indicated by the depletion of beneficial Actinobacteria and Bifidobacterium and increases in the abundances of the proinflammatory Proteobacteria and Rikenella. These microbial shifts correlated with impaired intestinal development and key growth indicators. Our findings delineate a pathological cascade wherein Zeb2 haploinsufficiency disrupts nutrient absorption and bone homeostasis, while concomitant dysbiosis likely exacerbates intestinal dysfunction, collectively driving growth retardation. The model we developed can provide a platform for exploring therapeutic interventions targeting nutritional support and microbiome modulation in MWS.
Similar content being viewed by others

Introduction
Mowat‒Wilson syndrome (MWS) is a rare multisystem disorder caused by loss-of-function heterozygous mutations or deletions in the ZEB2 gene. It is characterised by intestinal anomalies, distinctive facial features, moderate to severe intellectual disability, and frequent multisystem involvement, such as congenital heart defects and genitourinary and musculoskeletal defects1,2. A hallmark feature of MWS is postnatal growth retardation. Although most patients have normal birth parameters, more than 80% develop height and weight below the 50th percentile by early childhood, with progressive microcephaly emerging postnatally, defined as a head circumference at least 2 standard deviations below the mean3,4,5. Despite its clinical prevalence, the molecular mechanisms linking ZEB2 haploinsufficiency to growth retardation remain unclear.
The development of well-defined Zeb2 knockout mouse models is crucial for studying growth retardation mechanisms in MWS, given the rarity and clinical heterogeneity of this disorder. However, major challenges persist. First, the current models have limited representativeness. Several studies have utilised conditional knockout of the Zeb2 gene to reproduce partial MWS phenotypes in various organs. For example, mice with complete Zeb2 allele knockout in neural crest cells (NCC) exhibit craniofacial and gastrointestinal malformations similar to those observed in MWS patients, whereas heterozygous knockout in NCC does not result in these phenotypes6. Moreover, homozygous Zeb2 knockout mice exhibit high mortality, complicating growth and development observations. Second, there is insufficient research on whether these models accurately reflect MWS-related growth retardation. In 2015, a study by Tsuyoshi Takagi et al. demonstrated that Zeb2 Δex7/+ mice presented a progressive onset of growth retardation after multiple backcrosses with the inbred B57BL/6 strain and the closed-colony ICR strain. However, a comprehensive description of the phenotype and underlying mechanisms have not been provided7. Recent CRISPR/Cas9 advances now enable precise Zeb2 heterozygous knockout models that better mimic MWS genetics and phenotypes, providing a robust tool for unravelling MWS pathomechanisms and advancing therapeutic strategies8.
The small intestine is crucial for nutrient absorption, handling approximately 85% of carbohydrates, 66–95% of proteins, and all fats. Its dysfunction can severely impede growth and development9,10. Surgical data highlight the profound impact of small bowel resection on nutrient absorption. Extensive resection in neonates can lead to short bowel syndrome, which is characterised by severe malnutrition, weight loss, diarrhoea, steatorrhea, and potentially fatal outcomes11. Nutrient malabsorption resulting from intestinal dysfunction can significantly impair growth and development. For example, individuals with pancreatic exocrine insufficiency (PEI) experience impaired digestion and absorption of fats, proteins, and carbohydrates and present with symptoms such as abdominal pain, weight loss, and malnutrition12. Genetic mutations can also disrupt nutrient digestion and absorption. Mice lacking the monoacylglycerol acyltransferase-2 gene demonstrate reduced fat absorption in the proximal small intestine, resulting in lower levels of dietary fat entering the bloodstream. Similarly, mice with intestine-specific knockout of the apolipoprotein B (ApoB) gene exhibit defects in chylomicron production and fat malabsorption13,14.
Emerging evidence has underscored the pivotal role of gut microbiota‒intestine crosstalk in modulating nutrient assimilation. For instance, certain microbes such as Clostridium bifermentans have been shown to enhance the expression of Dgat2 in the duodenum and jejunum, thereby directly influencing lipid absorption15. Moreover, dysbiosis in the small intestine can impair nutrient absorption. Small intestinal bacterial overgrowth (SIBO), characterized by an excess of colon-derived Gram-negative bacteria, is frequently associated with diarrhea and malnutrition16. These findings highlight the significant impact of the gut microbiota on nutrient absorption, suggesting that a comprehensive understanding of growth retardation in MWS patients necessitates a detailed exploration of their intestinal function and microbiota.
Here, we utilised CRISPR/Cas9 technology to generate global Zeb2 heterozygous knockout (Zeb2+/−) mice to investigate the mechanisms of growth retardation in MWS. Through comprehensive phenotyping, transcriptomic analysis, and gut microbiota profiling, we revealed a pathological network linking Zeb2 haploinsufficiency to growth defects via nutrient malabsorption, reduced bone mass and gut microbiome imbalance.
Results
Zeb2 haploinsufficiency leads to postnatal growth retardation in mice
To model the growth phenotypes of Mowat–Wilson syndrome (MWS), we established global Zeb2 heterozygous mice (Zeb2+/−) via CRISPR/Cas9 editing (Fig. S1a, b). RNA sequencing analysis of duodenum revealed a 68.4% reduction in Zeb2 transcript levels (calculated by FPKM) in the mutant mice compared to their wild-type (WT) littermates (Fig. S1c). Comprehensive growth analysis revealed persistent developmental deficits in Zeb2+/− mice from postnatal Day 7 (P7) through weaning to adolescence (P35). Compared with WT, Zeb2+/− mice presented significantly reduced body weight (Fig. 1a) and cumulative weight gain (Fig. 1b), with differences evident as early as P7. A significantly reduced body length throughout the observation period was also observed in Zeb2+/− mice (Fig. 1c). Representative animal images confirmed these findings (Fig. 1d). Craniofacial analysis revealed specific morphological changes, with Zeb2+/− mice having a shorter skull length than WT mice while maintaining a normal skull width (Fig. 1e–f). Notably, these growth parameters showed no sex-dependent variations (Fig. S2).

Zeb2 haploinsufficiency leads to postnatal growth retardation in mice. Body weight (a), weight gain (b), and body length (c) curves of the WT (n = 12) and Zeb2+/− groups (n = 12) from postnatal Day 7 (P7) or 14 (P14) to P35. (d) Representative body images of WT and Zeb2+/− mice at P35, Scale bar, 1 cm. (e) Representative skull shape images of WT and Zeb2+/− mice, Scale bar, 1 cm. (f) Statistical data of skull length and width in WT and Zeb2+/− mice at P35.
We then investigated the effects of changes in food intake on growth retardation in Zeb2+/− mice. Our results reveal that Zeb2+/− mice exhibit significantly reduced cumulative and daily food intake compared to WT mice (Fig. S3a, b). However, the relative food intake (food intake/body weight) shows an increase at P21-P28, before reaching levels comparable to those of WT mice by P31 (Fig. S3c). This trend suggests that the initial weight loss observed from P21 to P28 may be disproportionate to the reduction in food intake, potentially due to non-ingestive factors such as malabsorption or hypermetabolism. By P31, the sustained low absolute food intake, coupled with stabilized relative intake, indicates that weight regulation in Zeb2+/− mice is likely multifactorial.
Zeb2 haploinsufficiency leads to abnormal intestinal development
Given the frequent gastrointestinal manifestations observed in MWS patients, such as chronic constipation and Hirschsprung disease, we hypothesised that intestinal hypoplasia might underlie their systemic growth retardation. Morphological analysis of Zeb2+/− mice revealed pronounced intestinal shortening, with a 10.7% reduction in the length of the small intestine compared with that of WT controls (Fig. 2a, b). Haematoxylin and eosin (H&E)-stained sections revealed marked villus blunting in Zeb2+/− mice (Fig. 2c). Quantitative analysis confirmed significant shortening of the small intestinal villus length and crypt depth in Zeb2+/− mice (Fig. 2d, e). These findings indicate impaired intestinal absorption in Zeb2+/− mice.

Zeb2 haploinsufficiency leads to abnormal intestinal development. (a) Representative image of the small intestines of WT and Zeb2+/− mice. (b) Quantification of small intestine length in both WT and Zeb2+/− mice. (c) Representative H&E staining of the small intestines of WT and Zeb2+/− mice. Scale bar, 100 μm. (d–e) Quantification of villus height and crypt depth in the duodenum of WT and Zeb2+/− mice.
Zeb2 haploinsufficiency disrupts nutrient absorption pathways
To explore the link between intestinal hypoplasia and systemic growth retardation in MWS, we conducted RNA sequencing on duodenal tissues from P35 Zeb2+/− mice. Unsupervised clustering revealed transcriptomic changes in Zeb2+/− mice, with 505 genes significantly downregulated and 54 genes upregulated (|log2FC|≥ 1.5, FDR < 0.05; Fig. 3a). KEGG pathway analysis revealed significant dysregulation of three key nutrient absorption pathways, namely, fat digestion and absorption, protein digestion and absorption, and starch and sucrose metabolism, in Zeb2+/− mice (Fig. 3b). Heatmap highlighting pathway-specific gene dysregulation. Notably, critical lipase genes, including Plpp3, Pnliprp1, Clps, Pla2g1b, Pnliprp2, and Pnlip, were markedly suppressed in the fat digestion and absorption pathway (Fig. 3c). Over 29 genes, including Col1a1 and Cela2a, were broadly downregulated in the protein digestion and absorption pathways (Fig. 3d). Additionally, five genes involved in starch and sucrose metabolism, namely, Hk3, Amy2a4, Amy2a3, Amy2a5, and Amy2b, were also downregulated (Fig. 3e). qRT-PCR confirmed the significant downregulation of lipid absorption-related pancreatic lipase family genes, including Pnliprp2, Pnlip, and Pnliprp1, in line with the RNA-seq data (Fig. 3f–h). These results emphasise the critical role of impaired nutrient absorption in contributing to growth retardation.

Zeb2 haploinsufficiency disrupts nutrient absorption pathways. (a) Volcano plot showing differentially expressed genes (DEGs) between duodenal tissues from Zeb2+/− and Zeb2+/+ mice. (b) KEGG pathway enrichment analysis based on RNA transcriptome sequencing data, plotted as a bubble chart displaying the top 20 pathways. Heatmaps showing the expression levels of DEGs associated with KEGG pathways related to protein digestion and absorption (c), fat digestion and absorption (d), and starch and sucrose metabolism (e). qPCR analyses of key genes responsible for nutrient absorption in the small intestine, namely, Pnliprp1 (f), Pnlip (g), and Pnliprp2 (h).
Zeb2 +/− mice exhibit lower fat mass
The molecular phenotype of nutrient absorption in Zeb2+/− mice showed significant physiological changes, including a 13.3% reduction in body fat mass relative to body weight and a 14.6% increase in total body water content, with muscle mass comparable to that of WT littermates (Fig. 4a). Consistent with these systemic changes, Zeb2+/− mice presented marked reductions in visceral adipose depot weights, including epididymal (eWAT, 17.3% decrease), perigonadal (pWAT, 27.7% decrease), and mesenteric (mWAT, 26.9% decrease, p = 0.015) white adipose tissues, compared with those of WT mice (Fig. 4b). Inguinal white adipose tissue showed a noticeable decreasing trend, although it was not statistically significant (Fig. 4b). Histological examination of adipose tissues revealed striking morphological differences. Specifically, Zeb2+/− adipocytes (eWAT, pWAT, and mWAT) exhibited significant reductions in size compared to the hypertrophic adipocytes observed in WT mice (Fig. 4c). Quantitative measurements further corroborated these findings (Fig. 4d). These findings indicate that impaired nutrient absorption leads to reduced whole-body adiposity, contributing to the postnatal growth retardation observed in Zeb2+/− mice.

Zeb2+/− mice exhibit lower fat mass. (a) Body composition of WT and Zeb2+/− mice adjusted for body weight at P35. (b) Fat mass of WT and Zeb2+/− mice, including inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), perigonadal white adipose tissue (pWAT) and mesenteric white adipose tissue (mWAT), P35. (c) H&E staining of eWAT, pWAT and mWAT from WT and Zeb2+/− mice. (d) Lipid droplet size was quantified by measuring the cross-sectional area in randomly selected adipocytes. For each sample (WT and Zeb2+/− mice), we analyzed four randomly chosen fields of view, with 10 adipocytes measured per field.
Zeb2 +/− mice exhibit reduced bone mass
Given that extensive small bowel resection and a reduced absorptive surface area are known to precipitate fat-soluble vitamin deficiencies, particularly vitamin D, A, K, and E deficiencies, in individuals with clinical malabsorption syndrome, we hypothesised that Zeb2+/− individuals would similarly exhibit impaired vitamin D absorption. To test this hypothesis, we measured the bone mass of Zeb2+/− mice. Micro-CT microarchitectural analysis revealed significant skeletal deficits in Zeb2+/− mice (Fig. 5a, Fig. S4 and Table. S3). The 3-D reconstructed images revealed a significant reduction in the trabecular bone volume (BV/TV) within the distal femur of Zeb2+/− mice compared with that of WT mice, with a 38.4% decrease from 17.7 ± 3.6% in WT mice to 10.6 ± 0.38% in Zeb2+/− mice (Fig. 5b). This finding was supported by a significant increase in the structural model index (SMI), indicating degraded structural integrity, from 2.1 ± 0.1 in WT mice to 2.5 ± 0.04 in Zeb2+/− mice (Fig. 5c). Additionally, Zeb2+/− mice exhibited compromised mineralization, as evidenced by a decrease in cortical bone mineral density (Ct. BMD) from 0.82 ± 0.004 mg HA/cm3 in WT mice to 0.8 ± 0.005 mg HA/cm3 in Zeb2+/− mice (Fig. 5d). The trabecular number (Tb.N) was also reduced, from 2.7 ± 0.2/mm in WT mice to 1.8 ± 0.01/mm in Zeb2+/− mice (Fig. 5e), whereas the intertrabecular spacing (Tb.Sp) expanded, increasing from 0.19 ± 0.005 mm in WT mice to 0.23 ± 0.008 mm in Zeb2+/− mice (Fig. 5f). Notably, no significant differences in trabecular thickness (Tb.Th) were detected between Zeb2+/− and WT mice (Fig. 5g), suggesting that Zeb2 gene mutation primarily affects trabecular proliferation and maintenance rather than matrix deposition. These results collectively indicate that Zeb2 haploinsufficiency and intestinal nutrient malabsorption lead to substantial trabecular bone loss, which not only reduces bone mass but also slows growth and development rates. The observed skeletal deficits significantly contributed to the developmental retardation observed in Zeb2+/− mice.

Zeb2+/− mice exhibit reduced bone mass. (a) Representative three-dimensional reconstructed images of the distal femur obtained from micro-CT scans, showing samples from each group of mice. Histomorphometric analysis of bone tissue in the region of interest (ROI) of the femur from different groups of mice (n = 3). (b) Trabecular bone volume over total volume (BV/TV); (c) structure model index (SMI); (d) cortical bone mineral density (Ct.BMD); (e) trabecular number (Tb.N); (f) trabecular separation (Tb.Sp); (g) trabecular thickness (Tb.Th).
Alterations in the microbial composition of Zeb2 +/− mice
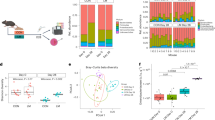
To investigate the effects of alterations in the gut microbiota on the growth retardation and impaired nutrient absorption observed in Zeb2+/− mice, we performed 16S rRNA gene sequencing on faecal samples from WT and Zeb2+/− littermates at P35. Our analysis revealed substantial restructuring of the gut microbiota in Zeb2+/− mice, characterised by the loss of 373 established amplified sequence variants (ASVs) and the emergence of 481 novel ASVs (Fig. 6a). While alpha-diversity metrics (Chao1 index) remained comparable between genotypes (Fig. 6b), beta-diversity analysis (PCoA) demonstrated significant divergence in overall microbial community structure (Fig. 6c). At the phylum level, Zeb2+/− mice exhibited a decrease in the relative abundance of Actinobacteria, while the abundance of Proteobacteria increased (Fig. 6d, e, h). We identified 142 predominant bacterial genera, among which Rikenella, Dubosiella, and Bifidobacterium were among the top 20 most abundant in both groups (Fig. 6f, g). Specifically, Zeb2+/− mice showed an increased abundance of Rikenella but decreased levels of Dubosiella and Bifidobacterium (Fig. 6i). These findings suggest that significant alterations in the community composition of the gut microbiota potentially exacerbated the growth retardation phenotype observed in Zeb2+/− mice.

Alterations in microbial composition in Zeb2+/− mice. (a) Venn diagram illustrating shared amplified sequence variants (ASVs) between the WT (n = 12) and Zeb2+/− mice (n = 12) at P35. (b) The alpha diversity of the gut microbiota in WT and Zeb2+/−mice was assessed using the Chao1 richness estimator and Shannon index. (c) Principal coordinate analysis (PCoA) based on the Bray‒Curtis distance between WT and Zeb2+/− mice at P35. (d) Bar plot depicting the relative abundance of taxa at the phylum level of individual samples from WT (n = 12) and Zeb2+/− (n = 12) mice at P35. (e) Bar plot displaying the relative abundance of the top taxa at the phylum level between WT and Zeb2+/− group. (f) Bar plot illustrating the relative abundance of taxa at the genus level of individual samples from WT (n = 12) and Zeb2+/− (n = 12) mice at P35. (g) Bar plot displaying the relative abundance of the top taxa at the genus level between WT and Zeb2+/− group. (h) The abundance of taxa at the phylum level with significant differences between WT and Zeb2+/− group. (i) The abundance of taxa at the genus level with significant differences between WT and Zeb2+/− group. Alpha diversity was analysed using the Kruskal‒Wallis test. Beta diversity was analysed via Adonis analysis. Box plot explanation for (b): The centerline represents the median (second quartile), and the box extends from the first quartile (box start) to the third quartile (box end).
Correlation between reduced postnatal growth-related indicators and the gut microbiota
Linear discriminant analysis effect size (LEfSe) analysis further revealed that Zeb2+/− mice were enriched in Proteobacteria and had a reduction in Actinobacteriota at the phylum level. These shifts were accompanied by genus-level enrichments of Rikenella, Rodentibacter, Olsenella, Bradyrhizobium, Parasutterella, Neisseria, Citrobacter and Barnesiella (LDA score > 2, Fig. 7a, b). At the phylum level, the depletion of Actinobacteriota, a taxon essential for maintaining gut barrier integrity and producing anti-inflammatory metabolites, was significantly positively correlated with postnatal growth metrics, including body weight, body length, skull length, food intake and adipose tissue mass (eWAT, pWAT, and mWAT). In contrast, Proteobacteria, a phylum strongly linked to dysbiosis and proinflammatory responses, exhibited inverse correlations with these parameters (Fig. 7c and S5a). At the genus level, the enrichment of Rikenella, a taxon implicated in mucosal barrier disruption and inflammatory bowel disease, was significantly negatively correlated with body weight, pWAT and food intake, suggesting its role in exacerbating malabsorption and gut dysfunction (Fig. 7d and S5b). Conversely, Bifidobacterium, a genus pivotal for nutrient metabolism and gut homeostasis, and Turicibacter, a taxon implicated in lipid metabolism and intestinal immune modulation, were both positively associated with body weight,fat deposition and food intake (Fig. 7e, f and S5c-d). The results indicated that the decline in Bifidobacterium and Turicibacter, coupled with the increase in Rikenella, were strongly associated with growth retardation. These changes likely act synergistically to exacerbate intestinal tissue damage, suppress anti-inflammatory signaling, and impair nutrient absorption efficiency.

Correlation between reduced postnatal growth-related indicators and the gut microbiota in Zeb2+/−mice. (a) LEfSe Cladogram depicting key gut microbiota differences between WT and Zeb2+/− mice. (b) LDA score indicating significant differences in the microbiota between WT and Zeb2+/− mice. (c) Correlations between the gut microbiota at the phylum level and growth indices in WT and Zeb2+/− mice. (d) Spearman correlation analysis of the correlations between the abundance of Rikenella and body weight or pWAT. (e) Spearman correlation analysis of the correlations between the abundance of Bifidobacterium and body weight or pWAT. (f) Spearman correlation analysis of the correlations between the abundance of Turicibacter and body weight or pWAT.
Discussion
Previous studies have reported that MWS is frequently associated with growth retardation, which is typically characterised by height and weight below the 50th percentile in 72–80% of participants with MWS3,5. Consistent with these observations, our research also revealed significant anomalies in head circumference among individuals with MWS, which aligns with findings from previous studies4,5. However, the exact causes of growth retardation in MWS are still unclear. In this study, we independently developed a Zeb2 heterozygous knockout mouse model to replicate the clinical features of MWS and explore its molecular basis. These Zeb2+/− mice exhibited reduced postnatal growth, with lower body weights, body lengths, and skull lengths, mirroring the clinical phenotype observed in paediatric MWS patients. These findings underscore the critical role of the Zeb2 gene as a transcription factor in regulating growth. The Zeb2+/− mouse model we developed offers a valuable tool for studying MWS pathomechanisms and developing therapeutic strategies, potentially leading to improved patient outcomes.
MWS patients often present with gastrointestinal issues such as chronic constipation and Hirschsprung disease. In our Zeb2+/− mice, we observed similar intestinal defects, including a shortened small intestine and villus atrophy. Since the small intestine is crucial for postnatal growth and is the primary site for the digestion and absorption of ~ 90% of nutrients10, we explored how these defects lead to growth retardation. Our mechanistic studies revealed significant downregulation of nutrient absorption pathways, especially lipid digestion/absorption pathways, with suppressed expression of key lipase genes (Pnliprp2, Pnlip, Pnliprp1). These changes are correlated with systemic metabolic disturbances, manifested as a reduced body fat percentage and smaller white adipose tissue depots (eWAT, pWAT, and mWAT). Our findings show that Zeb2 haploinsufficiency disrupts systemic energy homeostasis by impairing the intestinal nutrient absorption capacity, thereby contributing to growth retardation. This study reveals novel insights into the roles of Zeb2 in regulating intestinal development and metabolic coordination, offering critical insights into growth retardation in MWS.
Clinically, conditions such as short bowel syndrome (SBS) can lead to deficiencies in fat-soluble vitamins (D, A, K, E) due to impaired intestinal absorption. We hypothesise that Zeb2+/− mice, which exhibit intestinal shortening, villus atrophy, and defective lipid absorption, may similarly develop vitamin D deficiency, exacerbating skeletal abnormalities. Consistent with this hypothesis, low bone density was observed in Zeb2+/− mice. During childhood growth, skeletal development depends on a delicate balance between osteoblast-mediated bone formation and osteoclast-driven resorption. Previous studies have identified Zeb2 as a target of both miR-145a-5p and miR-493-5p17,18, linking it to the complex gene regulatory networks governing osteogenesis. Accordingly, we suggest that reduced Zeb2 expression may impede osteoblast differentiation19. Collectively, Zeb2 deficiency may disrupt both vitamin D absorption and osteoblast function, synergistically contributing to skeletal defects and growth retardation in MWS.
Our study revealed significant alterations in the gut microbiota of Zeb2+/− mice, which contributed to intestinal barrier dysfunction and subsequent growth impairments. These changes are marked by reduced Actinobacteriota, Bifidobacterium and Turicibacter, alongside increased Proteobacteria and Rikenella, which are strongly correlated with growth delay indicators such as lower body weight, shorter body and skull lengths, and reduced adipose tissue mass. Research in DSS-induced colitis models has demonstrated that the abundance of Proteobacteria is positively correlated with the severity of intestinal inflammation20. Intervention experiments with D-amino acids have confirmed that alleviation of experimental colitis and cholangitis is accompanied by inhibition of Proteobacteria growth20. In TLR5-deficient mice, Proteobacteria exacerbate chronic inflammation and intestinal barrier damage through abnormal immune responses21. Additionally, IBD patients with impaired barrier function often exhibit reduced abundance of Actinobacteriota, particularly Bifidobacterium, which further deteriorates gut barrier integrity22. Thus, we speculate that Actinobacteriota, Bifidobacterium, and Proteobacteria play analogous roles in Zeb2+/− mice, likely exacerbating intestinal inflammation and barrier damage, which in turn further impairs intestinal nutrient absorption and exacerbates the growth retardation phenotype. However, validating the causality between microbiota changes and the observed phenotypes in Zeb2+/− mice will require future interventional studies, such as fecal microbiota transplantation in germ-free models. Additionally, studies on MWS patients and their gut microbiota are currently limited. Conditions that are frequently associated with MWS, such as Hirschsprung disease, often exhibit significant microbiota dysbiosis23. This correlation suggests that MWS patients might also experience gut microbiota imbalances, similar to the microbiota disruptions observed in Zeb2+/− mice, which deserves further research in future.
In summary, our study explored the impact of Zeb2 haploinsufficiency on growth and metabolism in Mowat–Wilson syndrome (MWS) using a Zeb2+/− mouse model. The Zeb2+/− mice recapitulated key MWS features, including reduced body weight, shorter body length, and craniofacial hypoplasia. These defects were linked to intestinal abnormalities, such as shortened small intestine and villus atrophy, leading to impaired nutrient absorption, systemic metabolic dysfunction and reduced bone mass. Furthermore, Zeb2+/− mice exhibited severe gut microbiota dysbiosis, which exacerbates intestinal dysfunction, collectively driving growth retardation. Our findings suggest that therapeutic strategies targeting nutritional support and microbiome modulation might alleviate growth retardation in MWS.
Methods
Mice
Wild-type C57BL/6JGpt mice were obtained from GemPharmatech Co., Ltd. (Nanjing, China). Global Zeb2 heterozygous knockout (Zeb2+/−) mice were generated using CRISPR-Cas9 technology by GemPharmatech Co., Ltd., on a C57BL/6JGpt background. In brief, Cas9 mRNA and sgRNA were coinjected into zygotes to produce F0 mice. The sgRNA guided Cas9 to cleave between introns 3–4 and 6–7 of the Zeb2 gene, inducing double-strand breaks and subsequent homologous recombination leading to Zeb2 gene deletion. The genotype of Zeb2+/− mice was confirmed by PCR analysis of genomic DNA isolated from mouse tails using the following primers: forward: 5′-TCCACCAACCACAGCAGTGTATTATG-3′ and reverse: 5′-AGAAACGGTGTGACAACTGCTAGG-3′, resulting in a 253 bp product (Fig. S1). F1 generation mice, which were obtained by crossing F0 Zeb2+/− mice with wild-type mice, were used for the experiments.
Wild-type (WT) littermates served as controls in all experiments involving Zeb2+/− mice to ensure genetic and environmental consistency. Prior to weaning, all mice were co-housed within the same litter. After weaning (at postnatal day 21(P21)), mice were separated and group-housed according to genotype and sex. Mice were maintained in a specific pathogen-free (SPF) facility under a 12-h light/dark cycle, with ambient temperature regulated at 22–25 °C and relative humidity at 40–60%. Autoclaved chow and water were provided ad libitum. Weekly cage changes were performed under laminar flow hoods to ensure sterility.
The body weight, body length, and skull length of the mice were recorded from P7 or P14 to P35 (twelve biological replicates per group). Representative photographs of body shape and skull morphology were taken. Body fat mass and lean mass (FFM) were measured and calculated using magnetic resonance spectroscopy (MRS) with an EchoMRI Body Composition Analyser (Houston, TX). At P35, the mice were euthanised. Fat mass, including inguinal white adipose tissue (iWAT), epididymal white adipose tissue (eWAT), perigonadal white adipose tissue (pWAT), and mesenteric white adipose tissue (mWAT), was recorded. Tissues such as duodenal and fat tissues were partially formalin fixed for histochemistry experiments and partially cryopreserved in liquid nitrogen for molecular biology experiments.
All animal experiments were performed in accordance with relevant guidelines and regulations. This study is reported in accordance with ARRIVE guidelines (https://arriveguidelines.org). The animal experiments were approved by the medical ethics committee of the Capital Institute of Paediatrics (SHERLL2022023), and all the experimental procedures strictly adhered to the guidelines for Institutional Animal Care and Use. Mice were euthanized via carbon dioxide (CO2) asphyxiation in accordance with the guidelines of the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals. Briefly, mice were placed in a sealed euthanasia chamber, and CO2 gas was introduced at a flow rate of 5.5 L/min for 2–5 min until respiration stopped and death was evident (e.g., no heartbeat, pale conjunctiva).CO2 flow continued for another minute to ensure humane euthanasia.No additional chemical agents were used in the euthanasia process.
Transcriptome sequencing and data analysis
RNA was extracted from the duodenal tissues of WT and Zeb2+/− mice, with four mice per group serving as biological replicates. The RNA quality was assessed using the RNA Nano 6000 Assay Kit on a Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, California, USA). mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was achieved with divalent cations at elevated temperatures in First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesised using random hexamer primers and M-MuLV Reverse Transcriptase (RNase H-). Subsequently, second-strand cDNA synthesis was performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. Adenylation of the 3’ ends of DNA fragments was followed by ligation of adaptors with hairpin loop structures to facilitate hybridization. To select cDNA fragments 370–420 bp in length, the library fragments were purified using the AMPure XP system (Beckman Coulter, Beverly, USA). PCR was conducted with Phusion High-Fidelity DNA polymerase, universal PCR primers, and Index (X) Primer. Finally, the PCR products were purified with the AMPure XP system, and library quality was assessed using the Agilent Bioanalyzer 2100 system. Sequencing was performed on an Illumina NovaSeq platform (Illumina, San Diego, California, USA), generating 150 bp paired-end reads.
Differential expression analysis of two groups (four biological replicates per group) was performed using the DESeq2 R package (1.20.0). A significance threshold of P value (Padj) < 0.05 and |log2-fold change|≥ 1.5 was applied to identify significantly differentially expressed genes. Additionally, KEGG pathway enrichment analysis was performed using the clusterProfiler R package. The pathway map was generated using the KEGG database (Kanchisa Laboratories)24,25. Heatmaps illustrating changes in gene expression levels were generated to visualise the results.
Quantitative real-time PCR
Total RNA was extracted from the duodenal tissues of WT and Zeb2+/− mice via TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesised using a reverse transcription kit (TaKaRa, Dalian, Liaoning, China). Quantitative real-time PCR was conducted on an ABI 7500 system (Applied Biosystems, Foster, CA, USA) with UltraSYBR mixture (CWBIO, Jiangsu, China). The primer sequences are listed in Table S1 of the Supporting Information, with β-actin primers sourced from commercial suppliers at Sangon Biotech Co., Ltd., Shanghai. The target genes were quantified via the 2−ΔΔCt method, with β-actin used as the reference gene.
H&E staining
Tissues were fixed in formalin and embedded in paraffin. Sections (5 μm thick) were prepared from the paraffin blocks and stained with haematoxylin and eosin (H&E). Images were captured using an Olympus microscope (Hamburg, Germany). All villus and crypt measurements were conducted on longitudinal sections of the proximal duodenum. In each H&E-stained section of the duodenum from WT and Zeb2+/− mice, the length or depth of 14 well-oriented villi and crypts were measured using ImageJ software (with four biological replicates per group).
Bone mass measurement-microcomputed tomography (micro-CT)
Micro-CT images of distal femurs fixed in 4% paraformaldehyde were obtained using an ex vivo X-ray micro-computed tomography system (Quantum GX2, micro-CT). Image reconstruction was performed using Avizo software (Thermo Scientific), with consistent thresholds applied to all the samples (1500–5000 Hounsfield units). The region of interest (ROI) was defined as 0.5 mm below the growth plate at the distal femur, encompassing 50 slices (10 μm per slice) for trabecular analysis. Femoral morphometric parameters were analysed using CTAn software. The key trabecular bone parameters included BV/TV (bone volume/total volume), SMI (structure model index), Tb.Th (trabecular thickness), Tb.N (trabecular number), and Tb.Sp (trabecular separation). Cortical bone analysis focused on Ct. BMD (cortical bone mineral density).
16S rDNA sequencing and bioinformatics analysis
Total DNA was extracted from faecal samples collected from WT and Zeb2+/− mice on Day 35 via the cetyltrimethylammonium bromide method. The 16S rDNA genes were subsequently amplified using primers targeting the V4 region: 515F and 806R with a barcode. The PCR cycling program included initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, extension at 72 °C for 30 s, and a final extension at 72 °C for 5 min. The purity and concentration of the DNA were assessed with 2% agarose gel electrophoresis. The PCR products were purified with a GeneJET Gel Extraction Kit (Thermo Scientific, Waltham, Massachusetts, USA). The sequencing libraries were prepared with the Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, San Diego, California, USA) and sequenced on an Illumina NovaSeq 6000 platform.
Microbial community analysis was performed using QIIME2 software. Alpha diversity was assessed using metrics including the Chao1 index to estimate microbial richness in the samples. Beta diversity was evaluated using unweighted UniFrac Principal Coordinates Analysis (PCoA), with the unweighted UniFrac distance matrix calculated using the R package. Significant differences in microbial community composition between groups were analyzed using the Wilcoxon rank-sum test. The linear discriminant analysis effect size (LEfSe) method was employed to compare the taxonomic abundance profiles. Pairwise correlations between the relative abundance of microbial taxa and continuous phenotypic variables were computed using Spearman’s rank correlation coefficient (ρ).
Statistical analysis
Statistical analysis and graphical representations were performed with GraphPad Prism version 10 (GraphPad Software, Inc., San Diego, California, USA) or IBM SPSS Statistic 27. All data were presented as means ± standard deviations (SDs). The normality of data distribution was assessed using the Shapiro–Wilk test, while homogeneity of variances was evaluated by the Levene test. For statistical comparisons between groups, Student’s t-tests (two-sided) were employed (Figs. 1f, 2b and e, 4a–b and d, b–g, Fig. S1c, Fig. S2d). For data that did not follow a normal distribution, the nonparametric Mann–Whitney test was used (Figs. 2d, 3f–h, 6h–i, Fig. S3). Two-way analysis of variance (ANOVA) was applied for multiple comparisons (Fig. 1a–c, Fig. S2a–c). A significance level of P < 0.05 was considered statistically significant, with the following notations: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns for non-significant results.
Data availability
Paired raw sequencing data (16S rRNA sequences) have been deposited in the Genome Sequence Archive in National Genomics Data Center26, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences, under the accession number CRA017714. Paired raw sequencing data (RNA-Seq) have been deposited in Gene Expression Omnibus (GEO) under accession code GSE272386.
References
Mowat, D. R. et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet 35, 617–623. https://doi.org/10.1136/jmg.35.8.617 (1998).
Birkhoff, J. C., Huylebroeck, D. & Conidi, A. ZEB2, the Mowat-Wilson syndrome transcription factor: Confirmations, novel functions, and continuing surprises. Genes (Basel). https://doi.org/10.3390/genes12071037 (2021).
Ivanovski, I. et al. Phenotype and genotype of 87 patients with Mowat-Wilson syndrome and recommendations for care. Genet Med 20, 965–975. https://doi.org/10.1038/gim.2017.221 (2018).
Ivanovski, I. et al. Mowat-Wilson syndrome: Growth charts. Orphanet J Rare Dis 15, 151. https://doi.org/10.1186/s13023-020-01418-4 (2020).
Wu, L. et al. Physical, language, neurodevelopment and phenotype-genotype correlation of Chinese patients with Mowat-Wilson syndrome. Front Genet 13, 1016677. https://doi.org/10.3389/fgene.2022.1016677 (2022).
Van de Putte, T., Francis, A., Nelles, L., van Grunsven, L. A. & Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum Mol Genet 16, 1423–1436. https://doi.org/10.1093/hmg/ddm093 (2007).
Takagi, T., Nishizaki, Y., Matsui, F., Wakamatsu, N. & De Higashi, Y. novo inbred heterozygous Zeb2/Sip1 mutant mice uniquely generated by germ-line conditional knockout exhibit craniofacial, callosal and behavioral defects associated with Mowat-Wilson syndrome. Hum Mol Genet 24, 6390–6402. https://doi.org/10.1093/hmg/ddv350 (2015).
Sander, J. D. & Joung, J. K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32, 347–355. https://doi.org/10.1038/nbt.2842 (2014).
Wang, Y. et al. Single-cell transcriptome analysis reveals differential nutrient absorption functions in human intestine. J Exp Med https://doi.org/10.1084/jem.20191130 (2020).
Heizer, W. D. Normal and abnormal intestinal absorption by humans. Environ Health Perspect 33, 101–106. https://doi.org/10.1289/ehp.7933101 (1979).
Walker, A. A. & Cole, C. R. Ultra-short bowel syndrome during infancy: Improving outcomes and novel therapies. Curr Opin Pediatr 31, 177–181. https://doi.org/10.1097/MOP.0000000000000738 (2019).
Vujasinovic, M. et al. Pancreatic exocrine insufficiency after bariatric surgery. Nutrients https://doi.org/10.3390/nu9111241 (2017).
Yen, C. L. et al. Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nat Med 15, 442–446. https://doi.org/10.1038/nm.1937 (2009).
Young, S. G. et al. A genetic model for absent chylomicron formation: mice producing apolipoprotein B in the liver, but not in the intestine. J Clin Invest 96, 2932–2946. https://doi.org/10.1172/JCI118365 (1995).
Chang, E. B. & Martinez-Guryn, K. Small intestinal microbiota: the neglected stepchild needed for fat digestion and absorption. Gut Microbes 10, 235–240. https://doi.org/10.1080/19490976.2018.1502539 (2019).
Bushyhead, D. & Quigley, E. M. M. Small intestinal bacterial overgrowth-pathophysiology and its implications for definition and management. Gastroenterology 163, 593–607. https://doi.org/10.1053/j.gastro.2022.04.002 (2022).
Li, M. et al. CircRNA Lrp6 promotes cementoblast differentiation via miR-145a-5p/Zeb2 axis. J Periodontal Res 56, 1200–1212. https://doi.org/10.1111/jre.12933 (2021).
Zhai, Z. et al. High glucose inhibits osteogenic differentiation of bone marrow mesenchymal stem cells via regulating miR-493–5p/ZEB2 signalling. J Biochem 167, 613–621. https://doi.org/10.1093/jb/mvaa011 (2020).
Tani, S. et al. Stem cell-based modeling and single-cell multiomics reveal gene-regulatory mechanisms underlying human skeletal development. Cell Rep 42, 112276. https://doi.org/10.1016/j.celrep.2023.112276 (2023).
Umeda, S. et al. D-amino acids ameliorate experimental colitis and cholangitis by inhibiting growth of proteobacteria: Potential therapeutic role in inflammatory bowel disease. Cell Mol Gastroenterol Hepatol 16, 1011–1031. https://doi.org/10.1016/j.jcmgh.2023.08.002 (2023).
Carvalho, F. A. et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 12, 139–152. https://doi.org/10.1016/j.chom.2012.07.004 (2012).
Florio, M. et al. Disentangling the nutrition-microbiota liaison in inflammatory bowel disease. Mol Aspects Med 102, 101349. https://doi.org/10.1016/j.mam.2025.101349 (2025).
Chantakhow, S. et al. Alterations of gut bacteria in hirschsprung disease and hirschsprung-associated enterocolitis. Microorganisms https://doi.org/10.3390/microorganisms9112241 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Chen, T. et al. The genome sequence archive family: Toward explosive data growth and diverse data types. Genom Proteom Bioinf 19, 578–583. https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (82400215) and the Research Foundation of Capital Institute of Pediatrics (JCYJ-2023-04) to YG. ZZ was supported by Beijing Municipal Administration of Hospitals Incubating Program (PX2022054). QL was supported by the Beijing Municipal Administration of Hospitals Incubating Program (PX2020054). QJ was supported by the National Natural Science Foundation of China (82370522, 82070532), the Research Unit of Minimally Invasive Pediatric Surgery on Diagnosis and Treatment, Chinese Academy of Medical Sciences (2021RU015), Beijing Finance Bureau (CIP2024-0040) and the Research Foundation of Capital Institute of Pediatrics (CXYJ-2021-05).
Author information
Authors and Affiliations
Contributions
Yangyang Ge: Formal analysis, Investigation, Methodology, Validation, Visualization,Writing – original draft. Lingya Liu: Methodology, Validation, Writing – original draft. Lihua Wu: Methodology, Validation, Writing – original draft. Xiaofan Liu: Investigation, Methodology, Validation. Yingao Hao: Investigation, Validation. Shixu Wang: Investigation, Validation. Yi Xiong: Software. Zi Yang: Investigation. Zhen Zhang: Funding acquisition, Resources. Qi Li: Funding acquisition, Resources. Bo Li: Formal analysis, Writing – review & editing. Jianxin Wu: Formal analysis, Writing – review & editing. Guangxu Ren: Formal analysis, Writing – review & editing. Qian Jiang: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ge, Y., Liu, L., Wu, L. et al. Impaired nutrient absorption, reduced bone mass and alterations in the gut microbiome contribute to postnatal growth retardation in a mouse model of MWS. Sci Rep 15, 30890 (2025). https://doi.org/10.1038/s41598-025-16542-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16542-z
