
Abstract
Ciliopathies, caused by defective cilia biogenesis or function, comprise a genetically and clinically diverse group of diseases. Primary cilia play pivotal roles in the regulation of a multitude of signalling pathways during development and tissue homeostasis. Cilia assembly, maintenance and signalling depend on intraflagellar transport (IFT). Tubby-like protein 3 (TULP3) functions as an adapter protein for the ciliary trafficking of diverse membrane cargos via an interaction with the IFT-A complex. Recently, we and others have shown that individuals carrying pathogenic TULP3 variants suffer from progressive liver, kidney and heart disease. In line with these findings, adult Tulp3 knockout zebrafish displayed liver fibrosis and kidney cyst phenotypes. In the present study, we analysed the functional consequences of Tulp3 deficiency during zebrafish embryogenesis. Tulp3 deficiency resulted in well-known ciliopathy-associated phenotypes including pronephric cysts, body curvature and altered left-right asymmetry. Our analysis of urotensin 2-related peptide (Urp) signalling, which is required for proper spine morphogenesis, revealed reduced expression of urp1 in Tulp3 knockout embryos. We also observed scoliosis in a significant number of adult Tulp3 knockout zebrafish. Analysis of ciliogenesis revealed a reduced cilia number and ciliary length in Tulp3 deficient embryos. In addition, Tulp3 deficiency resulted in upregulation of cilia-dependent profibrotic Wnt and Jak/Stat signalling components. Furthermore, we demonstrate that loss of Tulp3 causes upregulation of genes related to liver fibrosis. In conclusion, our data highlights a role of Tulp3 in proper cilia formation and function to maintain healthy tissue architecture during zebrafish embryogenesis, and provides further insight into the spectrum of cilia-related phenotypes in adult zebrafish depleted for Tulp3 functions.
Similar content being viewed by others


Introduction
Primary cilia, microtubule-based sensory organelles protruding from the apical surface of almost all vertebrate cells, play key roles in cell signal transduction and thereby in development and tissue homeostasis1,2,3,4. Defects in the biogenesis, maintenance or function of cilia lead to a large group of inherited diseases, collectively termed ciliopathies5,6,7. Ciliopathies are characterized by the presence of renal cysts, but can also manifest in other organs including the brain, liver, pancreas, heart and eye. Frequently, respective clinical management is challenged by diagnostic ambiguity, variable disease expression and incomplete penetrance8,9,10. Therefore, disease classification highly depends on a profound understanding of the molecular mechanisms that are affected. Furthermore, animal models provide us with the opportunity to follow disease progression from embryogenesis to adulthood, and thereby offer essential information in disease development11. Over the last two decades the ciliary transition zone (TZ) has gained specific interest because many disease-causing genes have been localized to this evolutionary conserved subdomain at the proximal end of the ciliary axoneme6,12. The ciliary TZ regulates the membrane specific distribution of certain phospholipids, and is thereby attributed a diffusion barrier function13,14. In vitro and in vivo models showed that Tubby-like protein 3 (TULP3) acts as a critical adapter protein for the ciliary trafficking of transmembrane proteins (e.g., polycystins, fibrocystin and a subset of G protein-coupled receptors (GPCRs)), and membrane-associated proteins (e.g., ADP-ribosylation factor-like protein 13B (ARL13B) and inositol polyphosphate-5-phosphatase E (INPP5E))15,16,17,18,19,20. TULP3-dependent transport of proteins to the ciliary membrane requires its binding to the intraflagellar transport protein complex A (IFT-A) and phosphoinositide 4,5-bisphosphate (PI(4,5)P2)16,18,21. While PI(4,5)P2 is restricted to the ciliary base, PI(4)P localizes to the ciliary axoneme22,23. Loss of the Joubert syndrome-associated INPP5E leads to mislocalization of PI(4,5)P2 along the cilium. Subsequently, the disruption of the ciliary barrier function results in accumulation of TULP3 and its binding partner GPR161 to the ciliary membrane24,25. Homozygous loss of TULP3 in mice causes defective neural tube closure, polydactyly and eventually results in embryonic lethality26. Furthermore, Tulp3 knockout mice display an expansion in Hedgehog (HH) signalling related gene expression revealing that TULP3 functions as a negative regulator of the HH signalling pathway27,28,29. Mice carrying a hypomorph missense mutation in Tulp3 develop late embryonic kidney cysts, skeletal deformities and neural patterning defects17. While cilia formation and morphology in these mice are not affected, the ciliary axoneme transport of Polycystin-2 and ARL13B is reduced. In individuals affected by progressive degenerative liver fibrosis, fibrocystic kidney disease and hypertrophic cardiomyopathy, we and others have previously identified bi-allelic deleterious variants in TULP330,31. Ciliary trafficking of TULP3 cargo proteins was significantly reduced in TULP3 patient derived cells. While previous reports demonstrated a negative regulatory function of TULP3 on HH signalling, in a patient carrying a hypomorph mutation in TULP3 we found increased wingless/integrated (WNT) and Janus kinase/signal transduction and transcription activation (JAK/STAT) signalling27,28,30. Notably, recent research indicates that dysfunctional cilia act as important mediators of fibrosis in various tissues and organs via upregulation of distinct signalling pathways (HH, WNT, TGFβ, NOTCH and JAK/STAT signalling)32,33,34,35,36,37,38,39.
To study TULP3 in vivo functions in greater detail, we here used the zebrafish as a vertebrate model organism. We show that the knockdown of Tulp3 resulted in well-known ciliopathy-related phenotypes as well as defective ciliogenesis during zebrafish embryogenesis40. We could further validate these embryonic phenotypes in a previously generated CRISPR/Cas9-induced maternal-zygotic (MZ) tulp3 knockout zebrafish line30. We have recently shown that homozygous tulp3 knockout zebrafish developed into adulthood, but in this phase developed liver fibrosis and kidney cysts, a phenomenon similar to that observed in human patient phenotypes30. We now additionally documented spinal deformity in adult tulp3 mutant zebrafish. Our analysis in MZtulp3 embryos revealed no obvious defects in the formation of the cilia-dependent Reissner fiber (RF), a structure that is implicated in proper body axis morphogenesis in zebrafish41,42. However, we identified dysregulation of RF-induced expression of urotensin 2-related peptide 1 (urp1) in ventral cerebrospinal fluid contacting neurons (CSF-cNs) in MZtulp3 embryos, encoding for a neuropeptide that is involved in zebrafish axis straightness42,44,45,46,47,48,49. Our expression analyses of various cilia-related signalling pathway components revealed upregulation of profibrotic Wnt and Jak/Stat signalling, while Hh signalling was unaffected. Furthermore, in Tulp3 depleted embryos we observed upregulation of genes related to liver fibrosis50. Altogether, we here show that the loss of Tulp3 causes defects in ciliary function during early zebrafish development that eventually lead to liver fibrosis, cystic kidney disease and scoliosis in adult animals.
Results
Knockdown of Tulp3 results in ciliopathy-associated phenotypes during zebrafish embryogenesis
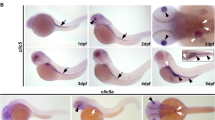
We used zebrafish as a vertebrate model organism to study a potential ciliary role of Tulp3 during embryonic development. While tulp3 expression by whole-mount in situ hybridization (WISH) was not present, our previous analysis by semi-quantitative RT-PCR did identify tulp3 expression during zebrafish embryogenesis30. We performed a Morpholino (MO)-based knockdown approach by using a translation-blocking MO (TB-MO) and two different splicing-blocking MOs (SB-MO) targeting the initiation codon of zebrafish tulp3 mRNA and different exon-intron boundaries of zebrafish tulp3 pre-mRNA, respectively (Fig. 1A-C). Embryos injected with either TB-MO tulp3, SB1-MO tulp3 or SB2-MO tulp3 revealed well-known ciliopathy-associated phenotypes such as pronephric cyst formation, otolith deposition defects, varying degrees of ventral body curvature and defective left-right (LR) asymmetry (analysed by the position of the heart looping) at 2 days post-fertilization (dpf) (Fig. 1D-O). None of these phenotypes were observed in embryos injected with a Control-MO. Co-injection of human TULP3 mRNA and either TB-MO tulp3, SB1-MO tulp3 or SB2-MO tulp3 significantly prevented the observed phenotypes of Tulp3 morphant embryos, thus confirming MO specificity. Taken together, these results indicate a specific function for Tulp3 in cilia formation and/or function in zebrafish embryogenesis.

Tulp3 knockdown analyses of cilia-related phenotypes during zebrafish embryogenesis. (A) Exon-intron structure (drawn to scale) of tulp3 in zebrafish (Ensembl Transcript ID: ENSDART00000093236.6). Translation start codon (ATG), termination codon (TGA), TB-MO tulp3, SB1-MO tulp3 and SB2-MO tulp3 are indicated. Black and blue half arrows indicate primer pairs used for analysis of SB1-MO tulp3 and SB2-MO tulp3 efficiency, respectively. (B, C) Expression analysis of tulp3 using semi-quantitative RT-PCR on cDNA of Co-MO (1ng) or SB1-MO tulp3 (0,5 or 1ng) injected embryos (B) and Co-MO (2ng) or SB2-MO tulp3 (2ng) injected embryos, respectively (C); while an exon exclusion is not detectable upon injection of SB1-MO tulp3 or SB2-MO tulp3, respectively, the highly reduced wildtype PCR products compared to the control indicate an (partial) intron insertion through cryptic splice site activation that might not be detectable by utilized PCR conditions. Black arrows point to tulp3 and ef1α PCR products, respectively; Primer dimer (PD). H2O served as negative control and ef1α as loading control; dividing lines in (B) indicate different contrast from different parts of the same gel image. (D-G) Bright-field images of zebrafish embryos at 2dpf injected with Co-MO (2ng) (D) or TB-MO tulp3 (2ng) (E-G). In comparison to Co-MO injected embryos, injection of TB-MO tulp3 leads to different degrees of ventral body curvature. (H–K) Knockdown of Tulp3 leads to pronephric cyst formation (white stars in (J)) and otolith deposition defects (white arrows in (K)) at 2dpf as shown in a dorsal view with anterior to the left of a TB-MO tulp3 (2ng) injected li1Tg embryo (J), and an embryo shown in a bright-field image (K), respectively, in comparison to Co-MO (2ng) injected embryos (H, I); expression of EGFP fluorescence labels glomerulus (G), neck (N) and proximal convoluted tubule (PCT). (L) Quantification of pronephric cyst formation in 2dpf zebrafish embryos injected with Co-MO (2ng) (4 independent experiments; n = 31, n = 63, n = 52 and n = 82 analysed embryos, respectively), TB-MO tulp3 (2ng) (n = 35, n = 79, n = 57 and n = 81), TB-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 54, n = 71, n = 57 and n = 73) or Co-MO (0,5 ng) (3 independent experiments; n = 46, n = 46 and n = 47), SB1-MO tulp3 (0,5 ng) (n = 41, n = 44 and n = 46), SB1-MO tulp3 (0,5 ng) + HTULP3 mRNA (5pg) (n = 46, n = 46 and n = 46) or Co-MO (2ng) (3 independent experiments; n = 42, n = 39 and n = 37), SB2-MO tulp3 (2ng) (n = 31, n = 37 and n = 38), SB2-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 41, n = 41 and n = 42). (M) Quantification of otolith deposition defects in 2dpf zebrafish embryos injected with Co-MO (2ng) (4 independent experiments; n = 42, n = 70, n = 50 and n = 82 analysed embryos, respectively), TB-MO tulp3 (2ng) (n = 44, n = 79, n = 71 and n = 81), TB-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 56, n = 71, n = 56 and n = 78). (N) Quantification of ventral body curvature in 2dpf zebrafish embryos injected with Co-MO (2ng) (4 independent experiments; n = 41, n = 70, n = 50 and n = 82 analysed embryos, respectively), TB-MO tulp3 (2ng) (n = 40, n = 80, n = 72 and n = 84), TB-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 57, n = 71, n = 56 and n = 79) or Co-MO (0,5ng) (3 independent experiments; n = 46, n = 46 and n = 46), SB1-MO tulp3 (0,5ng) (n = 43, n = 46 and n = 46), SB1-MO tulp3 (0,5ng) + HTULP3 mRNA (5pg) (n = 46, n = 46 and n = 46) or Co-MO (2ng) (3 independent experiments; n = 42, n = 39 and n = 34), SB2-MO tulp3 (2ng) (n = 42, n = 40 and n = 36), SB2-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 38, n = 40 and n = 36). (O) Quantification of altered heart looping in 2dpf zebrafish embryos injected with Co-MO (2ng) (3 independent experiments; n = 41, n = 70 and n = 50 analysed embryos, respectively), TB-MO tulp3 (2ng) (n = 43, n = 79 and n = 71), TB-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 56, n = 70 and n = 55) or Co-MO (0,5ng) (3 independent experiments; n = 46, n = 46 and n = 46), SB1-MO tulp3 (0,5ng) (n = 43, n = 35 and n = 46), SB1-MO tulp3 (0,5ng) + HTULP3 mRNA (5pg) (n = 46, n = 46 and n = 46) or Co-MO (2ng) (3 independent experiments; n = 42, n = 39 and n = 34), SB2-MO tulp3 (2ng) (n = 41, n = 39 and n = 40), SB2-MO tulp3 (2ng) + HTULP3 mRNA (5pg) (n = 39, n = 40 and n = 36); total number of embryos used for analyses are shown above respective bar. Unprocessed gel images (B and C, respectively) are presented in Suppl. Figure 7.
Tulp3 knockout zebrafish present with ciliopathy-associated phenotypes during embryogenesis and adulthood
We previously analysed and described a CRISPR/Cas9-induced tulp3 zebrafish knockout (tulp3uf3/uf3) presenting with severe liver and kidney phenotypes in adulthood30. We now focused on the effects of Tulp3 depletion on potential ciliopathy-associated phenotypes during zebrafish embryogenesis. Our analysis identified statistically significant pronephric cyst formation, defective LR-asymmetry (analysed by the position of the pancreas) and varying degrees of ventral body curvature in MZ tulp3 knockout embryos at 2dpf; but we did not observe a statistical difference in otolith deposition (Fig. 2A-L). Injection of human TULP3 mRNA into one-cell stage MZtulp3 embryos significantly prevented the observed phenotypes, confirming Tulp3 knockout specificity. Recent studies in zebrafish revealed that proper body axis morphogenesis relies on cilia-dependent formation of the RF, an acellular and filamentous structure that is present in the CSF42,43. In addition, Urp1 and Urp2, neuropeptides that are expressed in CSF-cNs, play a crucial role in signalling downstream of the RF in zebrafish43,44,45,46,47,48,49. Hence, we performed double whole-mount immunostainings on 2dpf old MZtulp3 embryos presenting with either a straight body axis or with a curvature phenotype, and respective control embryos using antibodies for RF and for the ciliary marker acetylated Tubulin. Our results revealed no obvious defects in RF formation in MZtulp3 embryos compared to the control at 2dpf (Fig. 3A). However, expression analyses of urp genes by WISH and quantitative real-time PCR (qPCR) studies revealed significantly reduced expression of urp1 in MZtulp3 embryos compared to the control at 28 h post-fertilization (hpf) and 2dpf, respectively, while urp2 expression was not significantly affected (Fig. 3B, C). In addition, analyses of adult tulp3uf3/uf3 zebrafish revealed ciliopathy-related spinal deformity compared to the respective heterozygous tulp3 knockout or control siblings (Fig. 3D, E). In summary, these results indicate a role of Tulp3 in ciliogenesis, and most Tulp3 knockout data is in line with our Tulp3 knockdown data.

Tulp3 knockout analyses of cilia-related phenotypes during zebrafish embryogenesis. (A-D) Bright-field images of MZtulp3 embryos (B-D) and respective control embryo (A) at 2dpf. In comparison to the control embryos, MZtulp3 embryos display different degrees of ventral body curvature. (E–K) Knockout of Tulp3 leads to pronephric cyst formation (white star in (F)) and altered positioning of the exocrine pancreas (white arrow in (H)) at 2dpf as shown in a dorsal view with anterior to the left of a MZtulp3; li1Tg embryo, respectively, in comparison to control embryos (E, G); expression of EGFP fluorescence labels glomerulus (G), neck (N), proximal convoluted tubule (PCT) and exocrine pancreas. (I) Quantification of pronephric cyst formation in control embryos (3 independent experiments; n = 39, n = 59 and n = 59 analysed embryos, respectively), MZtulp3 embryos (n = 59, n = 59 and n = 59) and MZtulp3 embryos + HTULP3 (n = 59, n = 59 and n = 59) at 2dpf. (J) Quantification of altered positioning of the exocrine pancreas in control embryos (3 independent experiments; n = 39, n = 59 and n = 59 analysed embryos, respectively), MZtulp3 embryos (n = 59, n = 59 and n = 59) and MZtulp3 embryos + HTULP3 (n = 59, n = 59 and n = 59) at 2dpf. (K) Quantification of otolith deposition defects in control embryos (3 independent experiments; n = 101, n = 83 and n = 83 analysed embryos, respectively) and MZtulp3 embryos (n = 76, n = 92 and n = 112) at 2dpf. (L) Quantification of ventral body curvature in control embryos (3 independent experiments; n = 39, n = 59 and n = 59 analysed embryos, respectively), MZtulp3 embryos (n = 59, n = 59 and n = 59) and MZtulp3 embryos + HTULP3 (n = 59, n = 59 and n = 59) at 2dpf; total number of embryos used for analyses are shown above respective bar.

Tulp3 knockout results in decrease of urp1 expression and scoliosis during zebrafish development. (A) Representative confocal images of MZtulp3 embryos (without curvature phenotype (normal), mild and severe ventral body curvature) and respective control embryos at 2dpf immunostained with anti-RF and anti-acetylated Tubulin as a ciliary marker. Loss of Tulp3 results in no obvious defects in RF formation compared to the control. Numbers represent embryos displaying RF disorganization and embryos that have been analysed in total from 3 independent experiments (control: n = 33, n = 42 and n = 26; MZtulp3 (normal): n = 11, n = 38 and n = 24; MZtulp3 (mild): n = 30, n = 28 and n = 36; MZtulp3 (severe): n = 22, n = 28 and n = 25). Scale bar: 10 μm. (B) WISH analysis reveals reduced urp1 expression in ventral CSF-cNs (black arrowheads) of MZtulp3 embryos (presenting with a ventral curvature phenotype) compared to the control at 28hpf. Expression levels of urp2 (black arrow) and pkd2l1 (black arrowheads; serves as a control as its expression in CSF-cNs has been shown to be unaffected in ciliary mutants43,48) are comparable between MZtulp3 and control embryos at 28hpf. (C) qPCR analysis reveals unaltered expression of urp2 and pkd2l1 while urp1 expression was significantly reduced in MZtulp3 embryos compared to the respective control at 2dpf. (D, E) Representative images (lateral and dorsal views) and quantification of adult (18 months) tulp3 mutant zebrafish displaying scoliosis phenotypes (black arrows) in comparison to the respective control analysed from 3 independent breedings (control: n = 15, n = 13 and n = 0; heterozygous tulp3 mutant: n = 29, n = 6 and n = 8; homozygous tulp3: n = 15, n = 40 and n = 7); total number of adult fish used for analyses are shown above respective bar.
Tulp3 deficiency results in defective ciliogenesis and disrupted cilia-related signalling
Previous data demonstrated reduced ciliary length in various Tulp3 deficient or depleted cell types20. In order to analyse cilia formation in Tulp3 morphant and mutant zebrafish embryos we performed acetylated Tubulin immunostaining to visualize the cilia in the Kupffer’s vesicle (the organ of laterality in zebrafish) at the stage of 8 somites. Quantification revealed statistically significant reduced ciliary length in Tulp3 deficient and depleted embryos compared to the respective controls (Fig. 4A-H). In addition, the number of cilia was significantly reduced in MZtulp3 embryos compared to the control (Fig. 4I). Transmission electron microscopy (TEM) studies of motile cilia in the pronephric tubules revealed normal ciliary architecture with nine outer doublet microtubules surrounding a central pair of singlet microtubules in control as well as MZtulp3 embryos (Fig. 4J-L). We also performed qPCR and WISH analyses to analyse cilia-dependent signalling pathways in Tulp3 deficient and depleted embryos at 1dpf. While Hh signalling was unaffected, we observed an upregulation of Wnt and Jak/Stat signalling pathway components in Tulp3 morphant and mutant embryos compared to the respective controls (Fig. 5A-C). In addition, we performed RNA sequencing (RNA-Seq) analyses on MZtulp3 embryos in comparison to the control at 2dpf. Our results revealed upregulation of Wnt signalling associated pathways in MZtulp3 embryos, while Hh signalling was unaffected (Fig. 5D, Suppl. Figures 1 and 2).

Tulp3 deficiency results in defective cilia formation and function in zebrafish. (A-C) Representative confocal images of the Kupffer’s vesicle of Co-MO (2ng) (A), TB-MO tulp3 (2ng) (B) or SB1-MO tulp3 (0,5ng) (C) injected embryos at 8 somites immunostained with anti-acetylated Tubulin as a ciliary marker. Scale bar: 10 μm. (D, E) Quantification of the ciliary length (D) and cilia number (E) in the Kupffer’s vesicle of Co-MO (3 independent experiments; n = 9, n = 7 and n = 7 analysed embryos, respectively), TB-MO tulp3 (n = 6, n = 4 and n = 7) or SB1-MO tulp3 (n = 4, n = 6 and n = 8) injected embryos at 8 somites; total number of embryos used for analyses are shown above respective bar. (F, G) Representative confocal images of the Kupffer’s vesicle of MZtulp3 embryo (G) and respective control embryo (F) at 8 somites immunostained with anti-acetylated Tubulin. Scale bar: 10 μm. (H, I) Quantification of the ciliary length (H) and cilia number (I) in the Kupffer’s vesicle of control embryos (3 independent experiments; n = 7, n = 7 and n = 8 analysed embryos, respectively) and MZtulp3 embryos (n = 6, n = 4 and n = 4) at 8 somites; total number of embryos used for analyses are shown above respective bar. (J-L) TEM of pronephric tubule microvilli and motile cilia of control embryos (J) and MZtulp3 (K, L) embryos (with severe ventral body curvature) at 5dpf. Scale bars: 500 nm (J, L); 1 μm (K).

Tulp3 deficiency results in the upregulation of cilia-dependent signalling pathway components in zebrafish. (A) qPCR analysis reveals unaltered expression of Hh signalling components (gli1, ptc1) and Wnt signalling component axin2 while Wnt signalling components wnt8a and lef1 were upregulated upon TB/SB1-MO tulp3 mediated knockdown compared to the respective control at 1dpf. (B) qPCR analysis reveals unaltered expression of Hh signalling components (gli1, ptc1) and Wnt signalling component axin2 while Wnt signalling components wnt8a and lef1 or Jak/Stat signalling components jak1 and stat1b were upregulated in MZtulp3 embryos compared to the respective control at 1dpf. (C) WISH analyses indicate unaltered expression of Hh signalling component ptc1 and Wnt signalling component axin2 while Wnt signalling components wnt8a and lef1 were upregulated in MZtulp3 embryos compared to the respective control at 1dpf. (D) Heatmap of Wnt signalling pathway associated gene ontology (GO)-terms (biological process, BP) of MZtulp3 embryos compared to control embryos at 2dpf. Columns represent up- or downregulated processes in MZtulp3 mutant embryos compared to controls.
Loss of Tulp3 leads to upregulation of genes related to liver fibrosis
Our previous data demonstrated that tulp3uf3/uf3 knockout zebrafish developed liver fibrosis during adulthood30. We now aimed to study whether signs of liver fibrosis are already detectable during zebrafish embryogenesis upon loss of Tulp3. Therefore, we performed qPCR and WISH to analyse the expression levels of genes related to liver fibrosis, inflammatory/damage and liver function in MZtulp3 embryos compared to the control at 4dpf50. Our results revealed significant upregulation of the fibrotic (hand2 and acta2), inflammatory/damage (tgfß and sdf1a) and function (gc and serpina1) related genes while expression of liver fibrosis-related gene col1α1 was not affected (Fig. 6A, B). In addition, our RNA-Seq analyses indicated an increase in fibrosis-related extracellular matrix (ECM) organization and upregulation of Tgfβ-signalling pathway in MZtulp3 mutant embryos compared to the control at 2dpf (Fig. 6C, D and Suppl. Figures 3 and 4).

Loss of Tulp3 results in the upregulation of genes related to fibrosis during zebrafish embryogenesis. (A) qPCR analysis reveals unaltered expression of col1α1 while genes related to liver-fibrosis (acta2, hand2), inflammatory/damage (tgfβ, sdf1a) and liver function (gc, serpina1) were significantly upregulated in MZtulp3 embryos compared to the respective control at 4dpf. (B) WISH analyses indicate upregulated expression of gc and serpina1 in the liver (black arrow) in MZtulp3 embryos compared to the respective control at 4dpf (for each condition a lateral and dorsal view is shown). (C, D) Heatmaps of ECM organization (C) and Tgfβ signalling pathway (D) indicate an increase in related biological processes (GOBP) in MZtulp3 mutant compared to control embryos at 2dpf. Shown are respective up- or downregulated BPs.
Discussion
In humans, bi-allelic deleterious variants in TULP3 are the cause of a progressive degenerative disease affecting the liver, kidney and heart tissue30,31. Disease onset, presenting with first signs of liver disease, mostly varies from early childhood to the mid-twenties. The progressive course of TULP3-dependent fibrocystic liver and kidney disease emphasizes that, for the identification of potential targets for therapeutic actions to circumvent or delay organ dysfunction, a detailed understanding of the disease mechanism is very important.
Previous mouse models showed that the loss of TULP3 function results in neural tube closure defects, craniofacial abnormalities, polydactyly and embryonic lethality26,27,28,29. In contrast to Tulp3 knockout mice, zygotic tulp3 knockout zebrafish survive into adulthood even though they developed fibrocystic liver and kidney disease phenotypes30. In addition, we now document a scoliosis phenotype in zygotic adult tulp3 mutants. To further study the progressive character of the disease, we have examined Tulp3 loss of function phenotypes during embryonic stages in zebrafish by using morpholino (MO)-mediated knockdown and CRISPR/Cas9-mediated knockout approaches. Notably, inconsistencies of phenotypes observed either by MO-mediated knockdown or gene-editing-based knockout in zebrafish have been documented by recent reports51. A subsequent study identified the activation of a genetic compensation response in mutants but not in morphants52. Following up on these observations, a further study reported that genetic compensation is triggered by the generation of unstable mRNAs53. Of note, compensation mechanisms and their investigation are likely to be complex and far from being completely understood54. Hence, it is widely accepted that gene functional analyses in zebrafish are ideally based on two or more different approaches, complementing knockout strategies with additional genetic tools55. We strongly agree with this view and therefore used the MO-mediated knockdown as well as knockout approach to study Tulp3 function in zebrafish embryogenesis. By the majority (except otolith deposition defects), we observe similar phenotypes with differing penetrance/severity between the morphant and mutant data, confirming specificity of our experimental data. Our analyses revealed less severe phenotypes in the MZtulp3 mutant embryos compared to tulp3 morphants. Therefore, we addressed a possible genetic compensation in MZtulp3 mutant embryos by analysing gene expression of tulp3 related family members and known interactors by RNA-Seq analysis. However, our results revealed no significant upregulation of selected compensatory candidate genes (Suppl. Figure 5). We cannot exclude that other possible genetic compensation mechanisms and/or transcriptional adaptation might be active in MZtulp3 mutants and thus is a matter of future investigations. In addition, we cannot exclude the possibility that the MOs induce an off-target effect that enhances the on-target phenotypes.
We here report that Tulp3 deficiency results in various well-known cilia-associated phenotypes. Previous data revealed a reduction in ciliary length in various Tulp3 knockout cells while the percentage of cilia was normal20. In line with these findings, our results revealed a significant reduction in ciliary length in Tulp3 deficient embryos, but also showed that the number of cilia is significantly reduced in tulp3 knockout embryos. It is noteworthy that our TEM analyses identified an unaffected ciliary architecture in tulp3 knockout embryos. We also demonstrate that Tulp3 deficiency results in ventral body curvature defects, a phenotype that is linked to dysfunctional cilia. Recent studies identified the importance of cilia-driven cerebrospinal fluid (CSF) flow in transmitting adrenergic signals to CSF-contacting neurons that subsequently promote the synthesis and secretion of Urp1 and Urp2. Subsequently, Urp1/Urp2 bind to and activate their receptor Uts2r3 on dorsal somitic muscles to promote axis straightening. Thus, signals from CSF finally direct dorsal muscle fiber contraction and control proper body axis straightening during early development42,44,45,48. Our results revealed significant downregulation of urp1 in MZtulp3. How Tulp3 deficiency affects urp1 expression still remains unknown. While we detect perturbation in urp1 expression and axial curvature phenotypes, our analyses of immunostained embryos followed by confocal microscopy revealed no obvious RF formation defects. Despite the formation of a continuous RF, it is still possible that the CSF signalling is affected in MZtulp3 embryos. Notably, a recent study demonstrated a scoliotic model with motile cilia and CSF flow defects but intact RF formation and unaffected urp gene expression showing that the curvature onset occurs independently of RF loss in this model56. In addition, neuro-inflammation has been described in a small subset of zebrafish IS models57,58. Conclusively, recent studies identified a variety of factors that lead to scoliosis but several aspects in respect to the regulatory mechanisms underlying spinal deformity still remain unknown and need to be addressed in future studies. Recent analyses of cilia-dependent signalling pathways in Tulp3 mutant mice identified TULP3 as a negative regulator of the HH signalling pathway27,28,29. In MZtulp3 mutant zebrafish embryos we did not detect defects in Hh target genes gli1 or ptc1 and RNA-Seq analyses showed no dysregulation in key target genes of Hh signalling. Notably, we observed a significant increase in the ciliary TZ protein encoding gene ttc23 in MZtulp3 mutant embryos that will need further validation. In addition, TTC23 has been described to participate in Hh signalling59. In Tulp3 morphant and MZtulp3 mutant embryos we found a significant increase in expression of profibrotic Wnt and Jak/Stat signalling components. Furthermore, our RNA-Seq analysis underlined the upregulation of Wnt associated signalling pathways in MZtulp3 mutant embryos. Notably, these in vivo results are in perfect line with our previous in vitro data of a TULP3 variant carrying individual that demonstrated unaltered HH signalling but an upregulation of WNT and JAK/STAT signalling components30. Thus, the contradictory results indicate that TULP3 has different functions in the context of cilia-related signalling pathways that have been analysed in human and the model organisms zebrafish and mouse. Dysregulation of WNT and JAK/STAT signalling have been linked to fibrosis of major organs and cystic kidney disease, phenotypes that we observed in adult homozygous tulp3 mutants30,60,61,62. We also examined if signs of liver fibrosis are already detectable in Tulp3 deficient embryos. Indeed, we observed that genes related to liver fibrosis are upregulated in MZtulp3 embryos, which is in line with our observation of liver fibrosis in adult homozygous tulp3 mutants30. Noteworthy, our RNA-Seq analyses further indicated an increase of various oncogenic and profibrotic cell processes, comparable to results that we previously reported in a TULP3-affected individual (Suppl. Figure 6)30.
Conclusively, we here present cilia-related phenotypes during zebrafish embryogenesis that are caused by deficiency of Tulp3 function. These results indicate a progressive disease character not only in humans but also in zebrafish. Therefore, our data demonstrates that the Tulp3 loss of function zebrafish model represents a suitable disease model to study progressive fibrocystic liver and kidney disease.
Materials and methods
Zebrafish lines and embryo maintenance
The fish used in this study were maintained at the Zebrafish Facility of the Medical Center of the University of Freiburg. All animal work has been conducted according to relevant national and international guidelines. The study was approved by the Institutional Animal Care of the Medical Center of the University of Freiburg and the Regional Council Freiburg (permit ID G-16/89). All methods were carried out in accordance with ARRIVE guidelines. Zebrafish were maintained and the embryos were staged as previously described63. The following strains were used: AB/TL wildtype (WT), li1Tg (https://zfin.org/ZDB-ALT-071127-1)64 and tulp3uf3/uf3; li1Tg (AB/TL) (https://zfin.org/ZDB-FISH-230405-2)30.
mRNA and morpholino (MO) injections
For synthesis of mRNA, full-length human Tulp3 was cloned into pCS2 + MxN, linearized with NsiI, and transcribed using SP6 mMessage mMachine Kit (Ambion). mRNA and Morpholino oligonucleotide (MO) injections were performed as described65. To attenuate possible off target effects, a p53 MO was co-injected 1.5-fold to the other MOs used66. The following translation/splicing-blocking (TB/SB) antisense MOs (Gene Tools) were used for zebrafish: TB-MO tulp3 5′-CTCTTCACCGTCTCCATGTCGAG-3′, SB1-MO tulp3 5′-TTGTTCTGTGTGTGTCTTACCGCGT-3′, SB2-MO tulp3 5’-TGTGCAGTTGGACTGACGTAGCATA-3’, TB-MO p53 5’-GCGCCATTGCTTTGCAAGAATTG-3’66 and a Standard Control (Co)-MO. The Co-MO is thought to have no target and only very little biological activity (https://www.gene-tools.com/custom_morpholinos_controls_endmodifications).
Reverse-transcription polymerase chain reaction (RT-PCR) analysis
Semiquantitative RT-PCR was performed to determine expression of zebrafish tulp3. Total RNA from entire 1dpf old zebrafish embryos was extracted with the RNeasy Kit (Qiagen), followed by complementary DNA (cDNA) synthesis with the ProtoScript First Strand cDNA Synthesis Kit (Promega). Analysis of zebrafish ef1α was used as a loading control. The following primers were used for PCR analysis: tulp3 (forward-1: 5ʹ-TCTGCTGGAGCAGAAGCAG-3ʹ, reverse-1: 5ʹ-GTGGATTTAGTGCTGGATGCAGAC-3ʹ; for analysis of SB1-MO tulp3 efficiency); tulp3 (forward-2: 5ʹ-CATCCAGCACTAAATCCACTAC-3ʹ, reverse-2: 5ʹ-GTAAGACTGTGTGTCATCGTTC-3ʹ; for analysis of SB2-MO tulp3 efficiency); ef1α (forward: 5ʹ-ATCTACAAATGCGGTGGAAT-3ʹ, reverse: 5ʹ-ATACCAGCCTCAAACTCACC-3ʹ).
Synthesis of antisense RNA and in situ analysis
Zebrafish urp1, urp2, pkd2l1 and lef1 probes were amplified from zebrafish cDNA with primers (urp1, forward: 5ʹ-ACATTCTGGCTGTGGTTTG-3ʹ, reverse: 5ʹ-TGTATGGGGAAAACAAAGG-3ʹ)67; (urp2, forward: 5ʹ-CGACGCGAGCATTAGATGAA-3ʹ, reverse: 5ʹ-TGTTGGTTTTCTTGGTTGACG-3ʹ); (pkd2l1, forward: 5ʹ-GTGACTGTTTCGATGTGTAC-3ʹ, reverse: 5ʹ-ACGATCTCACAGCCGATGAT-3ʹ); (lef1, forward: 5ʹ-CTGGACCCCACGCCACAGGAAT-3ʹ, reverse: 5ʹ-GGCCTGTAGCTGCTGTCTTTGCTT-3ʹ). Zebrafish gc and serpina1 probes were amplified from zebrafish cDNA with primers that have been used previously in quantitative real-time PCR analyses50. Amplification products were cloned into TOPO (Invitrogen), followed by sequence verification and linearization with corresponding restriction enzymes for synthesis of antisense RNA. We used already described pBS-ptc1, pCRII-axin2 and pGEMT-wnt8a for synthesis of antisense RNA68,69,70. Whole-mount in situ hybridization (WISH) analysis using Digoxigenin-labelled probes was performed as described using NBT (blue) (Roche) as substrate71.
Quantitative real‑time PCR (qPCR)
qPCR was performed as previously described72. For the expression analysis of urp1, urp2 and pkd2l1, total RNA was obtained from 30 maternal zygotic (MZ) tulp3 mutant embryos presenting with ventral body curvature or from respective control embryos at 2dpf using the RNeasy Kit (Qiagen). The following primers were used for qPCR analysis: ef1α (forward: 5ʹ-TGCCAACTTCAACGCTCAGGTC-3ʹ, reverse: 5ʹ-TCAGCAAACTTGCAGGCGATG-3ʹ); urp1 (forward: 5ʹ-ACATTCTGGCTGTGGTTTG-3ʹ, reverse: 5ʹ-GTCCGTCTTCAACCTCTGCTAC-3ʹ); urp2 (forward: 5ʹ-AGAGGAAACAGCAATGGACG-3ʹ, reverse: 5ʹ-TGTTGGTTTTCTTGGTTGACG-3ʹ); pkd2l1 (forward: 5ʹ-GTGACTGTTTCGATGTGTAC-3ʹ, reverse: 5ʹ-CTTGATAAAACCCTGCTCCG-3ʹ)43. For the analysis of Hh, Wnt and Jak/Stat signalling pathway components, total RNA was obtained from 30 Co-MO or TB/SB1-MO tulp3 injected zebrafish embryos and 30 unbiasedly pooled MZtulp3 mutant or from respective control embryos, all at 1dpf using the RNeasy Kit (Qiagen). The following primers were used for qPCR of analysis: ef1α (forward: 5ʹ-TGCCAACTTCAACGCTCAGGTC-3ʹ, reverse: 5ʹ-TCAGCAAACTTGCAGGCGATG-3ʹ); gli1 (forward: 5ʹ-TCAGACGTCCTCTCGCCTTA-3ʹ, reverse: 5ʹ-AGCTCATGTCTCCGATTGCC-3ʹ); ptc1 (forward: 5ʹ-GGGTCCTGAATGGACTGGTG-3ʹ, reverse: 5ʹ-CCGCTGGAGATACCTCAGGA-3ʹ); axin2 (forward: 5ʹ-ACCCTCGGACACTTCAAGGA-3ʹ, reverse: 5ʹ-GTGCAGTCATCCCAGACCTC-3ʹ); wnt8a (forward: 5ʹ-ATTCGTGGATGCGCTTGAGA-3ʹ, reverse: 5ʹ-TTACAGCCAAACGTCCAGCTT-3ʹ); lef1 (forward: 5ʹ-CAGACATTCCCAATTTCTATCC-3ʹ, reverse: 5ʹ-TGTGATGTGAGAACCAACC-3ʹ); jak1 (forward: 5ʹ-CCTGGAGGAGGGAAAGAGAC-3ʹ, reverse: 5ʹ-CAGGCTTTTGAAGTCGATCC-3ʹ); stat1b (forward: 5ʹ-CTCCAGGCACTTTCCTTCTG-3ʹ, reverse: 5ʹ-AATGGATCTTGGGTTCACCA-3ʹ). For the analysis of genes related to liver-fibrosis, total RNA was obtained from 30 MZtulp3 mutant or respective control embryos at 4dpf using the RNeasy Kit (Qiagen). For qPCR analysis we used the primers for ef1α and the primers that have been used previously50.
Immunostaining
Zebrafish whole-mount immunofluorescence (IF) was performed as previously described65. The following antibodies were used for IF: anti-acetylated αTubulin (clone 6-11B-1; Sigma Aldrich; 1:500) and anti-Reissner fiber (kind gift from Stéphane Gobron; 1:200). Cy3 (1:1000) and Alexa-488 (1:1000) labelled secondary antibodies were purchased from Jackson Immunoresearch and Molecular Probes (Invitrogen), respectively.
Microscopy and image acquisition
Embryos were analysed using a Leica MZ16F epifluorescent microscope. Images were obtained with a Leica DFC 450 C camera and processed with Leica Application Suite X (Version 1.4.5) (https://www.leica-microsystems.com/products/microscope-software/details/product/leica-las-x-ls/). Confocal images of whole-mount zebrafish immunostainings were generated with a Carl Zeiss LSM510 laser scanning microscope (ZEISS objectives: Achroplan NIR 40x/0.8 water-immersion). Confocal z-stacks were projected to one plane (maximum intensity projection). Confocal images of whole-mount zebrafish immunostainings were generated with a Carl Zeiss LSM510 laser scanning microscope. The transmission electron microscopy (TEM) procedure is described elsewhere73. All images were exported as TIFF files and imported into Adobe Photoshop Creative Suite 2 (Version 11) (https://www.adobe.com/de/products/photoshop.html) to arrange figures.
RNA sequencing and analysis
RNA sequencing was performed by Novogene. Total RNA was isolated from MZtulp3 (presenting with severe ventral body curvature) and respective control embryos at 2dpf using the RNeasy Kit (Qiagen). The raw RNA sequencing files were processed with the nf-core/rnaseq workflow (v3.18.0) including pre-processing with TrimGalore! to ensure sufficient read quality by removing adapters and reads in low-quality segment regions74. Subsequently, the reads were 2-pass aligned using the STAR aligner and quantified with RSEM. The Danio rerio genome GRCz10 was used as reference. Normalization and differential expression analysis was done with the R/Bioconductor package DESeq2 (v1.42.1)75,76,77. Genes were considered significant with an adjusted p-value (FDR corrected, according to Benjamini-Hochberg) < 0.05.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) of signalling pathways was performed as implemented in the R/Bioconductor package GAGE (Generally Applicable Gene-set Enrichment analysis), with signalling pathways from the Molecular Signatures Database (MSigDB)78,79. In particular the Gene ontology (GO) gene set Biological Processes (BP) (https://geneontology.org/docs/ontology-documentation/) and the MSigDB hallmark gene set collection were used80. Pathways were considered significant with an adjusted p-value (Benjamini-Hochberg) < 0.05. For better visualization, expression values of genes associated with specific GSEA terms were z-scaled.
Statistical analysis and quantification
For each quantification, the number of independent experiments and the number of embryos is specified in the respective figure legend. Total numbers of embryos used for analysis are indicated in the respective bar chart unless otherwise stated. Data were analysed by Student’s t-test (2-sided, unpaired); error bars represent the standard error of the mean (SEM). Measurement and quantification of ciliary length is described elsewhere81. qPCR data were analysed with GraphPad Prism (Version 10.5.0) (https://www.graphpad.com/updates/prism-10-5-0-release-notes) and one sample t-test; error bars represent the SEM.
Accession numbers
Corresponding GenBank accession number for human cDNA: Tulp3 (NM_003324.5) and zebrafish cDNA: tulp3 (XM_005164458.6).
Data availability
The RNA-Seq datasets generated and/or analysed during the current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE300126. All other datasets used and/or analysed during the current study are available from the corresponding authors on reasonable request.
Change history
12 January 2026
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-31559-0
References
Derderian, C., Canales, G. I. & Reiter, J. F. Seriously cilia: A tiny organelle illuminates evolution, disease, and intercellular communication. Dev. Cell. 58, 1333–1349. https://doi.org/10.1016/j.devcel.2023.06.013 (2023).
Mill, P., Christensen, S. T. & Pedersen, L. B. Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat. Rev. Genet. 24, 421–441. https://doi.org/10.1038/s41576-023-00587-9 (2023).
Gopalakrishnan, J. et al. Emerging principles of primary cilia dynamics in controlling tissue organization and function. EMBO J. 42, e113891. https://doi.org/10.15252/embj.2023113891 (2023).
Hilgendorf, K. I., Myers, B. R. & Reiter, J. F. Emerging mechanistic Understanding of cilia function in cellular signalling. Nat. Rev. Mol. Cell. Biol. https://doi.org/10.1038/s41580-023-00698-5 (2024).
Braun, D. A., Hildebrandt, F. & Ciliopathies Cold Spring Harb Perspect. Biol. 9 https://doi.org/10.1101/cshperspect.a028191 (2017).
Reiter, J. F. & Leroux, M. R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell. Biol. 18, 533–547. https://doi.org/10.1038/nrm.2017.60 (2017).
McConnachie, D. J., Stow, J. L. & Mallett, A. J. Ciliopathies and the kidney: A review. Am. J. Kidney Dis. 77, 410–419. https://doi.org/10.1053/j.ajkd.2020.08.012 (2021).
Bergmann, C. et al. Polycystic kidney disease. Nat. Rev. Dis. Primers. 4, 50. https://doi.org/10.1038/s41572-018-0047-y (2018).
Schrezenmeier, E., Budde, K. & Bergmann, C. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med 380, (2078). (2019) https://doi.org/10.1056/NEJMc1903250
Choi, M. et al. Interstitial nephritis: A change in diagnosis with Next-Generation sequencing. Kidney Int. Rep. 7, 1128–1130. https://doi.org/10.1016/j.ekir.2022.01.1061 (2022).
Quinlan, R. J., Tobin, J. L. & Beales, P. L. Modeling ciliopathies: primary cilia in development and disease. Curr. Top. Dev. Biol. 84, 249–310. https://doi.org/10.1016/S0070-2153(08)00605-4 (2008).
Park, K. & Leroux, M. R. Composition, organization and mechanisms of the transition zone, a gate for the cilium. EMBO Rep. 23, e55420. https://doi.org/10.15252/embr.202255420 (2022).
Garcia-Gonzalo, F. R. & Reiter, J. F. Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect. Biol. 9 https://doi.org/10.1101/cshperspect.a028134 (2017).
Goncalves, J. & Pelletier, L. The ciliary transition zone: finding the pieces and assembling the gate. Mol. Cells. 40, 243–253. https://doi.org/10.14348/molcells.2017.0054 (2017).
Mukhopadhyay, S. et al. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24, 2180–2193. https://doi.org/10.1101/gad.1966210 (2010).
Badgandi, H. B., Hwang, S. H., Shimada, I. S., Loriot, E. & Mukhopadhyay, S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell. Biol. 216, 743–760. https://doi.org/10.1083/jcb.201607095 (2017).
Legue, E. & Liem, K. F. Jr. Tulp3 is a ciliary trafficking gene that regulates polycystic kidney disease. Curr. Biol. 29, 803–812. https://doi.org/10.1016/j.cub.2019.01.054 (2019). e805.
Han, S. et al. TULP3 is required for localization of membrane-associated proteins ARL13B and INPP5E to primary cilia. Biochem. Biophys. Res. Commun. 509, 227–234. https://doi.org/10.1016/j.bbrc.2018.12.109 (2019).
Hwang, S. H. et al. Tulp3 regulates renal cystogenesis by trafficking of cystoproteins to cilia. Curr. Biol. 29, 790–802. https://doi.org/10.1016/j.cub.2019.01.047 (2019). e795.
Palicharla, V. R. et al. Interactions between TULP3 tubby domain and ARL13B amphipathic helix promote lipidated protein transport to cilia. Mol Biol Cell 34, ar18 (2023). https://doi.org/10.1091/mbc.E22-10-0473
Mukhopadhyay, S. et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic Hedgehog pathway via cAMP signaling. Cell 152, 210–223. https://doi.org/10.1016/j.cell.2012.12.026 (2013).
Nakatsu, F. A phosphoinositide code for primary cilia. Dev. Cell. 34, 379–380. https://doi.org/10.1016/j.devcel.2015.08.008 (2015).
Conduit, S. E. & Vanhaesebroeck, B. Phosphoinositide lipids in primary cilia biology. Biochem. J. 477, 3541–3565. https://doi.org/10.1042/BCJ20200277 (2020).
Chavez, M. et al. Modulation of ciliary phosphoinositide content regulates trafficking and Sonic Hedgehog signaling output. Dev. Cell. 34, 338–350. https://doi.org/10.1016/j.devcel.2015.06.016 (2015).
Garcia-Gonzalo, F. R. et al. Phosphoinositides regulate ciliary protein trafficking to modulate Hedgehog signaling. Dev. Cell. 34, 400–409. https://doi.org/10.1016/j.devcel.2015.08.001 (2015).
Ikeda, A., Ikeda, S., Gridley, T., Nishina, P. M. & Naggert, J. K. Neural tube defects and neuroepithelial cell death in Tulp3 knockout mice. Hum. Mol. Genet. 10, 1325–1334. https://doi.org/10.1093/hmg/10.12.1325 (2001).
Cameron, D. A., Pennimpede, T. & Petkovich, M. Tulp3 is a critical repressor of mouse Hedgehog signaling. Dev. Dyn. 238, 1140–1149. https://doi.org/10.1002/dvdy.21926 (2009).
Patterson, V. L. et al. Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic Hedgehog pathway. Hum. Mol. Genet. 18, 1719–1739. https://doi.org/10.1093/hmg/ddp075 (2009).
Norman, R. X. et al. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through Inhibition of Hedgehog signaling. Hum. Mol. Genet. 18, 1740–1754. https://doi.org/10.1093/hmg/ddp113 (2009).
Devane, J. et al. Progressive liver, kidney, and heart degeneration in children and adults affected by TULP3 mutations. Am. J. Hum. Genet. 109, 928–943. https://doi.org/10.1016/j.ajhg.2022.03.015 (2022).
Jafari Khamirani, H. et al. A pathogenic variant of TULP3 causes renal and hepatic fibrocystic disease. Front. Genet. 13, 1021037. https://doi.org/10.3389/fgene.2022.1021037 (2022).
Saito, S., Tampe, B., Muller, G. A. & Zeisberg, M. Primary cilia modulate balance of canonical and non-canonical Wnt signaling responses in the injured kidney. Fibrogenesis Tissue Repair. 8, 6. https://doi.org/10.1186/s13069-015-0024-y (2015).
Edeling, M., Ragi, G., Huang, S., Pavenstadt, H. & Susztak, K. Developmental signalling pathways in renal fibrosis: the roles of notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 12, 426–439. https://doi.org/10.1038/nrneph.2016.54 (2016).
Zhou, D. et al. Tubule-Derived Wnts are required for fibroblast activation and kidney fibrosis. J. Am. Soc. Nephrol. 28, 2322–2336. https://doi.org/10.1681/ASN.2016080902 (2017).
Tang, L. Y. et al. Transforming growth Factor-beta (TGF-beta) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 292, 4302–4312. https://doi.org/10.1074/jbc.M116.773085 (2017).
Bhattacharyya, D., Teves, M. E. & Varga, J. The dynamic organelle primary cilia: emerging roles in organ fibrosis. Curr. Opin. Rheumatol. 33, 495–504. https://doi.org/10.1097/BOR.0000000000000841 (2021).
Djenoune, L., Berg, K., Brueckner, M. & Yuan, S. A change of heart: new roles for cilia in cardiac development and disease. Nat. Rev. Cardiol. 19, 211–227. https://doi.org/10.1038/s41569-021-00635-z (2022).
Zhong, B. H. & Dong, M. The implication of ciliary signaling pathways for epithelial-mesenchymal transition. Mol. Cell. Biochem. https://doi.org/10.1007/s11010-023-04817-w (2023).
Liu, J., Wang, F. & Luo, F. The role of JAK/STAT pathway in fibrotic diseases: molecular and cellular mechanisms. Biomolecules 13 https://doi.org/10.3390/biom13010119 (2023).
Kramer-Zucker, A. G. et al. Cilia-driven fluid flow in the zebrafish pronephros, brain and kupffer’s vesicle is required for normal organogenesis. Development 132, 1907–1921. https://doi.org/10.1242/dev.01772 (2005).
Cantaut-Belarif, Y., Sternberg, J. R., Thouvenin, O., Wyart, C. & Bardet, P. L. The Reissner Fiber in the Cerebrospinal Fluid Controls Morphogenesis of the Body Axis. Curr Biol 28, 2479–2486 e2474 (2018). https://doi.org/10.1016/j.cub.2018.05.079
Troutwine, B. R. et al. The Reissner fiber is highly dynamic in vivo and controls morphogenesis of the spine. Curr. Biol. 30, 2353–2362. https://doi.org/10.1016/j.cub.2020.04.015 (2020). e2353.
Zhang, X. et al. Cilia-driven cerebrospinal fluid flow directs expression of Urotensin neuropeptides to straighten the vertebrate body axis. Nat. Genet. 50, 1666–1673. https://doi.org/10.1038/s41588-018-0260-3 (2018).
Orts-Del’Immagine, A. et al. Sensory Neurons Contacting the Cerebrospinal Fluid Require the Reissner Fiber to Detect Spinal Curvature In Vivo. Curr Biol 30, 827–839 e824 (2020). https://doi.org/10.1016/j.cub.2019.12.071
Lu, H., Shagirova, A., Goggi, J. L., Yeo, H. L. & Roy, S. Reissner fibre-induced Urotensin signalling from cerebrospinal fluid-contacting neurons prevents scoliosis of the vertebrate spine. Biol. Open. 9 https://doi.org/10.1242/bio.052027 (2020).
Cantaut-Belarif, Y. et al. Adrenergic activation modulates the signal from the Reissner fiber to cerebrospinal fluid-contacting neurons during development. Elife 9 https://doi.org/10.7554/eLife.59469 (2020).
Wu, M. Y. et al. Spinal sensory neurons project onto the hindbrain to stabilize posture and enhance locomotor speed. Curr Biol 31, 3315–3329 e3315 (2021). https://doi.org/10.1016/j.cub.2021.05.042
Bearce, E. A. et al. Urotensin II-related peptides, Urp1 and Urp2, control zebrafish spine morphology. Elife 11 https://doi.org/10.7554/eLife.83883 (2022).
Gaillard, A. L. et al. Urp1 and Urp2 act redundantly to maintain spine shape in zebrafish larvae. Dev. Biol. 496, 36–51. https://doi.org/10.1016/j.ydbio.2023.01.010 (2023).
van der Helm, D. et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Sci. Rep. 8, 16005. https://doi.org/10.1038/s41598-018-34351-5 (2018).
Kok, F. O. et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell. 32, 97–108. https://doi.org/10.1016/j.devcel.2014.11.018 (2015).
Rossi, A. et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. https://doi.org/10.1038/nature14580 (2015).
El-Brolosy, M. A. et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. https://doi.org/10.1038/s41586-019-1064-z (2019).
Peng, J. Gene redundancy and gene compensation: an updated view. J. Genet. Genomics. 46, 329–333. https://doi.org/10.1016/j.jgg.2019.07.001 (2019).
Blum, M., De Robertis, E. M., Wallingford, J. B., Niehrs, C. & Morpholinos Antisense and sensibility. Dev. Cell. 35, 145–149. https://doi.org/10.1016/j.devcel.2015.09.017 (2015).
Meyer-Miner, A., Van Gennip, J. L. M., Henke, K., Harris, M. P. & Ciruna, B. Resolving primary pathomechanisms driving idiopathic-like spinal curvature using a new katnb1 scoliosis model. iScience 25, 105028. https://doi.org/10.1016/j.isci.2022.105028 (2022).
Van Gennip, J. L. M., Boswell, C. W. & Ciruna, B. Neuroinflammatory signals drive spinal curve formation in zebrafish models of idiopathic scoliosis. Sci. Adv. 4, eaav1781. https://doi.org/10.1126/sciadv.aav1781 (2018).
Rose, C. D. et al. SCO-Spondin Defects and Neuroinflammation Are Conserved Mechanisms Driving Spinal Deformity across Genetic Models of Idiopathic Scoliosis. Curr Biol 30, 2363–2373 e2366 (2020). https://doi.org/10.1016/j.cub.2020.04.020
Breslow, D. K. et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet. 50, 460–471. https://doi.org/10.1038/s41588-018-0054-7 (2018).
Lancaster, M. A. & Gleeson, J. G. Cystic kidney disease: the role of Wnt signaling. Trends Mol. Med. 16, 349–360. https://doi.org/10.1016/j.molmed.2010.05.004 (2010).
Wang, Y., Zhou, C. J. & Liu, Y. Wnt signaling in kidney development and disease. Prog Mol. Biol. Transl Sci. 153, 181–207. https://doi.org/10.1016/bs.pmbts.2017.11.019 (2018).
Strubl, S. et al. STAT signaling in polycystic kidney disease. Cell. Signal. 72, 109639. https://doi.org/10.1016/j.cellsig.2020.109639 (2020).
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. & Schilling, T. F. Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310. https://doi.org/10.1002/aja.1002030302 (1995).
Perner, B., Englert, C. & Bollig, F. The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev. Biol. 309, 87–96. https://doi.org/10.1016/j.ydbio.2007.06.022 (2007).
Epting, D. et al. The Rac1 regulator ELMO controls basal body migration and Docking in multiciliated cells through interaction with Ezrin. Development 142, 174–184. https://doi.org/10.1242/dev.112250 (2015).
Robu, M. E. et al. p53 activation by knockdown technologies. PLoS Genet. 3, e78. https://doi.org/10.1371/journal.pgen.0030078 (2007).
Xie, H. et al. Ependymal Polarity defects coupled with disorganized ciliary beating drive abnormal cerebrospinal fluid flow and spine curvature in zebrafish. PLoS Biol. 21, e3002008. https://doi.org/10.1371/journal.pbio.3002008 (2023).
Concordet, J. P. et al. Spatial regulation of a zebrafish patched homologue reflects the roles of Sonic Hedgehog and protein kinase A in neural tube and Somite patterning. Development 122, 2835–2846. https://doi.org/10.1242/dev.122.9.2835 (1996).
Westphal, M., Panza, P., Kastenhuber, E., Wehrle, J. & Driever, W. Wnt/beta-catenin signaling promotes neurogenesis in the diencephalospinal dopaminergic system of embryonic zebrafish. Sci. Rep. 12, 1030. https://doi.org/10.1038/s41598-022-04833-8 (2022).
Lekven, A. C., Thorpe, C. J., Waxman, J. S. & Moon, R. T. Zebrafish wnt8 encodes two wnt8 proteins on a bicistronic transcript and is required for mesoderm and neurectoderm patterning. Dev. Cell. 1, 103–114. https://doi.org/10.1016/s1534-5807(01)00007-7 (2001).
Epting, D., Vorwerk, S., Hageman, A. & Meyer, D. Expression of rasgef1b in zebrafish. Gene Expr Patterns. 7, 389–395. https://doi.org/10.1016/j.modgep.2006.11.010 (2007).
Ott, E. et al. A novel role for the chloride intracellular channel protein Clic5 in ciliary function. Sci. Rep. 13, 17647. https://doi.org/10.1038/s41598-023-44235-y (2023).
Riedmann, H., Kayser, S., Helmstadter, M., Epting, D. & Bergmann, C. Kif21a deficiency leads to impaired glomerular filtration barrier function. Sci. Rep. 13, 19161. https://doi.org/10.1038/s41598-023-46270-1 (2023).
Ewels, P. A. et al. nf-core/rnaseq: community curated bioinformatics pipelines. Zenodo https://doi.org/10.5281/zenodo.1400710 (2016).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. ; Available online: (2022). https://www.R-project.org/
Gentleman, R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. https://doi.org/10.1186/gb-2004-5-10-r80 (2004).
Love, M. I., Huber, W. & Anders, S. Moderated Estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. https://doi.org/10.1186/s13059-014-0550-8 (2014).
Luo, W., Friedman, M. S., Shedden, K., Hankenson, K. D. & Woolf, P. J. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 10, 161. https://doi.org/10.1186/1471-2105-10-161 (2009).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. https://doi.org/10.1093/bioinformatics/btr260 (2011).
Liberzon, A. et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell. Syst. 1, 417–425. https://doi.org/10.1016/j.cels.2015.12.004 (2015).
Epting, D. et al. Loss of CBY1 results in a ciliopathy characterized by features of Joubert syndrome. Hum. Mutat. 41, 2179–2194. https://doi.org/10.1002/humu.24127 (2020).
Acknowledgements
We are grateful to the staff of the Aquatic Core Facility (AquaCore (RI_00544)) at the University Freiburg Medical Center - IMITATE, Germany, for the zebrafish care. The work of S.K. and M.H. was performed at the Core Facility for Electron Microscopy (EMcore (RI_00555)) at the University Freiburg Medical Center - IMITATE, Germany. We would like to thank the Life Imaging Center of the University Freiburg for the use of confocal microscopes and technical support. We thank Eric Barnsley for critical reading of the manuscript. M.B. receives support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through CRC 1479 (Project ID 441891347-S1), CRC 1160 (Project ID 256073931-Z02), CRC 1453 (Project ID 431984000–S1), TRR 167 (Project ID 259373024-Z01), TRR 359 (Project ID 491676693-Z01), TRR 353 (Project ID 471011418-SP02), FOR 5476 UcarE (Project ID 493802833-P7) and by the German Federal Ministry of Education and Research (BMBF) within the National Decade against Cancer program for PM4Onco (FKZ 01ZZ2322A) and SATURN3 (01KD2206L). C.B. holds a part-time faculty appointment at the University of Freiburg in addition to his employment with the Limbach Group. He heads and manages Limbach Genetics and the Medizinische Genetik Mainz. C.B. receives support from the DFG (BE 3910/8−1, BE 3910/8−2, BE 3910/9−1, and Project-ID 431984000 – Collaborative Research Center SFB 1453), BMBF (01GM2203I and 01GM1903G) and the European Union (EU HORIZON-HLTH-2022-DISEASE-06). E.O. receives support from the DFG (Project-ID 431984000 – Collaborative Research Center SFB 1453).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Experiments were designed by D.E., J.D., S.K., M.H. and E.O. Experiments were performed by D.E., J.D., S.K. and E.O. Data were interpreted by D.E., J.D., R.M., M.H., P.M., M.B., C.B. and E.O. The paper was written by D.E. and E.O.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional Information
The original online version of this Article was revised: The original version of this Article contained an error in the Reference list. Full information regarding the corrections made can be found in the correction for this Article.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Epting, D., Devane, J., Mertes, R. et al. Tulp3 deficiency results in ciliopathy phenotypes during zebrafish embryogenesis. Sci Rep 15, 32435 (2025). https://doi.org/10.1038/s41598-025-16584-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-16584-3
