
Abstract
The search for pathological biomarkers in biological fluids that can provide valuable insight into an individual’s health status, is a relevant area of research for multiple pathologies. Currently, the use of proteomics for the identification of differences in protein expression profiles between samples from healthy subjects and patients, has emerged as a powerful strategy to improve the current diagnosis of various pathologies or propose novel therapeutic approaches. Among the biological fluids from which new pathological biomarkers can be identified, tear secretion is highly attractive, since it can be collected non-invasively and could better concentrate proteins that sensitively reflect allergic responses, owing to their exposure to environmental factors and its connection to the respiratory system. Despite its potential, tear fluid remains underexplored, offering significant research opportunities. In this study, we collected human tear samples using the Shirmer Test from healthy and allergic individuals. Our optimized workflow, combining sample preparation and high-throughput proteomics using the data-independent acquisition (DIA) strategy, identified 2542 proteins and enabled the successful differentiation of the two groups. We identified 99 differentially expressed proteins. Our results show the feasibility of protein analysis in human tear samples, highlighting tears as a highly sensitive fluid for detecting health conditions. Data are available via ProteomeXchange with identifier PXD067099.
Similar content being viewed by others

Introduction
Early diagnosis constitutes a cornerstone of modern healthcare strategies, enabling better patient and family preparation, as well as timely therapeutic interventions that can delay or reduce disease symptoms1. This early diagnosis can be defined as the ability to identify patients who, although asymptomatic, exhibit molecular alterations critical to disease progression.
In this context, the search for molecular biomarkers in various biological fluids that can provide early information about health status constitutes a promising area of research with the potential to improve diagnostic accuracy2. Broadly speaking, molecular biomarkers are biomolecules such as proteins, RNAs, miRNAs, or metabolites, whose production or levels change in response to physiological or pathological conditions3. Advances in sensitive and high-throughput analytic techniques, such as proteomics, capable of detecting differences in protein expression profiles between samples from healthy subjects and patients, have enabled the identification of biomarkers across a wide range of pathologies4,5.
Mass spectrometry has rapidly advanced in the past five years, driven by the integration of innovative technologies. Notably, 4D proteomics, enabled by timsTOF mass spectrometers, combines trapped ion mobility spectrometry (TIMS) with parallel accumulation-serial fragmentation (PASEF). This synergy allows for unprecedented proteome resolution and coverage6,7. In this context, data-independent acquisition (DIA) strategies, particularly diaPASEF, have become established as powerful quantitative tools8. The ability of diaPASEF to separate ions based on their shape, size, and charge through ion mobility provides a more robust solution for the analysis of complex biological fluids such as plasma6.
The biological fluid most used for biomarker identification is plasma/serum, due to its easy access and the possibility of obtaining sufficient volume in a safe manner9. However, other fluids, with similar safety and non-invasive obtention may offer better protein profiles, such as tear secretion. Lacrimal or tear secretion fulfills lubricating and protective functions for the ocular surface against physical, chemical and biological insults, and is made up of aqueous and lipid layers10. Although primarily composed of water and salts, tear fluid also contains lipids, mucins and proteins, which differ significantly between healthy individuals and patients with ocular or systemic diseases, providing an opportunity for biomarker discovery11.
Proteomic analysis using liquid chromatography coupled to mass spectrometry (LC-MS), have identified 1300 to 1500 proteins in tear samples from healthy individuals12,13. Among these, the most abundant proteins are associated with anti-inflammatory and antibacterial activity14. Different studies have demonstrated that ocular alterations such as uveitis, dry eye syndrome and diabetic retinopathy alter the protein composition of tear secretion15,16,17,18.
Despite this evidence, tear secretion remains underexplored as a diagnostic or monitoring tool for non-ocular diseases, representing an important opportunity. This study aims to evaluate the feasibility of isolating enough high-quality proteins from tear fluid, using the Shirmer Test to collect them and to develop a robust proteomic analysis workflow for profiling protein expression.
We successfully isolated a substantial quantity of high-quality proteins from tear fluid in both healthy and allergic individuals, which were subsequently analyzed using mass spectrometry. In addition, we established an efficient and reliable strategy for high-throughput proteomic analysis based in DIA methodology that distinguishes between healthy and allergic individuals. Our findings highlight the potential of tear fluid as a valuable biological fluid for biomarker discovery and the detection of systemic and allergic conditions.
Results
Quantitative proteomic analyses in human tears samples
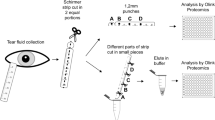
Tear samples from healthy and allergic adult subjects were collected using Shirmer Test and processed for further proteomic analysis (Fig. 1). The demographic and clinical characteristics of the participants are summarized in Table 1.

Experimental design to study the proteomic profile of the human tear’s samples using Nc-MS/MS timsTOF PRO 2.
Proteomic analysis of human tears samples was done to identify proteins altered in allergic subjects. Using mass spectrometry, 2542 proteins were identified in all individuals per group, with a false discovery rate (FDR) of 0.01 (Supplementary Files 1 and 2). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE19 partner repository with the dataset identifier PXD067099”. The quality of proteomic data was evaluated by assessing protein abundance in each human tear sample (Supplementary Fig. 1A) and analyzing the similarity of individual proteomic signatures using a Pearson correlation matrix (Supplementary Fig. 1B). The correlation coefficients ranged from 0.7 to 1.0, indicating a high degree of global similarity in the human tears’ proteome of healthy and allergic individuals. We also assessed reproducibility by visualizing the relationships between protein measurements across multiple replicates for each sample (Supplementary Fig. 1C).
To explore the differences in protein expression profiles between healthy controls and allergic subjects, a principal component analysis (PCA) was performed (Fig. 2A). The PCA results revealed two distinct sample clusters, indicating a clear separation between the tear proteomes of healthy and allergic individuals, where PC1 on the X-axis explained 56.12% of total variability, while PC2 on the Y-axis explained 14.25%. Differentially expressed proteins (DEPs) analysis identified 99 proteins with significant differences in expression between the two groups (Supplementary File 3). The PCA findings were further supported by unsupervised hierarchical clustering, visualized in a heatmap plot. This heatmap demonstrated a clear clustering of samples into healthy and allergic groups, as well as the relative expression levels of the 99 DEPs, across samples and groups (Fig. 2B). Of the 99 DEPs, 47 proteins were up-regulated and 52 were down-regulated in allergic tear samples compared to healthy controls. A volcano plot (Fig. 2C) was used to categorize the down and up-regulated proteins according to their fold change (Log2 FC) and -Log10 (p-value). Also, the volcano plot highlights the top 5 up-regulated and down-regulated proteins in allergic subject tear samples; the up-regulated proteins included Zinc Finger, FYVE Domain Containing 19 (ZFYVE19), HLA class II histocompatibility antigen, DRB5 beta chain (HLA-DRB5), TRNA Nucleotidyl Transferase 1 (TRNT1), IgGFc-binding protein (FCGBP), and Ras-related protein 25 (RAB25). While down-regulated proteins were keratin 80 (KRT80), filaggrin (FLG), DDB1 and CUL4 Associated Factor 7 (DCAF7), Inter-Alpha-Trypsin Inhibitor Heavy Chain 3 (ITIH3) and Elongator Acetyltransferase Complex Subunit 5 (ELP5).

Proteomic quality analysis. (A) Principal components analysis. The lilac and light blue colors show the allergic and healthy subjects, respectively. (B) Hierarchy heatmap representing the clustering of proteins from tears, showing the 99 differentially expressed proteins clustered into healthy controls and allergic subjects, each group consisting of 4 subjects. The heatmap plot was generated using Pearson correlation and Euclidean distance between proteins. (C) Volcano plot showing the distribution of proteins from healthy controls and allergic subjects tears. The x-axis represents the log2 of fold change (FC) and the y-axis the log10 p value provided by EdgeR. The red dots represent 47 up-regulated proteins, and the light blue dots represent 52 down-regulated proteins (Pvalue < 0.05, and FC 0.585 ≥ x ≥ -0.585).
Gene ontology enrichment analysis for pathways in tears human samples
The gene ontology analysis of the 99 significant proteins identified in human tear samples revealed their involvement in 14 enriched pathways, according to IPA software (Fig. 3). Ingenuity Pathway Analysis (IPA) results showed enrichment in seven pathways related to protein translocation, such as eukaryotic translation initiation, elongation, and termination; SRP-dependent co-translational protein targeting to membrane; ribosomal quality control; processing of CAP-introns-containing pre-mRNA, and response of EIF2AK4 to amino acid deficiency. In addition, IPA showed enrichment in two pathways involved in RNA metabolism, which included nonsense-mediated decay and rRNA processing; as well as one pathway related to selenoamino acid metabolism. However, an important difference with the results obtained by Reactome (data not shown) was that IPA was able to find four pathways related to immune system responses downregulated, such as binding and uptake of ligands by scavenger receptors, complement cascade, Fc epsilon receptor (FCERI) signaling, and immunoregulatory interactions between a lymphoid and a non-lymphoid cell (Fig. 3).

Enrichment analyses of pathways in tears proteome from allergic subjects. The bubble chart shows enriched proteins in several categories from 99 DEPs in allergic subjects using -log (p-value-adjust) with IPA analysis. The negative Z-score represents down-regulated pathways, and the positive Z-score represents up-regulated pathways. The up-regulated proteins were involved in 10 pathways categorized mainly in Traslocation processes. The downregulated pathways with the most abundant proteins were related to immune responses. Count: represents the protein abundance in each pathway.
Network analysis by IPA gave us information regarding potential master regulators and regulatory mechanisms involving the DEPs of our tear samples. Of the eight networks identified (Supplementary File 4), ordered from highest to lowest score, three were linked to immunological responses. The first one was related to dermatological, immunological and inflammatory diseases, the third network was related to immune cell trafficking, and the sixth one was related to immunological diseases.
Predicted transcription factors regulating the expression of DEPs
To investigate which transcription factors could be involved in the regulation of the 99 DEPs, we utilized the functional enrichment analysis tool FunRich. Our results identified two transcription factors, CREB1 and SPDEF, as significantly enriched (adjusted p-value < 0.05) in human tears samples from allergic subjects (Table 2). Furthermore, the top five transcription factors with the highest percentage of target proteins were SP1, KLF7, EGR1, SP4 and GABPA (Supplementary Table 1).
Discussion
Human tear secretion is a body fluid that has not been widely documented for its use as a biomarker source, although its recollection is simple and non-invasive. Our results show that the presented pipeline for collection, protein extraction, proteomic analysis by LC-MS/MS and bioinformatics data processing allows the detection of a greater number of proteins than previously identified12,13,15,16,17,18. Moreover, it facilitated the identification of distinct proteomic profile that enable segregation of allergic from healthy subjects through unsupervised analysis.
This study presents the development and optimization of a high-throughput, data-independent acquisition (DIA) methodology for efficient tear proteomic analysis. The optimized workflow achieves robust protein identification and quantification, characterized by a low incidence of missing values. Importantly, this method is scalable for the analysis of large cohorts, maintaining reasonable processing times.
We acknowledge that a limitation of our study is the small sample size, which restricts the ability to draw definitive conclusions or formulate hypotheses about the mechanisms associated with conjunctival allergy. The number of samples used in this study was limited, as it was part of an exploratory study aimed at observing the feasibility of isolating and characterizing proteins from human tear fluid and its potentiality. Besides, from an experimental and technical point of view, we have shown high reproducibility with our sample’s replicas and how bioinformatic analysis can accurately and clearly separate each group. This work demonstrates the robustness and efficacy of our workflow, showcasing its capacity to detect significant variations in protein expression levels, sufficient to discriminate between study groups through unsupervised analysis. It is crucial to expand the sample size in future investigations to conclusively determine whether the observed changes are directly related to the pathology in question, or whether they could be influenced by injuries or inflammatory processes associated with other conditions.
Our analysis identified 99 DEPs. Among the top 5 upregulated we find ZFYVE19, HLA-DRB5, TRNT1, FCGBP, and RAB25. ZFYVE19 regulates cytokinesis checkpoint, allowing proper cell division. Loss of function mutations produces ciliopathies that compromises cell division20,21.
HLA-DRB5 is part of the major histocompatibility complex class II, which plays a crucial role in the presentation of antigens to CD4 T cells. Polymorphism associated with HLA-DRB5 has been linked to antibiotics allergy22 and uveitis23. In addition, very recently, a study has described its upregulation can promote immune thrombocytopenia24. Interestingly, HLA-DRB5 expression has been detected before in the eye by RT.PCR from cytology samples, finding reduced levels in dry-eye patients25. TRNT1 is an enzyme that participates in tRNA processing, by adding CAA trinucleotide at its 3’ end, which is required for aminoacylation and correct positioning on the ribosome. Some case studies have related TRNT1 mutations with B cell deficiency and type I interferonopathy, a condition characterized by overactivation of interferon pathways26,27. FCGBP is a mucin-like glycoprotein expressed in different mucous membranes of the organism, that plays a role in mucosal epithelial defense28fulfilling cytoprotective and anti-inflammatory roles29. It has been recently identified increased in the ocular surface of patients with dry eye, where it is assumed to exert a protective a role30. RAB25 is a small GTPase that regulates intracellular trafficking pathways. Although the Rab family is known for controlling multiple key immunological processes, such as phagocytosis and phagosome maturation, neutrophile and macrophage degranulation31specifically for Rab25 there is only evidence linking decreased expression in skin lesions from atopic dermatitis patients, negatively correlating with its severity32.
Downregulated proteins, on the other hand, were KRT80, FLG, DCAF7, ITIH3 and ELP5. The KRT80 is a filament protein involved in the differentiation and structural arrangement of epithelial barrier. Although it has not a direct role in immune responses its expression has been related to higher infiltration of immune cells in various cancer types33,34and also in the context of tumor cells its inhibition has been shown to decrease the pro-tumoral and anti-inflammatory TGF-β/SMAD signaling pathway35. FLG is an epidermal protein that interacts with keratin fibers to provide strength and elasticity to epithelial barriers. Loss of functions mutation for flg gene are related to impairment of barrier function and risk factors for atopic dermatitis, asthma, allergic rhinitis and food allergies36,37. Interestingly, downregulation of FLG detected in tear samples form allergic individuals seems to reflect what happens with FLG systemically and not in the eye, as another study has been previously found FLG is upregulated in inflamed sclera tissue38. DCAF7, also known as WDR68/HAN11 is a scaffold protein that enables the assembly of multiprotein complexes involved in kinase signaling. It has been shown to participate in nucleotide excision repair39 and ventral/dorsal patterning of craniofacial embryonic development40. We could not find evidence of immune related functions for DCAF7 in the literature. ITIH3 is part of a family of plasma protease inhibitors that interacts with hyaluronic acid acting as matrix stabilizers. No direct link has been established before with allergic responses, but it has been proposed that ITIH3 could be a mediator of thrombo-inflammation in gestational hypertension and preeclampsia41. Finally, ELP5 is part of a protein complex that regulates transcription by modifying tRNA and acetylating histones42which is a global process that can indirectly affect immune responses. However, so far there are no reports linking ELP5 directly with allergic responses or inflammatory pathways.
IPA analysis of 99 DEPs in samples from allergic individuals provided insight into potential molecular pathways that could be altered in this group. For Translocation pathway, which was found upregulated, we identified enrichment of ribosomal protein S19 (RPS19), shown to modulate the immune response during inflammatory processes by decreasing neutrophil activity and promoting monocyte activity43. Recently studies described its role in inhibiting the tumor-suppressor activity of the immune system through interaction with the complement receptor C5aR144. Thus, the increase of RPS19 in the tears of allergic individuals may represent a counter-regulatory response to prevent neutrophils recruitment and avoid acute inflammation. In the skin, downregulation of EIF3F in atopic dermatitis has been shown to reduce inflammation and immune cell recruitment45.
IPA analysis also showed enrichment of General Control Non-Derepressible Protein 1 (GCN1), signal recognition particle 68 (SRP68), eukaryotic translation elongation factor 1 epsilon 1 (EEF1E1), and TRNT1. GCN1 is a key factor necessary for the activation of amino acid starvation sensing by GCN2, regulating co-translational protein quality control46. One study has shown that GCN1 is also necessary to mediate the anti-inflammatory responses elicit by amino acid restriction47proposing it could also participate in suppressing inflammation.
For the Selenenoamino acid metabolism pathway, which was also upregulated we found eukaryotic translation elongation factor 1 epsilon 1 (EEF1E1), also known as AIMP3/p18, in addition to proteins enriched in the translocation pathway. EEF1E1 facilitates the assembly of Methionyl-tRNA synthetase (MARS) during the translation elongation step48. Immunologically, increased EEF1E1 expression correlates with immune cell infiltration in hepatocellular carcinoma49. Moreover, in ankylosing spondylitis, an inflammatory rheumatic disease, EEF1E1 upregulation suggest it may promote pro-inflammatory cytokine production, similar to other proteins of the EEF1E1 family, such as AIMP1, which promote the expression of pro-inflammatory genes in macrophages and dendritic cells50.
Interestingly, IPA analyses found four pathways directly related to immunological responses that were not identified by Reactome. These included pathways involved in the recognition and endocytosis of foreign molecules by macrophages (binding and uptake of ligands by scavenger receptors), complement cascade, mastocyte activation in response to antigen-IgE interactions (Fc epsilon receptor (FCERI) signaling), and immunoregulatory interactions between a lymphoid and a non-lymphoid cell. The downregulation in these pathways involves ferritin light chain protein (FTL), complement component 7 (C7) and immunoglobulins Heavy Variable 1–69 (IGHV1-69), Kappa Variable 2D-30 (IGKV2D-30), Lambda Variable 2–18 (IGLV2-18), and Lambda Variable 3–16 (IGLV3-16). FTL is a sub-unit of ferritin, the primary intracellular iron storage protein. It has been shown that FTL overexpression decreases the production of pro-inflammatory mediators such as TNF-α, IL-1β, nitric oxide and prostaglandin E2 in response to lipopolysaccharide (LPS)51while in the context of allergy, FTL downregulation may impair anti-inflammatory response, exacerbating the inflammatory storm. Although no specific literature on C7 was found regarding allergies, complement responses are tightly controlled in the eye to prevent continuous inflammatory processes which could lead to vision loss52,53so it makes sense that proteins participating in complement activation pathways are downregulated.
Future perspectives
Tear proteomics holds immense promise as a minimally invasive strategy for advancing diagnostics and personalized medicine, particularly in allergy management. By identifying disease-specific biomarkers, this methodology enables the early detection of allergic conditions, paving the way for customized treatment strategies, including targeted anti-inflammatory therapies. This technique could become invaluable for monitoring immune status across various pathological conditions.
Understanding the molecular mechanisms of critical proteins such as FLG, and EIF3F could lead to the development of innovative therapeutic interventions. These therapies might focus on restoring barrier function or modulating inflammatory responses, benefiting not only individuals with allergies but also those with other inflammatory diseases.
Tear proteomics also offers a unique platform to investigate the influence of environmental and lifestyle factors, such as allergens and pollutants, on both ocular and systemic immune health. This application could drive preventive healthcare initiatives by identifying environmental triggers and their effects on the immune system. To further advance the field, integrating metabolomics and transcriptomic analyses with proteomic studies of tears could provide a more comprehensive understanding of the molecular pathways underlying allergies and related conditions. Such integrative approaches may unveil novel therapeutic targets and enhance our understanding of immune regulation.
In conclusion, the insights gained from tear proteomics emphasize its potential to revolutionize diagnostics, drive therapeutic innovations, and expand the scope of biomedical research. The translational opportunities presented by this field underscore its significance in addressing both allergic diseases and broader health challenges.
Materials and methods
Study design and sample
The following study is exploratory in nature, with a cross-sectional design, aiming to standardize the isolation of tear secretion proteins from Schirmer Test strips, and to standardize a protein analysis method for discriminating differentially expressed proteins in healthy versus allergic persons.
Our population of interest corresponds to healthy men and women between 20 and 30 years old, and individuals suffering from seasonal allergies which affected they eyes, noted as irritation or watery eyes of the same age range, who can provide a Schirmer test > 10 mm in 5 min. The ‘healthy’ group reported that they did not have any allergies or other eye complications at the time of participating in the study. Exclusion criteria included any diagnosed ocular pathology, signs of ophthalmological inflammation or infectious disease in the last 6 months, ophthalmological surgical procedures in the last 12 months, pregnancy, and autoimmune diseases. The absence of these affections was self-reported through a short questionnaire, and the absence of eye alterations were also corroborated by the medical technologist of the research team.
The sample was taken by convenience and corresponds to a total of 8 fourth-year medical technology students with specialization in Ophthalmology and Optometry, who as part of their clinical training must apply the technique of Schirmer Test. All students gave their written consent to use their tear secretion before tear collection.
This study was approved by the Ethics Scientific Committee of Universidad San Sebastián (CEC-USS 140 − 23). All research was performed in accordance with the guidelines provided by the Declaration of Helsinki and the regulation of the CEC. Informed consent was obtained from all participants.
Protein collection from tear secretion
Tear secretion was collected by the Schirmer Test, a routinely used test in ophthalmology to measure basal and reflex tear secretion without topical anesthesia. Subject were asked to look up and the lower eyelid of each eye was delicately moved downward to position the head of a graduated paper strip (Schirmer-Plus® Strip Biotech Vision Care PVT LTD, Gujarat, India) on the lower eyelid temporal zone. The subject was asked to maintain this position for 5 min. Once this time has elapsed, the test strips were removed from the patient’s eyelids using tweezers and placed into sterile 1.7 ml tubes, free of DNAse and RNAse54. The length of the wet region of the Schirmer strip was recorded in mm. Immediately after collection, 400 µl of sterile 1X PBS (pH 7.4) with protease inhibitor cocktail (Roche, Basel, Switzerland) was added to each tube containing a test strip and stored at -80° C until analysis.
Protein extraction
We extracted proteins from the Shirmer strip according to the protocol described by Gijs and collaborators54. Briefly, the tubes containing the Schirmer strips at -80 °C were thawed on ice and the strips were cut into small 1 mm fragments with sterile scissors and placed in the same tube containing buffer. Then, strip pieces were incubated in the buffer for 90 min at 4 °C on a shaker at 900 rpm. With the help of a micropipette, the pieces of Schirmer strip were transferred to a new 0.5 ml tube with a hole at its end, made by a 25G needle. This 0.5 ml tube was positioned inside the initial 1.7 ml tube and centrifuged at 12,000 rpm for 10 min at 4 °C. At the end of the centrifugation, the 0.5 ml tube with the strip fragments was discarded and the liquid in the lower tube was stored at -80 °C until further analysis.
Protein yield and quality assessment
Protein concentration for each eye tear of each patient was determined using the Qubit protein assay (ThermoFisher Scientific). Additionally, 20 µg of protein were seeded into an SDS-PAGE gel for Coomassie staining to reveal protein integrity.
Protein digest to nLC-MS/MS
Proteins were denatured using 100 µl of lysis buffer (50 mM HEPES, pH 8, 1% (wt/vol) Triton X-100, 1% (vol/vol) NP-40, 1% (vol/vol) Tween 20, 1% (wt/vol) deoxycholate, 5 mM EDTA, 50 mM NaCl, 1% (vol/vol) glycerol, 1X Complete protease inhibitor, and 5 mM DTT), then they were incubated for half an hour at 60° C, then the sample was homogenized by ultrasound for 2 min with cycles of 10 s at 40% intensity, subsequently it was alkylated using 20 mM of iodoacetamide in 25 mM of ammonium bicarbonate and incubate in darkness for 30 min. The protein extract was subjected to cleaning liquid/liquid extraction using the chloroform/methanol method. 1 vol of the protein extract was incorporated into 5 volumes of 100% methanol and a volume of 100% chloroform (vol/vol) was added then 3 volumes of milliQ water, centrifuged at 15,000 x g for 5 min, observing a protein disc, the soluble phase is eliminated and then the disc is washed with 400 µl of 100% methanol 4 times, and the protein pellets were dried in a rotary concentrator at 2000 rpm for 10 min at 40 °C.
The samples were resuspended in a maximum of 100 µL of 8 M Urea and 25 mM ammonium bicarbonate. The mixture was incubated for 5 min at 25 °C and vortexed several times. The samples were sonicated on ice with 10-second on/off pulses at 90% amplitude and incubated on ice for 5 min. Centrifugation was performed at 19,000 x g for 15 min at 4 °C, and the supernatant was transferred to a new tube. The proteins were immediately quantified using the Qubit Protein Assay reagent (#Q33212, Invitrogen). The samples were diluted 8X, and then, digestion was carried out with sequencing-grade trypsin (#V5071, Promega) at a 1:50 protease/protein (w/w) ratio and incubated for 16 h at 37 °C. Digestion was stopped by adding 10% formic acid. The samples were then cleaned using EVOTips following the supplier’s instructions.
Peptide fractionation and library construction
High pH reversed-phase fractionation was performed on an ÄKTA Avant25 (General Electric) coupled to a refrigerated fraction collection. Purified peptides were separated on a reversed-phase column BHE 2.1 cm x 5 cm (Waters) at a flow rate of 0.2 ml/min at pH 10. The binary gradient started from 3% buffer B (90% ACN in 5 mM Ammonium Formate pH 10), followed by linear increases to first 40% B within 30 min, to 60% B within 15 min, and finally to 85% B within 5 min. Each sample was fractionated into 24 fractions in 400 µl volume intervals. The fractions were dried in a vacuum-centrifuge and reconstituted in water with 2% ACN and 0.1% FA and concatenated in 8 fractions.
nLC-MS/MS TimsTOF PRO 2 analysis
Generation of spectral library: Each fraction was injected into a nanoELUTE nano liquid chromatography system (Bruker Daltonics), Peptides (200 ng of digest) were separated within 60 min at a flow rate of 400 nL/min on a reversed-phase column Aurora Series CSI (25 cm x 75 μm i.d. C18 1.6 μm) (ionopticks Australia) with 50 °C. Mobile phases A and B were water and acetonitrile with 0.1 vol% FA, respectively. The % of B was linearly increased from 2 to 17% within 37 min, followed by an increase to 25% B within 15 min and further to 35% within 8 min, followed by a washing step at 85% B and re-equilibration. All fractions were analyzed on a hybrid trapped ion mobility spectrometry (TIMS) quadrupole time-of-flight mass spectrometer (TIMS-TOF Pro, Bruker Daltonics) via a CaptiveSpray nano-electrospray ion source. The MS was operated in data-dependent mode for the ion mobility-enhanced spectral library generation. We set the accumulation and ramp time was 100ms each and recorded mass spectra in the range from m/z 100–1700 in positive electrospray mode. The ion mobility was scanned from 0.8 to 1.3 Vs/cm2. The overall acquisition cycle of 0.5 s comprised one full TIMS-MS scan and 4 parallel accumulation-serial fragmentation (PASEF) MS/MS scans.
DiaPASEF analysis
The peptides from all voluntaries were separated injected into a EVOSEP ONE nano liquid chromatography system (EVOSEP System), Peptides (200 ng of digest) were separated within 30SPD gradient on a reversed-phase column Bruker pepsep 15 (15 cm x 150 μm i.d. C18 1.6 μm) (Bruker, Daltonics) with 50 °C. Mobile phases A and B were water and acetonitrile with 0.1 vol% FA, respectively. When performing DIA, we define quadrupole isolation windows as a function of the TIMS scan time to achieve seamless and synchronous ramps for all applied voltages. We defined up to 16 windows for single 100 ms TIMS scans according to the m/z-ion mobility plane. During PASEF MSMS scanning, the collision energy was ramped linearly as a function of the mobility from 59 eV at 1/K0 = 1.6 Vs/cm2 to 20 eV at 1/K0 = 0.6 Vs/cm2.
Database searching
For spectral library generation: We used FragPipe computational platform (version 15) with MSFragger (version 3.2), Philosopher (version 3.4.13), and EasyPQP (https://github.com/grosenberger/easypqp; version 0.1.9) components to build spectral libraries. Peptide identification from tandem mass spectra (MS/MS) was done using MSFragger search engine, using raw (.d) files as input. Protein sequence databases H. sapiens (UP000005640) from UniProt (reviewed sequences only; downloaded on Feb. 15, 2021) and common contaminant proteins, containing 20,421 (H. sapiens) sequences were used. Reversed protein sequences were appended to the original databases as decoys. For the MSFragger analysis, both precursor and (initial) fragment mass tolerances were set to 20 ppm. Enzyme specificity was set to ‘stricttrypsin’, and fully enzymatic peptides were allowed. Up to two missed trypsin cleavages were allowed. Oxidation of methionine, acetylation of protein N-termini, -18.0106 Da on N-terminal Glutamic acid, and − 17.0265 Da on N-terminal Glutamine and Cysteine were set as variable modifications. Carbamidomethylation of Cysteine was set as a fixed modification. The maximum number of variable modifications per peptide was set to 3. The final spectral library was filtered to 1% protein and 1% peptide-level FDR.
The processing of diaPASEF raw data was performed with DIA-NN v1.7.15 software55which uses neural networks to perform library-based protein identification and intensity-based library previously generated by the MSFragger tool. The parameters of maximum mass accuracy tolerances were set to a default average of 10 ppm for both the MS1 and MS2 spectra. The samples were analyzed with match-between-runs (MBR) enabled; quantification mode was set to “any LC (high accuracy)”. All other settings were left default. The DIA-NN output was filtered at a precursor q value < 1% and a global protein q value < 1%.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE19 partner repository with the dataset identifier PXD067099.
Bioinformatics analysis
Quantitative output .csv files generated by DIA-NN, containing protein identifiers and intensities per sample, were exported for downstream analysis in R, specifically for LFQ analysis. First, the dataset was split into samples groups (Healthy and Allergic). Missing values were imputed for each group using the missForest algorithm, requiring at least 60% presence within each group. Subsequently, intensity values for each run were adjusted to the median using quantile normalization to reduce technical variation. Significant differential protein expressions were determined using a Bayesian moderated t-test implemented in the limma package v.3.54.2. Any protein associated with a p-value < 0.05 was considered significant. Exploratory data analysis and visualization were created using R v.3.6.0 with EnhancedVolcano, ComplexHeatmap v.2.0.0, and base packages. Pathway enrichment analysis was conducted using ReactomePA v.1.42.0, querying the human database with logFC and p-value data for each statistical comparison. for transcriptional enrichment analysis we used FunRich Tool Vol 3.1.356.
Data availability
Data are available via ProteomeXchange with identifier PXD067099.
References
Word Health Organization. Strengthening diagnostics capacity. Resolution from executive board 152nd session, Agenda item 5. (2023).
Hartl, J. et al. Quantitative protein biomarker panels: a path to improved clinical practice through proteomics. EMBO Mol. Med. 15, e16061. https://doi.org/10.15252/emmm.202216061 (2023).
Bodaghi, A., Fattahi, N. & Ramazani, A. Biomarkers: Promising and valuable tools towards diagnosis, prognosis and treatment of Covid-19 and other diseases. Heliyon 9, e13323. https://doi.org/10.1016/j.heliyon.2023.e13323 (2023).
Aerqin, Q. et al. Omics-based biomarkers discovery for Alzheimer’s disease. Cell. Mol. Life Sci. 79, 585. https://doi.org/10.1007/s00018-022-04614-6 (2022).
Xiao, Y., Bi, M., Guo, H. & Li, M. Multi-omics approaches for biomarker discovery in early ovarian cancer diagnosis. EBioMedicine 79, 104001. https://doi.org/10.1016/j.ebiom.2022.104001 (2022).
Meier, F. et al. DiaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods. 17, 1229–1236. https://doi.org/10.1038/s41592-020-00998-0 (2020).
Mun, D. G. et al. Four-dimensional proteomics analysis of human cerebrospinal fluid with trapped ion mobility spectrometry using PASEF. Proteomics 23, e2200507. https://doi.org/10.1002/pmic.202200507 (2023).
Bader, J. M., Albrecht, V. & Mann, M. MS-based proteomics of body fluids: the end of the beginning. Mol. Cell. Proteomics: MCP. 22, 100577. https://doi.org/10.1016/j.mcpro.2023.100577 (2023).
Geyer, P. E., Holdt, L. M., Teupser, D. & Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 13, 942. https://doi.org/10.15252/msb.20156297 (2017).
Pflugfelder, S. C. & Stern, M. E. Biological functions of tear film. Exp. Eye Res. 197 https://doi.org/10.1016/j.exer.2020.108115 (2020).
Zhan, X., Li, J., Guo, Y. & Golubnitschaja, O. Mass spectrometry analysis of human tear fluid biomarkers specific for ocular and systemic diseases in the context of 3P medicine. EPMA J. 12, 449–475. https://doi.org/10.1007/s13167-021-00265-y (2021).
Aass, C., Norheim, I., Eriksen, E. F., Thorsby, P. M. & Pepaj, M. Single unit filter-aided method for fast proteomic analysis of tear fluid. Anal. Biochem. 480, 1–5. https://doi.org/10.1016/j.ab.2015.04.002 (2015).
Dor, M. et al. Investigation of the global protein content from healthy human tears. Exp. Eye Res. 179, 64–74. https://doi.org/10.1016/j.exer.2018.10.006 (2019).
de Souza, G. A., Godoy, L. M. & Mann, M. Identification of 491 proteins in the tear fluid proteome reveals a large number of proteases and protease inhibitors. Genome Biol. 7, R72. https://doi.org/10.1186/gb-2006-7-8-R72 (2006).
Amorim, M. et al. Putative biomarkers in tears for diabetic retinopathy diagnosis. Front. Med. 9 https://doi.org/10.3389/fmed.2022.873483 (2022).
Benitez-Del-Castillo, J. M. et al. Quantification of a panel for dry-eye protein biomarkers in tears: A comparative pilot study using standard ELISA and customized microarrays. Mol. Vis. 27, 243–261 (2021).
Das, N. et al. Proteomics analysis of tears and saliva from Sjogren’s syndrome patients. Front. Pharmacol. 12, 787193. https://doi.org/10.3389/fphar.2021.787193 (2021).
Kumar, N. R. et al. Tear biomarkers in dry eye disease: progress in the last decade. Indian J. Ophthalmol. 71, 1190–1202. https://doi.org/10.4103/IJO.IJO_2981_22 (2023).
Perez-Riverol, Y. et al. The PRIDE database at 20 years: 2025 update. Nucleic Acids Res. 53, D543–d553. https://doi.org/10.1093/nar/gkae1011 (2025).
Teles, L. M. et al. Comparison of the neutrophil proteome in trauma patients and normal controls. Protein Pept. Lett. 19, 663–672. https://doi.org/10.2174/092986612800493977 (2012).
Yang, J. et al. ZFYVE19 deficiency: a ciliopathy involving failure of cell division, with cell death. J. Med. Genet. 61, 750–758. https://doi.org/10.1136/jmg-2023-109779 (2024).
Gueant, J. L. et al. HLA-DRA variants predict penicillin allergy in genome-wide fine-mapping genotyping. J. Allergy Clin. Immunol. 135, 253–259. https://doi.org/10.1016/j.jaci.2014.07.047 (2015).
Davoudi, S. et al. Association of genetic variants in RAB23 and ANXA11 with uveitis in sarcoidosis. Mol. Vis. 24, 59–74 (2018).
Ye, Q., Ying, Q., Chen, Y., Liao, C. & Li, A. HLA-DRB5 promotes immune thrombocytopenia via activating CD8(+) T cells. Open Med. 19, 20240955. https://doi.org/10.1515/med-2024-0955 (2024).
Bradley, J. L., Edwards, C. S. & Fullard, R. J. Adaptation of impression cytology to enable conjunctival surface cell transcriptome analysis. Curr. Eye Res. 39, 31–41. https://doi.org/10.3109/02713683.2013.823213 (2014).
Chakraborty, P. K. et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood 124, 2867–2871. https://doi.org/10.1182/blood-2014-08-591370 (2014).
Slade, A., Kattini, R., Campbell, C. & Holcik, M. Diseases associated with defects in tRNA CCA addition. Int. J. Mol. Sci. 21 https://doi.org/10.3390/ijms21113780 (2020).
Gorman, H., Moreau, F., Dufour, A. & Chadee, K. IgGFc-binding protein and MUC2 mucin produced by colonic goblet-like cells spatially interact non-covalently and regulate wound healing. Front. Immunol. 14, 1211336. https://doi.org/10.3389/fimmu.2023.1211336 (2023).
Liu, Q. et al. Role of the mucin-like glycoprotein FCGBP in mucosal immunity and cancer. Front. Immunol. 13, 863317. https://doi.org/10.3389/fimmu.2022.863317 (2022).
Bräuer, L., Wille, T., Schicht, M., Garreis, F. & Paulsen, F. P. Detection of the glycoprotein FCGBP (IgG Fc binding protein) at the ocular surface. Investig. Ophthalmol. Vis. Sci. 64, 703–703 (2023).
Prashar, A., Schnettger, L., Bernard, E. M. & Gutierrez, M. G. Rab GTPases in immunity and inflammation. Front. Cell. Infect. Microbiol. 7, 435. https://doi.org/10.3389/fcimb.2017.00435 (2017).
Jeong, H. et al. RAB25 coordinates filaggrin-containing keratohyalin granule maturation and affects atopic dermatitis severity. Allergy 78, 1007–1019. https://doi.org/10.1111/all.15582 (2023).
Yun, W. J. et al. The facilitating effects of KRT80 on chemoresistance, lipogenesis, and invasion of esophageal cancer. Cancer Biol. Ther. 25, 2302162. https://doi.org/10.1080/15384047.2024.2302162 (2024).
Shi, K. H. et al. KRT80 expression works as a biomarker and a target for differentiation in gastric cancer. Histol. Histopathol. 39, 117–130. https://doi.org/10.14670/hh-18-618 (2024).
Tong, Y., Chen, X., Feng, Z., Xu, C. & Li, Y. Keratin 80 promotes migration and invasion of non-small cell lung cancer cells by regulating the TGF-beta/SMAD pathway. Evid.-based Complement. Altern. Med. 2022, 2630351. https://doi.org/10.1155/2022/2630351 (2022).
Drislane, C. & Irvine, A. D. The role of Filaggrin in atopic dermatitis and allergic disease. Ann. allergy Asthma Immunol. 124, 36–43. https://doi.org/10.1016/j.anai.2019.10.008 (2020).
Kalb, B. et al. Filaggrin loss-of-function mutations are associated with persistence of egg and milk allergy. J. Allergy Clin. Immunol. 150, 1125–1134. https://doi.org/10.1016/j.jaci.2022.05.018 (2022).
Vergouwen, D. P. C. et al. Scleral proteome in noninfectious scleritis unravels upregulation of filaggrin-2 and signs of neovascularization. Invest. Ophthalmol. Vis. Sci. 64, 27. https://doi.org/10.1167/iovs.64.3.27 (2023).
Kawara, H., Akahori, R., Wakasugi, M., Sancar, A. & Matsunaga, T. DCAF7 is required for maintaining the cellular levels of ERCC1-XPF and nucleotide excision repair. Biochem. Biophys. Res. Commun. 519, 204–210. https://doi.org/10.1016/j.bbrc.2019.08.147 (2019).
Alvarado, E. et al. Wdr68 mediates dorsal and ventral patterning events for craniofacial development. PloS One. 11, e0166984. https://doi.org/10.1371/journal.pone.0166984 (2016).
de Almeida, L. G. N. et al. Proteomics and metabolomics profiling of platelets and plasma mediators of thrombo-inflammation in gestational hypertension and preeclampsia. Cells. 11. https://doi.org/10.3390/cells11081256 (2022).
Close, P. et al. DERP6 (ELP5) and C3ORF75 (ELP6) regulate tumorigenicity and migration of melanoma cells as subunits of Elongator *. J. Biol. Chem. 287, 32535–32545. https://doi.org/10.1074/jbc.M112.402727 (2012).
Yamamoto, T. Roles of the ribosomal protein S19 dimer and the C5a receptor in pathophysiological functions of phagocytic leukocytes. Pathol. Int. 57, 1–11. https://doi.org/10.1111/j.1440-1827.2007.02049.x (2007).
Markiewski, M. M. et al. The ribosomal protein S19 suppresses antitumor immune responses via the complement C5a receptor 1. J. Immunol. 198, 2989–2999. https://doi.org/10.4049/jimmunol.1602057 (2017).
Andoh, T., Yoshihisa, Y., Rehman, M. U., Tabuchi, Y. & Shimizu, T. Berberine induces anti-atopic dermatitis effects through the downregulation of cutaneous EIF3F and MALT1 in nc/nga mice with atopy-like dermatitis. Biochem. Pharmacol. 185, 114439. https://doi.org/10.1016/j.bcp.2021.114439 (2021).
Tatara, Y. et al. Emerging role of GCN1 in disease and homeostasis. Int. J. Mol. Sci. 25 https://doi.org/10.3390/ijms25052998 (2024).
Kim, Y. et al. Aminoacyl-tRNA synthetase inhibition activates a pathway that branches from the canonical amino acid response in mammalian cells. Proc. Natl. Acad. Sci. 117, 8900–8911. https://doi.org/10.1073/pnas.1913788117 (2020).
Cristiano, L. The pseudogenes of eukaryotic translation elongation factors (EEFs): role in cancer and other human diseases. Genes Dis. 9, 941–958. https://doi.org/10.1016/j.gendis.2021.03.009 (2022).
Han, R. et al. A novel HCC prognosis predictor EEF1E1 is related to immune infiltration and may be involved in EEF1E1/ATM/p53 signaling. Front. Oncol. 11, 700972. https://doi.org/10.3389/fonc.2021.700972 (2021).
Fan, X., Qi, B., Ma, L. & Ma, F. Screening of underlying genetic biomarkers for ankylosing spondylitis. Mol. Med. Rep. 19, 5263–5274. https://doi.org/10.3892/mmr.2019.10188 (2019).
Fan, Y. et al. The effect of anti-inflammatory properties of ferritin light chain on lipopolysaccharide-induced inflammatory response in murine macrophages. Biochim. Biophys. Acta. 1843, 2775–2783. https://doi.org/10.1016/j.bbamcr.2014.06.015 (2014).
Clark, S. J. & Bishop, P. N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 40, 65–74. https://doi.org/10.1007/s00281-017-0649-6 (2018).
Jha, P., Bora, P. S. & Bora, N. S. The role of complement system in ocular diseases including uveitis and macular degeneration. Mol. Immunol. 44, 3901–3908. https://doi.org/10.1016/j.molimm.2007.06.145 (2007).
Gijs, M. et al. Association of tear fluid amyloid and Tau levels with disease severity and neurodegeneration. Sci. Rep. 11, 22675. https://doi.org/10.1038/s41598-021-01993-x (2021).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 17, 41–44. https://doi.org/10.1038/s41592-019-0638-x (2020).
Fonseka, P., Pathan, M., Chitti, S. V., Kang, T. & Mathivanan, S. FunRich enables enrichment analysis of omics datasets. J. Mol. Biol. 433, 166747. https://doi.org/10.1016/j.jmb.2020.166747 (2021).
Funding
This study was supported by CLA-025025-1 FISAR funding (Melisa Institute)and by Vicerrectoría de Investigación y Doctorados de la Universidad San Sebastián – Fondo USS-FIN-25-APCS-35.
Author information
Authors and Affiliations
Contributions
E.S, C.C and S.A participated in the recruitment of research subjects and the tear samples collection.C.C, S.A and A.V participated in tear sample extraction protocol. M. H processed tear samples for proteomic analysis. M. H, P. S and G. N participated in proteomic data processing and analysis. A.V, C. C, R. E and K. O participated in data interpretation and generation of final figures. K. O and C. V supervised the experimental work. K. O conceived the idea and generated the first draft, with inputs and revisions from A.V, C.C, R.E and M.H. E. K and C. V provided funding for proteomic analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vera-Montecinos, A., Pardo, C.C., Hernández, M. et al. High throughput tear proteomics with data independent acquisition enables biomarker discovery in allergic conditions. Sci Rep 15, 31181 (2025). https://doi.org/10.1038/s41598-025-17105-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-17105-y
