
Abstract
The hyaluronan-mediated motility receptor (HMMR) is widely expressed across various species and plays a crucial role in cancer progression. However, its role in nasopharyngeal carcinoma (NPC) remains unexplored. Here, we identified a novel HMMR-FAM83D-β-catenin axis, which drives NPC progression through β-catenin signaling. Using bulk RNA sequencing, single-cell RNA-sequencing, and immunohistochemistry, we determined that HMMR is highly expressed in NPC tissues, correlating with poor survival in NPC patients. In vitro assays demonstrated that modulating HMMR expression influenced NPC cell proliferation, migration, and invasion. In vivo, HMMR knockdown significantly inhibited tumor growth and metastasis in NPC models. HMMR was significantly upregulated in NPC tissues and correlated with worse patient survival. Mechanistically, HMMR was found to interact with the Wnt/β-catenin signaling pathway, affecting both pathway activation and β-catenin expression, as evidenced by RNA-seq and western blotting. Co-immunoprecipitation, mass spectrometry, and western blot analyses further revealed that HMMR interacts with Family with Sequence Similarity 83 Member D (FAM83D), stabilizing its expression by inhibiting its ubiquitination. This interaction, in turn, modulates β-catenin levels, driving NPC progression. Our findings reveal a novel HMMR-FAM83D-β-catenin axis that promotes NPC cell progression through Wnt/β-catenin signaling by modulating β-catenin signaling. HMMR could serve as a potential prognostic biomarker and therapeutic target for NPC, deepening our understanding of its role in disease progression.
Similar content being viewed by others


Introduction
Nasopharyngeal carcinoma (NPC), a malignant head and neck tumor prevalent in southern China, is closely associated with Epstein–Barr virus (EBV) infection. Advances in intensity-modulated radiation therapy and chemoradiotherapy have significantly improved NPC treatment outcomes, with 5-year patient survival rates approaching 80%1,2,3,4. Nevertheless, patients face a 20–30% risk of subsequent distant metastasis or loco-regional recurrence following standard treatment5,6. The onset of progression complicates the treatment, resulting in a poor prognosis and posing a significant barrier to further therapeutic efforts7. Therefore, understanding the mechanisms underlying NPC progression and developing effective preventive and therapeutic strategies are of utmost importance.
The hyaluronan-mediated motility receptor (HMMR), also known as CD168, is an acidic coiled-coil protein that was originally identified as part of the hyaluronic acid-receptor complex in murine fibroblast-conditioned media8,9. HMMR lacks a hydrophobic signal peptide and potential hydrophobic transmembrane domains10 and it is present on and secreted from the cell surface8. This dynamic localization suggests its involvement in various physiological and pathological processes. HMMR plays a crucial role in maintaining tissue homeostasis and neural development and is involved in spindle assembly regulation during mitosis and meiosis10. HMMR is also overexpressed in various cancers and plays a regulatory role in tumor progression, including proliferation, metastasis, and chemoresistance11,12,13,14,15,16. Higher HMMR expression correlates with poorer survival rates in patients with head and neck squamous cell carcinoma (HNSC) and is associated with disease progression14. Given that NPC is a subtype of HNSC and is prevalent in China, understanding the role of HMMR in NPC progression is crucial to address the urgent challenge of managing metastatic NPC. Meanwhile, FAM83D, a known regulator of cell cycle and tumor progression in several cancers such as lung17colorectal18and ovarian cancer19has not yet been investigated in the context of NPC, highlighting the novelty of exploring its role in this disease.
The mechanisms underlying NPC progression remain elusive, making the investigation of HMMR functions and interactions within NPC a field of potentially significant impact. Therefore, we aimed to explore the molecular mechanisms underlying HMMR during NPC progression.
Methods and materials
Data acquisition and bioinformatics analysis
The GSE12452 dataset (comprising 31 NPC tissues and 10 normal tissues) and the GSE61218 dataset (comprising 10 NPC tissues and 6 normal tissues) were used to detect differences in HMMR expression levels. In the GSE102349 dataset, samples were stratified into high-expression and low-expression groups based on the median value of HMMR expression, and progression-free survival analysis was performed using the grouped data (Sup Table 1). Additionally, HMMR’s role was explored using the GSE162025 scRNA-seq dataset (Sup Table 1). The filtering criteria were set as: nFeature > 200 and < 6000 and mitochondrial genes < 15%. Standard scRNA-seq data analysis was performed with the Seurat 5.1.0 R package20, while personalized analyses, such as active cell analysis and enrichment analysis, were conducted using inferCNV, AUCell, ClusterProfiler, and GSVA R packages21,22,23. (details showed in the supplementary data)
Specimens and IHC analysis
Human NPC tissues were collected from 262 patients who underwent nasopharyngoscopic biopsy at the Jiangxi Cancer Hospital between October 2014 and September 2017 (Sup Table 2). Informed consent was secured from all participants prior to their involvement in the study, which received formal approval from the Ethics Committee of Jiangxi Cancer Hospital. NPC tissue slides were treated with 0.3% H2O2 for 30 min to inhibit endogenous peroxidase activity, permeabilized with 0.5% Triton X-100, and incubated with citrate buffer to unmask antigens. Following PBS washes, slides were blocked with 5% normal goat serum, then incubated with HMMR antibody (1:100 dilution) for 2 h at room temperature, followed by a secondary HRP-conjugated anti-rabbit IgG incubation for 30 min. Nuclei were counterstained with DAPI. Staining intensity was graded on a scale of 0 to 3, representing no staining (0), weak (1), moderate (2), and strong (3). The H-score, with a maximum value of 300, was determined using the formula: (3 × percentage of strongly stained cells) + (2 × percentage of moderately stained cells) + percentage of weakly stained cells. Expression levels were categorized as negative (score: 0), weak (0–60), moderate (60–150), and strong (150–300). We also presented representative IHC results in the results based on the staining intensity of negative, low expression, moderate expression, and high expression. Next, patients were divided into high/low HMMR expression groups based on the median H-score, and the 5-year OS, PFS, DMFS, and LRRFS of NPC patients were analyzed using the Kaplan-Meier method. (details showed in the supplementary data)
Cell culture, reagents, and plasmids
Human NPC cell lines, SUNE1, HONE1, and CNE2 were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C with 5% CO2. All cell lines were authenticated and confirmed to be mycoplasma-free. The reagents and plasmids used in this study are listed in Sup Tables 3 and Sup Table 4, respectively.
Reverse transcription-polymerase chain reaction (PCR)
Total RNA was extracted from NPC cells using TRIzol reagent (Invitrogen) and reverse transcribed into cDNA with the Transcriptor First Strand cDNA Synthesis Kit (Roche) using random and oligo-dT primers. qPCR was performed with FastStart Universal SYBR Green Master (Rox) to assess relative mRNA expression levels. Primer sequences are listed in Sup Table 5.
Western blot and Co-IP
For western blotting, cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes. After blocking with 5% milk at room temperature for 1 h, membranes were incubated with primary antibodies overnight at 4 °C. The following antibodies were used: HMMR, E-cadherin, N-cadherin, vimentin, β-catenin, non-phospho(active)-β-catenin, and FAM83D (Sup Table 6). Membranes were then incubated with secondary antibodies at room temperature for 1 h and visualized using the ChemiDoc XRS + system.
For the Co-IP assay, NPC cell lysates were incubated with antibodies to test various protein interactions, including HMMR and β-catenin, HMMR and FAM83D, FAM83D and ubiquitin, and Flag-HMMR plasmid and FAM83D. Lysates were stored at 4 °C overnight, precipitated with protein G agarose, and analyzed by SDS-PAGE and immunoblotting. Membranes were blocked with 5% BSA, incubated with primary antibodies overnight at 4 °C, and visualized using the Tanon 5200 imaging system after secondary antibody incubation.
Immunofluorescence staining
Cells were grown to 70–80% confluence in six-well dishes with cover slides, fixed with 4% paraformaldehyde for 30 min, and permeabilized with PBS containing 0.1% Triton X-100 for 1 h. Cells were then incubated with CoraLite® 594-conjugated E-cadherin and CoraLite® Plus 488-conjugated N-cadherin antibodies at 37 °C for 2 h. After DAPI staining, the slides were washed and examined under a laser scanning microscope.
Cell growth and proliferation assays
Colony formation assays involved seeding cells into six-well plates, followed by incubation at 37 °C with 5% CO2 for a period of 10 days. Post-incubation, colonies were treated with crystal violet for staining, rinsed with PBS, and then visualized. For EdU assays, cells were exposed to EdU reagent for 4 h, subsequently rinsed, and stained using Hoechst dye according to the manufacturer’s instructions (Sup Table 3).
Transwell migration and invasion assays
For the wound-healing assay, NPC cells were grown to confluence in six-well plates, a scratch was made using a micropipette tip, and images were taken at 0 and 24 h. For transwell migration assays, NPC cells were seeded in serum-free PRMI-1640 in the upper chamber of 24-well inserts, with 20% FBS PRMI-1640 in the lower chamber. After 24 h, migrated cells on the bottom surface were fixed in 4% paraformaldehyde and stained with crystal violet. For transwell invasion assays, cells were plated on Matrigel-coated transwell inserts, incubated for 48 h, and counted in five random fields.
Animal experiments
All animal experiments were performed following institutional guidelines and received approval from Nanchang Royo Biotech Co., Ltd. (Approval Number: RYE2024031001). For xenograft experiments, 5-week-old male BALB/c-nude mice were randomly assigned to two groups (n = 10 each) and subcutaneously injected with 4 × 10^6 SUNE1 cells stably infected with lentiviruses (vector or shHMMR). Tumor volumes were measured every other day and calculated as (width^2 × length) / 2. Mice were euthanized when tumors reached 15 mm in length or when moribund, and tumors were harvested for analysis. For tail vein injection assays, mice (n = 6 per group) were injected with 2 × 10^6 SUNE1 cells in PBS. After 4 weeks, mice were euthanized, and lung metastases were assessed using fluorescence imaging and counted. To ensure unbiased results, the investigator was blinded to group assignments. Detailed sample sizes and related information are provided in the figure legends, and no animals were excluded from the analysis. At the end of each experimental procedure, mice were sacrificed by cervical dislocation under anaesthesia (4% isoflurane, MedChemExpress).(details showed in the supplementary data).
RNA sequencing and LC-MS/MS analyses
Total RNA was isolated from SUNE1 cells transfected with shHMMR-3# or shControl lentivirus using the RNeasy Mini Kit (QIAGEN). Paired-end libraries were prepared using the NEBNext® Ultra™ RNA Library Prep Kit (Illumina) and sequenced on the Illumina Novaseq platform. Sequence files (fastq) were aligned to the GRCh38 reference genome using Hisat2. Gene expression was quantified as FPKM, and differential expression analysis was performed using DESeq2. Genes with |log2 fold change| > 1 and adjusted P-value < 0.05 were considered significant. Additionally, KEGG and GO enrichment analyses were performed on the differentially expressed genes24,25.
For LC-MS/MS analysis, SDS-PAGE gels were fixed, sensitized, and stained with silver nitrate. Differential bands were excised and identified by LC-MS/MS at GeneChem Co., Ltd. (Shanghai, China).
Statistical analysis
All experiments were conducted in triplicate. Statistical analyses were performed using GraphPad Prism 9.0, with data presented as mean ± SD. Additional software and R packages used are listed in Sup Table 7. For two-group comparisons, unpaired Student’s t-test or Mann–Whitney U test was applied. For multi-group comparisons, one-way or two-way ANOVA with Bonferroni post-hoc testing was used. Survival analysis was performed using the Kaplan–Meier method with log-rank test comparison. Multivariate Cox proportional hazards models were employed to assess the risk of distant metastasis or death. All tests were two-tailed, with statistical significance set at P < 0.05.
Results
HMMR expression is upregulated in NPC and closely related to progression
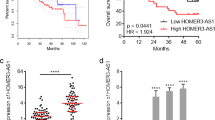
To explore the expression and significance of HMMR in NPC samples, we examined its expression using the Gene Expression Omnibus database. HMMR mRNA levels were significantly higher in NPC tissues compared to normal nasopharyngeal tissues across two GEO datasets (GSE12452 and GSE61228) and our own tissue samples (Fig. 1A). Furthermore, high HMMR expression was independently associated with worse progression-free survival (PFS; P = 0.044) in the GSE102349 dataset (Fig. 1B). Immunohistochemistry (IHC) was used to detect HMMR expression in NPC (Fig. 1C). Based on the H-score of HMMR expression in IHC, the patients were divided into overall survival (OS), PFS, distant metastasis-free survival (DMFS), and local regional failure-free survival groups. Kaplan–Meier analysis revealed that high HMMR expression was significantly associated with inferior 5-year OS (77.6% vs. 87.9%, P = 0.0038), PFS (64.3% vs. 79.4%, P = 0.0025), and DMFS (77.1% vs. 91.3%, P = 0.00059) compared to low-expression patients (Fig. 1E). Multivariate Cox regression analysis revealed that HMMR expression in IHC (hazard ratio [HR]: 1.75, P = 0.026) was an independent prognostic factor of OS in NPC (Fig. 1D) after adjusting for age, sex, T stage, N stage, number of chemotherapy cycles, and EBV expression status.

The expression of HMMR in NPC and is related to NPC progression. (A) The mRNA expression of HMMR upregulated in the NPC tissue compared to the normal tissue. (B) The NPC patients with high expression of HMMR shown the worse prognosis than the low expression. (C) Typical IHC images, the staining intensity was negative, low, median and high, respectively. (D) Forest map of clinical features. (E) Kaplan-Meier analysis for the OS, PFS, DMFS and LRFFS.
HMMR expression and potential function in single-cell level
We performed scRNA-seq analysis on 10 NPC samples using standard protocols (Sup Fig. 1). By integrating turbinate epithelial data from GSE121600, inferCNV identified high copy number variations in the NPC tissues (Sup Fig. 2A). After building the six clusters, we focused on the epithelial cells. First, we subdivided 18 subpopulations of epithelial cells (Fig. 2A). Based on normal epithelial markers (C20orf85 and CAPS) and inferCNV auxiliary analysis,26,27 cluster 15 was identified as normal epithelial cells (Fig. 2B and Sup Fig. 2B, C). HMMR mRNA expression levels in NPC cells were higher than those in normal epithelial cells, similar to the RNA sequence dataset (Fig. 2C, D). The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that, compared to normal epithelial cells, NPC tumors were associated with several important pathways, such as those in EBV infection, cellular senescence, and cell cycles (Fig. 2E). Malignant cells were subsetted and subjected to AUCell analysis based on HMMR expression (Fig. 2F). High- versus low-HMMR activity groups were defined, and differential enrichment was assessed. GSVA analysis demonstrated that high-HMMR activity cells exhibited significant enrichment in hallmark pathways and C2 categories, including G2M checkpoint, E2F target, Wnt/β-catenin, Notch signaling, and MAPK/ERK activation (Fig. 2G).

To explore the role of HMMR in NPC from a single-cell perspective. (A) The results of epithelial cell clustering. (B) inferCNV assisted analysis to identify tumor cells. (C) Both tumor and normal epithelial cells are depicted. (D) The expression of HMMR in tumor and normal cells. (E) KEGG enrichment analysis of tumor cells compared with normal cells. (F) AUCell method was used to analyze the group of HMMR in different activation states. (G) GSVA analysis for Hallmark and C2 gene set in different HMMR active groups.
HMMR promotes the progression of NPC cells in vitro
To investigate the role of HMMR in NPC, we assessed how its knockdown or overexpression impacts the proliferation, migration, and invasion of NPC cells. Compared to NP69 cells, HMMR mRNA and protein were highly expressed in the six NPC cell lines (Sup Fig. 3A, B). Next, we chose two medium-expressing cell (SUNE1 and CNE2) and one high-expressing cell (HONE1) lines for further experiments. We then manipulated HMMR expression by stably transfecting HMMR shRNA lentivirus (shHMMR) (Sup Fig. 3C-F) or overexpressing HMMR lentivirus (pcDNA3-HMMR) (Sup Fig. 3G, H). The depletion of HMMR expression reduced the proliferation capacity of SUNE1 and HONE1 cells, as demonstrated by colony-forming and 5-ethynyl-2′-deoxyuridine (EdU) assays (Fig. 3A, C and Sup Fig. 4A, B). Conversely, HMMR overexpression enhanced the proliferation of CNE2 cells (Fig. 3B, D). Scratch-wound healing and transwell assays revealed that the loss of HMMR suppressed the migration and invasion of SUNE1 and HONE-1 cells (Fig. 3E, G and Sup Fig. 4C, D). In contrast, HMMR overexpression enhanced the migration and invasion capabilities of CNE2 cells (Fig. 3F, H).

The ability of NPC cells to progress in vitro is influenced by HMMR. (A) Decreasing the expression of HMMR reduces the number of SUNE1 cell colonies. (B) Increasing the expression of HMMR elevates the number of CNE2 cell colonies. (C) Reducing HMMR expression decreases the percentage of Edu-positive SUNE1 cells. (D) Enhancing HMMR expression increases the percentage of Edu-positive CNE2 cells. (E) Downregulated HMMR expression suppresses the migration ability of SUNE1 cells as shown by wound healing assay. (F) Upregulated HMMR expression promotes the migration ability of CNE2 cells as demonstrated by wound healing assay. (G) Downregulated HMMR expression suppresses the migration and invasion abilities of SUNE1 cells as shown by transwell assay. (H) Upregulated HMMR expression promotes the migration and invasion abilities of CNE2 cells as demonstrated by transwell assay. (I) Modulating HMMR expression affects the expression of EMT-related biomarkers in SUNE1 cells as evidenced by immunofluorescence and western blot assays. (J) Modulating HMMR expression affects the expression of EMT-related biomarkers in CNE2 cells as evidenced by immunofluorescence and western blot assays.
Immunofluorescence and western blot assays showed that N-cadherin and vimentin proteins were downregulated after HMMR expression was knocked down, while E-cadherin was upregulated (Fig. 3I and Sup Fig. 4E). Conversely, HMMR overexpression resulted in a significant increase in N-cadherin and vimentin protein expression and decrease in E-cadherin protein expression (Fig. 3J). These results indicated that HMMR mediates epithelial-mesenchymal transition (EMT) in NPC cells.
Knocking down HMMR inhibits tumor growth and metastases in vivo
To investigate whether HMMR depletion inhibits NPC tumor growth in vivo, we generated stable SUNE1 cell lines expressing either shHMMR or shControl. In a subcutaneous xenograft model, the tumors in the shHMMR group displayed significantly reduced volume and weight compared to the control group, with minimal or no growth observed in the shHMMR group (Fig. 4A). Tail vein injection assay revealed that the fluorescence intensity of metastatic tumors and the number of metastatic lesions were significantly reduced in the shHMMR group compared to those in the control group (Fig. 4B). Subsequently, HE staining was performed on pathological sections of mouse lung tissue. The results revealed that the control group exhibited a higher number and larger size of metastatic foci, whereas the shHMMR group showed a significant reduction in metastatic foci (Fig. 4C).

The ability of progression in NPC cell in vivo is influenced by HMMR. (A) Subcutaneous tumor formation in mice. (B) Fluorescence intensity of lung metastases in mice (C) Representative HE staining results of lung metastases.
β-catenin May play an important role in HMMR-mediated NPC progression
HMMR influenced the progression of NPC cells in vitro and in vivo. To further explore the mechanism by which HMMR affects NPC progression, bulk RNA sequencing was performed using SUNE1-shHMMR and shControl cells. KEGG enrichment analysis showed that several vital signaling pathways, such as AMPK, TNF, p53, and Wnt signaling pathways, were associated with tumor progression (Fig. 5A). Gene Ontology (GO) enrichment analysis revealed that the Wnt signaling pathway had the highest counts and significantly adjusted P-values compared to those of the other pathways (Fig. 5A). Additionally, cell-cell signaling by the canonical Wnt signaling pathway showed high counts and significantly adjusted P-values (Fig. 5A). Therefore, HMMR affects the Wnt signaling pathway.

FAM83D regulated by HMMR and influence the Wnt signaling pathway. (A) KEGG and GO enrichment analysis of different expression genes. (B) Interaction between HMMR and β-catenin by Co-IP analyses in SUNE1 cells. (C) Protein binding to HMMR was detected by CO-IP/MS analysis. (D) Interaction between HMMR and FAM83D by Co-IP analyses in SUNE1 cells. (E) SUNE1-shHMMR cells transfected with pcDNA3-FAM83D was subjected to western blot for indicated proteins. (F) CNE2-pcDNA3-HMMR cells transfected with shFAM83D was subjected to western blot for indicated proteins.
Furthermore, experiments were conducted to explore whether HMMR promotes NPC progression via the Wnt signaling pathway. Western blot results showed that down expressed HMMR decreased the expression of β-catenin, N-cadherin, and vimentin and increased the expression of E-cadherin (Sup Fig. 5A). Meanwhile, the upregulation of β-catenin improves the expression of N-cadherin and vimentin, decreased the expression of E-cadherin and did not influence the expression of HMMR in HMMR-downregulated SUNE1 cells (Sup Fig. 5A). The wound healing and transwell experiments further showed that the downregulation of HMMR dramatically decreased the migration and invasion abilities of SUNE1 cells, whereas the upregulation of β-catenin rescued the decreased migration and invasion abilities induced by HMMR downregulation (Sup Fig. 5C, E). Upregulation of HMMR expression showed the opposite result in CNE-2 cells, as observed through western blotting and transwell assays (Sup Fig. 5B, D, F).
FAM83D is regulated by HMMR and influences the Wnt signaling pathway
After confirming that HMMR can regulate the Wnt/β-catenin signaling pathway, we aimed to further understand the regulatory molecular mechanism of HMMR. First, co-immunoprecipitation (Co-IP) assay revealed that HMMR does not bind with β-catenin (Fig. 5B). Next, using the liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay to detect the molecules that combine with HMMR, we found that FAM83D was the only molecule (except HMMR) in the three anti-HMMR groups compared to the anti-IgG groups (Fig. 5C). Therefore, Co-IP experiments confirmed that FAM83D interacts with HMMR (Fig. 5D). Furthermore, the expression of FAM83D and active-β-catenin were decreased in HMMR-downexpressing SUNE1 cells (Fig. 5E). Meanwhile, increasing the expression of FAM83D in HMMR- downexpressing SUNE1 cells improve the abundance of active β-catenin (Fig. 5E). Additionally, after upregulating HMMR expression, the expression of FAM83D and active β-catenin was increased (Fig. 5F). Furthermore, decreasing the abundance of FAM83D downregulated the abundance of active β-catenin in HMMR-upnregulated CNE2 cells (Fig. 5F).
HMMR stabilizes FAM83D expression through the ubiquitination of FAM83D in NPC cells
Next, we explored the mechanisms through which HMMR regulates FAM83D expression. HMMR upregulation and downregulation did not alter FAM83D mRNA expression; however, FAM83D protein expression was altered (Fig. 6A, B). HMMR reportedly inhibits the ubiquitination of the AURKA protein that interacts with HMMR11. Based on this, we speculated that HMMR expression may increase the abundance of FAM83D protein by inhibiting FAM83D ubiquitination. To test this, we treated HMMR-knockdown SUNE1 cells with the protein synthesis inhibitor cycloheximide and observed a marked reduction in FAM83D half-life compared to shControl cells (Fig. 6C). Furthermore, the shHMMR-mediated destabilization of FAM83D was reversed by the proteasome inhibitor MG132 (Fig. 6D). Therefore, HMMR silencing promotes the degradation of FAM83D through the ubiquitin-proteasome pathway. Consistent with these results, ubiquitinated FAM83D expression was dramatically increased in HMMR-silenced cells compared to that in shControl cells (Fig. 6E). These results indicate that HMMR plays an important role in FAM83D ubiquitination.

HMMR stabilizes FAM83D through inhibiting the ubiquitination of FAM83D. (A,B) The protein expression of FAM83D can be regulated by HMMR, but HMMR do not affect FAM83D mRNA level in SUNE1, HONE1 and CNE2 cells. (C) Knockdown of HMMR enhanced the degradation of FAM83D. HMMR-silenced cell and control cells were treated with cycloheximide (CHX; 20 µg/ml) for the indicated periods of time. FAM83D protein levels were analyzed by western blotting. (D) Knockdown of HMMR destabilized FAM83D. HMMR silencing, and control groups were treated with MG132 (10 µM) or DMSO for 24 h. Cell lysates were analyzed by western blotting. (E) HMMR stabilized FAM83D through inhibiting ubiquitination. SUNE1 and HONE1 cells with knockdown of HMMR were treated with MG132 (10 µM) for 24 h. Cell lysates were immunoprecipitated with either control IgG or antibody against FAM83D and analyzed by western blotting with a ubiquitin (Ub)-specific antibody. Bottom, input from cell lysates. (F) SUNE1 and HONE1 cells were transfected with different amounts of Flag-HMMR plasmid, and the level of HMMR binding to FAM83D protein was detected by CO-IP. (G) SUNE1 and HONE1 cells were transfected with different amounts of Flag-HMMR plasmid, and the level of Ub binding to FAM83D protein was detected by CO-IP.
Next, we transfected SUNE1 and HONE1 cells with different concentrations of the Flag-HMMR plasmid. Co-IP assay results showed that as exogenous levels of HMMR increased, HMMR-FAM83D levels increased, whereas levels of the ubiquitin-FAM83D complex gradually decreased (Fig. 6F, G). These results confirmed that HMMR and ubiquitin competitively bind to FAM83D and that HMMR overexpression reduces the formation of the ubiquitin-FAM83D complex, thereby increasing FAM83D expression.
Discussion
In this study, we revealed a novel mechanism by which HMMR promotes the expression of FAM83D by inhibiting its ubiquitination, thereby driving the progression of NPC cells. Firstly, HMMR was highly expressed in NPC, and high HMMR expression in patients correlated with poor survival. Second, HMMR increased FAM83D expression and promoted the progression of NPC cells by increasing the β-catenin expression. Finally, we demonstrated that HMMR functions as a modulator of decreased ubiquitination and subsequent degradation of FAM83D.
HMMR is highly expressed in several tumors and is involved in carcinogenesis28. Through in vivo and in vitro experiments, we confirmed that HMMR was significantly overexpressed in NPC and played a crucial role in its growth and metastasis. To our knowledge, this is the first study to show that HMMR expression plays an important role in the progression of NPC. However, the mechanism through which HMMR promotes NPC development remains unclear.
RNA sequencing analysis showed that HMMR affected the Wnt signaling pathway. scRNA-seq data showed that HMMR expression in NPC cells was higher than that in normal epithelial cells and that NPC cells with high-HMMR activity were more enriched in the Wnt signaling pathway than those with low-HMMR activity. Therefore, Wnt signaling pathway-related genes are considered candidate targets of HMMR. In this study, HMMR promoted EMT and NPC metastasis by upregulating active β-catenin. A previous study has shown that β-catenin, the downstream molecule of the Wnt/β-catenin signaling pathway, promotes EMT and tumor metastasis in head and neck squamous cell carcinoma29. HMMR has been shown to interact with β-catenin and inhibit its degradation in fibrosarcoma. Our results also showed that HMMR expression increased the protein levels of active β-catenin. However, the Co-IP analysis did not support a direct interaction between HMMR and β-catenin. Interestingly, our in vitro experiments revealed that knocking down HMMR almost completely inhibited tumor formation in mice. However, this effect was less pronounced in in vivo experiments compared to the in vitro observations. Moving forward, we plan to focus on further exploring the differences between the in vitro and in vivo experimental outcomes.
Our Co-IP, LC-MS/MS, and Western blot analyses revealed that FAM83D interacts with HMMR in NPC cells. FAM83D is involved in mitosis and directs the protein kinase CK1α into the mitotic spindle for proper spindle positioning30. Additionally, FAM83D promotes tumor progression via the Wnt/β-catenin pathway31,32. We found that FAM83D interacted with HMMR in NPC cells and played an important role in regulating the active β-catenin expression through HMMR. A previous study also confirmed that FAM83D combines with HMMR to govern metaphase pate organization and spindle orientation33.
FAM83D reportedly promotes β-catenin protein expression in pancreatic adenocarcinoma and increases β-catenin release into the nucleus in gastric cancer. However, the specific mechanism thereof remains unclear. Recently, a study on glioblastoma showed that FAM83D affects phosphorylated AKT, which in turn affects the phosphorylation of GSK-3β to increase active β-catenin levels32. We did not further analyze these potential molecular mechanisms in the present study. However, future research will explore how FAM83D promotes the increase in activated β-catenin expression in NPC.
An important finding of our study is the preliminary revelation of the mechanism by which HMMR promotes increased FAM83D protein levels by inhibiting the ubiquitin-mediated degradation of FAM83D. Although it has not yet been confirmed whether HMMR functions as a deubiquitinating enzyme, previous studies have shown that HMMR competes with ubiquitin to bind to AURKA, thereby stabilizing the AURKA protein11. This suggests that HMMR may be involved in ubiquitination and could regulate this process in various tumors. Additionally, literature reports indicate that HMMR plays a crucial role in the PLK1 (Polo-like kinase 1)-dependent spindle positioning pathway34while PLK1 has been widely recognized in several studies for its potential association with ubiquitination processes. For example, the C-terminal domain of Ubiquitin-Binding Protein 2-Like (UBAP2L) is thourght to mediates PLK1 function, and it has been observed that UBAP2L-depleted cells might be largely rescued by chemical inhibition of PLK135. Moreover, it is known that PLK1 could induce the ubiquitination and degradation of Smad436. Therefore, the underlying mechanisms regarding the role of HMMR in ubiquitination warrant further investigation and could be an area of focus for future studies.Our study has several limitations. First, although we found that HMMR regulates β-catenin expression through FAM83D, neither HMMR nor FAM83D are classical components of the Wnt/β-catenin signaling axis. Therefore, the precise mechanism by which HMMR modulates β-catenin expression via FAM83D requires further investigation. Second, while our study and previous research suggest that HMMR may play a role in ubiquitination, it remains unclear whether HMMR itself functions as a deubiquitinating enzyme or acts as a cofactor for other ubiquitination-related enzymes. Further studies are needed to elucidate its specific function in this process. Lastly, we observed that the inhibitory effect of HMMR knockdown on tumor progression was more pronounced in in vivo experiments compared to in vitro studies. This discrepancy suggests that HMMR may influence the tumor microenvironment, thereby affecting NPC progression, an aspect that warrants further exploration in future research.
In summary, we demonstrated that HMMR is significantly overexpressed in NPC and plays a crucial role in the growth and metastasis of NPC by regulating β-catenin expression. Additionally, we demonstrated that HMMR promotes NPC progression by interacting with FAM83D and inhibiting the ubiquitination and degradation of FAM83D, which activates the Wnt/β-catenin pathway (Fig. 7). The newly identified HMMR-FAM83D-β-catenin axis provides new insights for NPC therapy.

Scientific hypothesis graph (Created by Adobe Illustrator 2025, https://www.adobe.com/).
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) at the National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: PRJCA028220), and are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
References
Lin, S. et al. Update report of nasopharyngeal carcinoma treated with reduced-volume intensity-modulated radiation therapy and hypothesis of the optimal margin. Radiother Oncol. 110, 385–389 (2014).
Sun, X. et al. Long-term outcomes of intensity-modulated radiotherapy for 868 patients with nasopharyngeal carcinoma: an analysis of survival and treatment toxicities. Radiother Oncol. 110, 398–403 (2014).
Wu, M., Ou, D., Hu, C. & He, X. Comparing Long-Term survival and late toxicities of different sequential chemotherapy regimens with Intensity-Modulated radiotherapy in locoregionally advanced nasopharyngeal carcinoma. Transl Oncol. 13, 100765 (2020).
Zhang, Y. et al. Gemcitabine and cisplatin induction chemotherapy in nasopharyngeal carcinoma. N Engl. J. Med. 381, 1124–1135 (2019).
Sun, Y. et al. Induction chemotherapy plus concurrent chemoradiotherapy versus concurrent chemoradiotherapy alone in locoregionally advanced nasopharyngeal carcinoma: a phase 3, multicentre, randomised controlled trial. Lancet Oncol. 17, 1509–1520 (2016).
Guo, Q. et al. Depicting distant metastatic risk by refined subgroups derived from the 8th edition nasopharyngeal carcinoma TNM. Oral Oncol. 91, 113–120 (2019).
Zhou, H. et al. Effects of oral maintenance chemotherapy and predictive value of Circulating EBV DNA in metastatic nasopharyngeal carcinoma. Cancer Med. 9, 2732–2741 (2020).
Hardwick, C. et al. Molecular cloning of a novel hyaluronan receptor that mediates tumor cell motility. J. Cell. Biol. 117, 1343–1350 (1992).
Turley, E. A., Austen, L., Vandeligt, K. & Clary, C. Hyaluronan and a cell-associated hyaluronan binding protein regulate the locomotion of ras-transformed cells. J. Cell. Biol. 112, 1041–1047 (1991).
He, Z., Mei, L., Connell, M. & Maxwell, C. A. Hyaluronan mediated motility receptor (HMMR) encodes an evolutionarily conserved homeostasis, mitosis, and meiosis regulator rather than a hyaluronan receptor. Cells 9, 819 (2020).
Guo, K. et al. HMMR promotes prostate cancer proliferation and metastasis via AURKA/mTORC2/E2F1 positive feedback loop. Cell. Death Discov. 9, 48 (2023).
Zhang, H. et al. Hyaluronan-mediated motility receptor confers resistance to chemotherapy via TGFβ/Smad2-induced epithelial-mesenchymal transition in gastric cancer. FASEB J. 33, 6365–6377 (2019).
Berdiaki, A. et al. RHAMM/hyaluronan inhibit β-catenin degradation, enhance downstream signaling, and facilitate fibrosarcoma cell growth. Mol. Biol. Rep. 50, 8937–8947 (2023).
Lu, T. et al. High expression of Hyaluronan-Mediated motility receptor predicts adverse outcomes: A potential therapeutic target for head and neck squamous cell carcinoma. Front. Oncol. 11, 608842 (2021).
Ye, S. et al. TGFβ and Hippo pathways cooperate to enhance sarcomagenesis and metastasis through the Hyaluronan-Mediated motility receptor (HMMR). Mol. Cancer Res. 18, 560–573 (2020).
Tarullo, S. E. et al. Receptor for hyaluronan-mediated motility (RHAMM) defines an invasive niche associated with tumor progression and predicts poor outcomes in breast cancer patients. J. Pathol. 260, 289–303 (2023).
Yin, C. et al. FAM83D promotes epithelial-mesenchymal transition, invasion and cisplatin resistance through regulating the akt/mtor pathway in non-small-cell lung cancer. Cell. Oncol. (Dordr). 43, 395–407 (2020).
Mu, Y., Zou, H., Chen, B., Fan, Y. & Luo, S. FAM83D knockdown regulates proliferation, migration and invasion of colorectal cancer through inhibiting FBXW7/Notch-1 signalling pathway. Biomed. Pharmacother. 90, 548–554 (2017).
Zhu, H., Diao, S., Lim, V., Hu, L. & Hu, J. FAM83D inhibits autophagy and promotes proliferation and invasion of ovarian cancer cells via PI3K/AKT/mTOR pathway. Acta Biochim. Biophys. Sin (Shanghai). 51, 509–516 (2019).
Hao, Y. et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 42, 293–304 (2024).
Wu, T. et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb). 2, 100141 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 14, 1083–1086 (2017).
Kanehisa, M., Furumichi, M., Sato, Y. & Kawashima, M. Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Chen, Y. P. et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell. Res. 30, 1024–1042 (2020).
Hinneh, J. A., Gillis, J. L., Moore, N. L., Butler, L. M. & Centenera, M. M. The role of RHAMM in cancer: exposing novel therapeutic vulnerabilities. Front. Oncol. 12, 982231 (2022).
Ji, H. et al. FAM83A promotes proliferation and metastasis via Wnt/β-catenin signaling in head neck squamous cell carcinoma. J. Transl Med. 19, 423 (2021).
Fulcher, L. J. et al. FAM83D directs protein kinase CK1α to the mitotic spindle for proper spindle positioning. EMBO Rep. 20, e47495 (2019).
Hua, Y. Q. et al. Fam83D promotes tumorigenesis and gemcitabine resistance of pancreatic adenocarcinoma through the Wnt/β-catenin pathway. Life Sci. 287, 119205 (2021).
Wang, J., Quan, Y., Lv, J., Gong, S. & Ren, P. Inhibition of FAM83D displays antitumor effects in glioblastoma via down-regulation of the AKT/Wnt/β-catenin pathway. Environ. Toxicol. 37, 1343–1356 (2022).
Dunsch, A. K. et al. Dynein light chain 1 and a spindle-associated adaptor promote dynein asymmetry and spindle orientation. J. Cell. Biol. 198, 1039–1054 (2012).
Connell, M. et al. HMMR acts in the PLK1-dependent spindle positioning pathway and supports neural development. Elife 6, e28672 (2017).
Guerber, L. et al. UBAP2L-dependent coupling of PLK1 localization and stability during mitosis. EMBO Rep. 24, e56241 (2023).
Gao, P. et al. PELO facilitates PLK1-induced the ubiquitination and degradation of Smad4 and promotes the progression of prostate cancer. Oncogene 41, 2945–2957 (2022).
Funding
This study was supported by the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (No. 2020-PT320-004), Natural Science Foundation of Jiangxi Province (20212BAB216064, 20224BAB216053, 20224BAB206065, 20242BAB25517), National Natural Science Foundation of China (grant: 82103478, 82160710), Key Project of Jiangxi Provincial Department of Education (GJJ2203507), Jiangxi Province Key R&D Program (Key Program) (grant: 20232BBG70025), and The “Five-level Progressive” talent cultivation project of Jiangxi Cancer Hospital & Institute (WCDJ2024YQ01).
Author information
Authors and Affiliations
Contributions
JGL, FYZ, TZL, ML & DCL: Conceived and designed the research; FYZ, TZL, ML, DCL, JLW, AQY & HL: Conducted the cellular and molecular biological experiments; TZL & ML: Conducted the animal experiments; FYZ, TZL, YS, QD, JW, KH and LZ: Analyzed the data; FYZ, TZL and ML: Wrote the manuscript; ZLL, CSH, XXP, XCG and JGL helped to revise the manuscript. All authors approved the final version of this manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was conducted in accordance with the Declaration of Helsinki and approved by the Medical Ethics Committee of Jiangxi Cancer Hospital (No. 20220956). Written informed consent was obtained from each patient. All the animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and the Institutional Code of Ethics for Animal Experiments. Ethical approval for the animal research was obtained from Nanchang Royo Biotech Co., Ltd. (approval number: RYE2024031001). All procedures were conducted in full compliance with the ARRIVE guidelines.
Consent for publication
All authors have read and approved of its submission to this journal.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhong, F., Lu, T., Li, M. et al. HMMR mediates progression of nasopharyngeal carcinoma by inhibiting FAM83D ubiquitination and activating beta-catenin signaling pathway. Sci Rep 15, 35701 (2025). https://doi.org/10.1038/s41598-025-19641-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19641-z
