
Abstract
What drives inflammation-associated tumorigenesis and progression in pancreatic ductal adenocarcinoma (PDAC)? Doublecortin-like kinase 1 (DCLK1) is a central driver of inflammation-associated tumorigenesis, with elevated expression linked to worse clinical outcomes. Two isoforms of DCLK1 possess a unique extracellular domain (ECD). DCLK1 isoform 2 contains two microtubule-binding domains, while isoform 4, lacks the microtubule-binding domains but, plays a pivotal role in tumor progression. We identified novel D-peptides that selectively target this ECD, significantly suppressing PDAC cell proliferation in vitro and tumor growth in xenograft models without inducing cell death. In silico modeling and binding assays revealed DCLK1 isoform 4 interacts with pro-tumorigenic proteins like plasma gelsolin (pGSN), with D-peptides modulating these interactions. These findings underscore DCLK1’s non-kinase functions as a therapeutic target and highlight novel avenues for developing precision treatments aimed at halting cancer progression and improving patient outcomes.
Similar content being viewed by others


Introduction
Doublecortin-like kinase 1 (DCLK1) is a recognized marker of tumor stemness across many solid tumor cancers, including pancreatic ductal adenocarcinoma (PDAC)1. It plays a key role in epithelial-to-mesenchymal transition (EMT), facilitating metastasis and resistance to chemo-, radio-, and immunotherapies2,3,4. Recent studies have identified crystal structures of the DCLK1 kinase domain, spurring interest in developing specific kinase inhibitors5. However, DCLK1’s multi-functional nature, derived from its isoforms, DCLK1 isoform 1 (alpha-long) and DCLK1 isoform 4 (beta-short), adds complexity to understanding its precise roles in tumorigenesis6,7. DCLK1 isoform 4 lacks the microtubule-binding domains but includes a unique extracellular domain (ECD) and is strongly associated with metastasis and poor survival (Fig. 1A)8. This unique ECD is shared with DCLK1 isoform 2, which also contains the microtubule-binding domains.

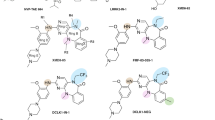
Isolation of D-peptides Targeting the Unique C-terminus of DCLK1 Isoform 4. (A) Alignment of human DCLK1 isoform 1 (α-long, NP_004725) and isoform 4 (β-short, NP_001182345) highlights structural differences. Isoform 1 contains the microtubule-binding domains DCX1 and DCX2 (orange), absent in isoform 4, along with the conserved kinase domain (red) and auto-inhibitory domain (blue). The unique NKEBD sequence is shaded purple with the CBT-15 peptide sequence (also used to develop CBT-15) is boxed and underlined. (B) Amino acid sequences of six Dpeptides selected by screening against the DCLK1 isoform 2/4 Cterminal region. Gray boxes denote the core pentapeptide motifs from which the library was constructed.
Crystal structure studies of the DCLK1 kinase domain that demonstrate binding to drug inhibitors show a complex regulatory relationship between the functional domains of DCLK19,10. Interestingly, the DCLK1 kinase domain is regulated by a C-terminal autoinhibitory domain (AID), the novel Hippocalcin like-1 protein (HPCAL1) binds to the AID and activates the DCLK1 kinase in a Ca2+-dependent manner11,12. Thus, C-terminal protein-protein interactions (PPI) control DCLK1 kinase activity and may provide for more nuanced forms of regulation. Surface-biotinylation studies have shown that DCLK1 is cell-surface exposed and that the DCLK1 isoforms 2/4 C-terminal domain is extracellular13,14. We previously reported that a monoclonal antibody (CBT-15 mAb) targeting a unique ECD region, termed the non-kinase extracellular binding domain (NKEBD), reduces PDAC tumor growth and disrupts tumorigenic signaling7,15. Here, we extend these findings by developing a novel D-peptide that binds the NKEBD of DCLK1 isoforms 2/4, inhibiting tumor growth in vitro and in vivo. Additionally, we identify potential interacting proteins through in silico modeling, revealing mechanisms of DCLK1-mediated pro-tumorigenic activity.
Results
Identification of D-peptides targeting the C-terminus of DCLK1 isoforms 2/4
Building on the success of CBT-15, we hypothesized that the unique C-terminus of DCLK1 isoform 2/4 has functional domains that can be targeted by smaller, more stable D-peptides. We screened a proprietary D-peptide library enriched in hydrophobic and aromatic residues for peptides that bound specifically to isoform 4, excluding those that bound isoform 1, and identified six candidates (Fig. 1B).
D-Peptide 1 suppresses PDAC cell proliferation and tumor growth
Functional assays were conducted in PDAC cell lines AsPC-1 and Capan-1. We chose AsPC-1 for its high DCLK1 expression16while CAPAN-1 is known to form DCLK1-dependent spheroids. Both D-peptides 1 and 5 significantly reduced colony formation (clonogenicity) at low micromolar concentrations (Fig. 2A,B). This blinded study revealed that D-peptides 2, 3, 4, and 6 were less active. Additionally, spheroid formation assays indicated that D-peptide 1 markedly impaired self-renewal capacity, suggesting a disruption of DCLK1-mediated stemness (Fig. 2C,D).

Anti-Tumor Effects of DCLK1 D-peptides on Pancreatic Cancer Cells (A-B) Colony Formation Assays: Boxplots show colony counts after treatment in (A) AsPC-1 and (B) Capan-1 pancreatic cancer cells. (C,D) PDAC Spheroid Self-Renewal: Boxplots display spheroid counts following treatment in (C) AsPC-1 and (D) Capan-1 cells. (E,F) In Vivo Efficacy of D-peptide 1: AsPC-1-derived xenografts were treated with D-peptide 1 or control peptide (D-peptide 3) every three days for six doses. (E) Representative tumor images after 36 days of treatment. (F) D-peptide 1 significantly reduced tumor size compared to control (Control n = 8, D-peptide 1 n = 9; *p < 0.01).
Given the inhibitory activity of D-peptide 1 in vitro, we investigated its effectiveness on tumor xenograft growth inhibition in vivo. Initial drug toxicity experiments in athymic mice found no apparent toxicity (Fig. S1). In athymic mice bearing AsPC-1 xenografts, D-peptide 1 treatment reduced tumor volume by 60% compared to controls (Fig. 2E,F). Afterwards the xenografts were excised and evaluated by gross inspection, no visible necrosis or hemorrhage was observed. These results suggest that tumor growth suppression occurred without inducing necrosis or apoptosis, indicating non-tumoricidal mechanisms.
DPeptide 1 targets a unique epitope on DCLK1 isoform 4 with high affinity
Building on our prior demonstration that mAb CBT-15 inhibits PDAC tumor growth7,15 we assessed whether Dpeptides compete with CBT-15 for DCLK1 isoform 4 binding. Dpeptide 1 was selected for its high potency in the bioassays, Dpeptide 3 for minimal activity and a singleresidue divergence from the D-peptide 1 sequence, lastly, we also examined Dpeptide 5 as it showed robust bioactivity, albeit lower potency. Peptides 2, 4, and 6 showed negligible effects and were excluded. In competition assays, none of the Dpeptides impeded CBT-15 mAb binding, whereas the CBT-15 peptide, a mimetic for the CBT-15 epitope, itself fully blocked the interaction (Fig. 3A), indicating that the Dpeptides target distinct epitopes from that of CBT-15. Consistent with its potent anticlonogenic effect and unique binding site, Dpeptide 1 exhibited high affinity for DCLK1 isoform 4 (KD = 396 nM; Fig. 3B). These findings establish Dpeptide 1 as a highaffinity, functionally discrete ligand of DCLK1 isoform 4, warranting its advancement into mechanistic and therapeutic evaluations.

DPeptide 1 Binds a Distinct Epitope on DCLK1 Isoform 4 with Nanomolar Affinity (A) D-peptide 1 interacts at a site distinct from CBT-15’s binding site. A competition assay demonstrates mAb CBT-15 binding to DCLK1. Data are presented as mean ± SEM; *p < 0.01. (B) Bio-layer interferometry (BLI) analysis shows binding of D-peptide 1 at four concentrations to DCLK1 isoform 4 immobilized on a biosensor to determine the KD of 396 nM.
Identification of DCLK1 binding partners
Given the functional response of D-peptide 1 on tumor growth, we identified potential pro-tumorigenic binding partners that have the potential to bind specifically to the C-terminal extracellular domain of DCLK1. While the unique C-termini of DCLK1 isoform 4 and the related longer isoform DCLK1 isoform 2 are largely disordered and omitted from crystal structure-based analysis9,17,18,19 a high-quality candidate partial structure exists20 that can be modeled to obtain a representative structure for the DCLK1 NKEBD. Using this representative structure and a modeled structure for D-peptide 1 we looked for proteins with structures like D-peptide 1 that had known roles in cancer stemness, inflammation, immune modulation, or EMT. Based on the ECD amino acid sequence corresponding to the D-peptide 1 region we performed an in silico 3-D confirmational binding analysis with a stringency of greater than 80% binding affinity and identified five top potential binding partners (Table 1) Based on our 3-D modeling and in silico top hit strategy (Fig. 4A) we chose to evaluate the key innate immunity modulatory protein plasma gelsolin (pGSN) which is extracellularly localized and has also been implicated in the tumorigenic processes in several solid tumor cancers including PDAC21,22. Immunohistochemistry revealed intermittent colocalization of DCLK1 and pGSN in PDAC tissues (Fig. 4B). However, in a cancer more affiliated with inflammation we found that the colocalization of DCLK1 and pGSN in colon cancer was more abundant. (Fig. 4C).

Gelsolin and FGF19 as Potential Extracellular DCLK1 Interactors (A) Identification Strategy for DCLK1 NKEBD Interactors: Step 1: BLAST peptide sequence against protein databases; Step 2: Analyze top-ranked proteins; Step 3: Upload and examine 3D structures; Step 4: Validate binding interactions experimentally. (B–E) Co-localization of DCLK1 with Gelsolin and FGF19: (B) DCLK1 and Gelsolin in PDAC; (C) DCLK1 and Gelsolin in CRC; (D) DCLK1 and FGF19 in PDAC; (E) DCLK1 and FGF19 in CRC. (F,G) SPR Binding Analyses: (F) DCLK1 isoform 4 binding to Gelsolin; (G) DCLK1 isoform 4 binding to FGF19. (H) Effects of Peptides on DCLK1-Gelsolin and DCLK1-FGF19 PPI.
We also examined another candidate DCLK1 interacting protein known to be extracellularly localized, FGF19. In both PDAC and normal adjacent tissue (NAT) tissue, no colocalization was observed between DCLK1 and FGF19 (Fig. 4D). In contrast we observed that DCLK1 and FGF19 were colocalized in both colorectal cancer and NAT tissues (Fig. 4E) supporting a role for their interaction in colon cancers but not PDAC.
Given the colocalization results we examined whether DCLK1 isoform 4 physically interacts with pGSN and FGF19 using surface plasmon resonance (SPR). We found that PPI between DCLK1 isoform 4 and Gelsolin or FGF19 via SPR data indicated that DCLK1 isoform 4 is associated with FGF19 with a 1-1.8 mM KD, whereas Gelsolin was ~ 100 nM KD (Fig. 4F and G, and Suppl. Table 1).
Biophysical characterization of DCLK1-Gelsolin Protein-Protein interactions
To assess the effects of DCLK1 NKEBD peptides on the binding of pGSN and FGF19 to DCLK1, we conducted ELISA-based binding assays. Initially, we observed consistent, linear binding between DCLK1 isoform 4 and pGSN across the gradient, with maximum binding at 2 µg/ml (Fig. S2). We used this concentration to test whether D-peptide 1, D-peptide 3, D-peptide 5, or the CBT-15 blocking peptide could disrupt pGSN or FGF19 binding to DCLK1. FGF19 bound to DCLK1 isoform 4 similarly to pGSN and the CBT-15 peptide effectively blocked their binding to DCLK1 isoform 4 (Fig. 4H, CBT peptide). Interestingly, the D-peptides, especially D-peptide 1, did not block the PPI interaction but seemed to improve pGSN binding to DCLK1 suggesting a potential synergistic effect (Fig. 4H, D-peptide 1). These data confirm that pGSN and FGF19 both engage the Cterminal ECD of DCLK1 isoform 4, whereas Dpeptide 1 binds a distinct site and allosterically enhances pGSN association, highlighting its potential for synergistic modulation of DCLK1 PPIs.
Discussion
This study identifies and characterizes a novel D-peptide, D-peptide 1, that targets the unique C-terminal region (NKEBD) of DCLK1 isoforms 2 and 4. Focusing on the NKEBD rather than the kinase domain uncovers its underappreciated role in modulating PPIs that drive tumor progression. Dpeptide 1 binds DCLK1 isoform 4 with high affinity (KD = 396 nM; Fig. 3B) and shows minimal crossreactivity with isoform 1. Competition assays confirm that its epitope is distinct from that of mAb CBT-15, suggesting opportunities for combinatorial or sequential targeting. Although the precise Dpeptide binding sites remain to be mapped, in silico modeling—supported by SPR and ELISA—also implicates plasma gelsolin (pGSN) as a key PPI partner of DCLK1 isoform 4 (KD = ~ 100 nM), and Dpeptide 1 enhances this interaction, consistent with allosteric modulation, rather than blockade, of downstream protumorigenic signaling23,24. Notably, while D-peptide 1 enhances pGSN binding, its weak effect with FGF19-DCLK1 association underscores the selective nature of these PPIs.
In vivo, Dpeptide 1–treated flank xenografts exhibited no gross necrosis, hemorrhage, or cavitation, consistent with a primarily cytostatic mechanism that aligns with modest in vitro proliferation decreases. Detailed histopathology, including H&E, cleaved caspase-3, and Ki-67 analyses, are needed to delineate apoptosis versus growth arrest. Despite their limited microenvironmental fidelity, these flank models validated peptide bioavailability, stability, and ontarget activity, as evidenced by tumor suppression without general toxicity in non–DCLK1–expressing tissues, and justify advancement to orthotopic and genetically engineered systems.
DCLK1 expression in PDAC has long been characterized as restricted to epithelial compartments—particularly tuft cells and the cancer stem–like subpopulation—while absent from stromal elements14. This epithelial specificity underpins our strategy to target DCLK1 isoform 4 in tumor cells without offtarget effects on fibroblasts or endothelial cells. Intriguingly, recent studies have identified a subset of macrophages that express DCLK1, suggesting a potential role in modulating the immune microenvironment25,26,27. These findings raise the possibility that Dpeptide 1 might also influence DCLK1⁺ macrophage function.
Peptides have emerged as effective tools to disrupt PPI in cancer biology, thanks to their small size, conformational flexibility, and high specificity28,29. Regulating PPI has been a longstanding goal in drug discovery30and peptides’ small size and conformational flexibility enhance their attractiveness for this strategy. Therapeutic peptides disrupting PPIs face challenges like protease degradation31, but advances in peptide chemistry have improved stability32. D-peptides, which resist degradation, offer promise for targeting PPIs33. Compared to conventional L-peptides, D-peptides offer greater in vivo stability, specificity, and reduced immunogenicity, and can be readily modified for imaging, diagnostics, improved oral absorption, and tissue-specific targeting34,35,36,37.
Multiple binding partners interacting with distinct motifs in the C-terminal domain likely contribute to DCLK1’s roles in both inflammation and cancer. D-peptide 1 effectively suppressed tumor growth and stemness in vitro and in vivo potentially by modulating pGSN or other PPI binding without complete disruption, underscoring the therapeutic potential of targeting the DCLK1 NKEBD. Future work will define Dpeptide 1’s precise epitope, optimize its stability and specificity for clinical use, and evaluate synergistic combinations with mAb CBT-15 to fully leverage DCLK1’s therapeutic potential.
Methods
Synthesis of D-peptide library
Solid-phase peptide synthesis was used to create the D-peptide probes using appropriate protection/deprotection steps38. The D-peptide library was synthesized (Peptides International, Louisville, KY), using a TentaGel S resin, NH2 (TentaGel beads). Except for glycine, which is an achiral molecule, all the amino acid residues in the D-peptides are of the D-configuration. The TentaGel beads have a polystyrene core with polyoxyethylene arms attached to the core; each arm has a primary amine functional group at its terminus. The resin contains 8.8 7 × 105 beads/gram, an average bead diameter of 130 microns, 0.2–0.3 med/gram capacity, and 280–330 pmol of primary amine groups/bead capacity. The amino acids were conjugated to the resin and deprotected using standard D-peptide synthetic chemistries. Glycine was attached to the resin to achieve about a 30% substitution of the available primary amine groups at the ends of the polyoxyethylene chains of the TentaGel beads. The amine groups to which glycine was not added were blocked by acetylation using acetic anhydride. A 30% substitution yields an average spacing of about 100 to 200 angstroms between D-peptides on the bead surface. This was done to optimize the binding of a single protein to a single D-peptide sequence and to reduce the likelihood that steric hindrance will prevent a protein molecule from binding to a D-peptide or that a protein molecule will bind to more than one D-peptide.
Following the blocking of the unreacted primary amine groups, the D-peptide library was built by the split synthesis method39. The resin mixture was divided equally into five portions and one of the amino acids, Glycine (G), D-Alanine (A), D-tryptophan (W), D-tyrosine (Y), or D-phenylalanine (F), was added by covalent coupling to each of the five portions of the G-substituted resin. The beads were then recombined, and again equally divided into five portions, and each used in reactions one of G, A, F, Y, or W. This procedure was repeated for five cycles to yield a library of pentapeptide sequences attached to the G residues of the resin. Each bead contained multiple copies of a single D-peptide sequence. Because five amino acids were used at each of the five amino acid-adding steps, the resulting bead library contained 3125 unique pentapeptide sequences. Following the final amino acid addition, the reaction mixtures were not recombined, which resulted in five sub-libraries of 625 different sequences, designated G, A, F, Y, or W, according to the last amino acid added. A sixth G amino acid residue was then added to all sub-libraries, before the addition of 2–3 amino acids with charged R groups, specifically aspartic acid (D) and lysine (K). This resulted in a total library of ~ 50,000 different D-peptide sequences.
Screening the D-peptide library
An aliquot from each sub-library, containing approximately 1000 beads, was added to a well of a 96-well polystyrene multi-well plate. From 1.5 to 2 ml of Superblock (Pierce Chemical Company, Rockford, IL.) reagent, 0.1% gelatin, or 1%(w/v) bovine serum albumin (BSA) in phosphate-buffered saline (PBS), pH 7.4, was added to each well, and the plates were incubated for one to two hours at room temperature (RT), with periodic or continuous mixing by gentle rocking. Purified DCLK1 isoform 4 protein was labeled with biotin using the biotinylating reagent NHS-LC-biotin (Pierce Chemical Company) according to the supplier’s instructions. Biotin-labeled protein was detected using AP-conjugated to neutravidin (Pierce Chemical Company) incubated with the beads for 30 min, after which the beads were washed three times with a Tris-buffered saline solution (pH 7.5), with the second wash being left in contact with the beads for 30 min. One-step NBT/BCIP (nitro-blue tetrazolium chloride/5-bromo-4-chloro-3’-indolyphosphate p-toluidine salt) (Pierce Chemical Co.) was then added and the beads observed under a low-power microscope until some of the beads had turned a dark purple color. The beads were washed with PBS twice, followed by 1% acetic acid and a final wash in water. Dark purple-black beads were removed using a small-bore pipette and subjected to amino acid sequence analysis.
Synthesis and purification of DCLK1 isoform 4
Purification of human DCLK1 isoform 4 was performed at the University of Oklahoma Protein Production and Characterization Core Facility (Norman, OK) using an E. coli strain BL21(DE3) pLPP containing the pET19-DCLK1 isoform 4 plasmid. For Bio-layer Interferometry (BLI) experiments, a C-terminal DCLK1 isoform 2/4 His-tagged protein was used (NovoPro, Shanghai, China). All His-tagged proteins were dialyzed using PBS/glycerol to remove imidazole.
BLI evaluation of D-peptide binding to DCLK1 NKEBD
Human His-tagged DCLK1 C-terminal recombinant protein from NovoPro was used. Binding to the sensor was carried out in PBS/0.05%Tween20/1%DMSO. Response is measured as a nm shift in the interference pattern and is proportional to the number of molecules bound to the biosensor’s surface. Initially, all peptides were tested at 5 and 30 µM. The top binders were tested again with eight different concentrations, starting at 30 µM and diluted in a 1:3 series to determine the dissociation constant (KD).
Proliferation assay
AsPC-1 and CAPAN-1 cells were grown as previously described with varying concentrations of candidate D-peptides40. Afterward, we treated cells with TACS MTT Reagent (RND Systems) at 37 C until a dark crystalline precipitate within the cells was observed. NH4OH in DMSO was added and incubated for 10 min while protected from light. We determined the OD550 and averaged it as a percentage of cell proliferation.
Self-renewal assay
AsPC-1 and CAPAN-1 cancer cells were treated with candidate D-peptides or vehicle control and suspended in growth factor-reduced Matrigel™ for 7 days or 0.3% agar for 3 weeks and monitored for spheroid growth. The size and number of spheroids were analyzed, counted, and then compared between the treatment groups41,42.
Ethical statement
The animal studies were approved by the University of Oklahoma Health Sciences Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with all relevant guidelines and regulations. In addition, this study adheres to the ARRIVE guidelines for reporting research involving live animals43. All anesthesia and euthanasia methods employed are consistent with current veterinary best practices.
Mice xenografts
Athymic nude (Foxn1nu/Foxn1nu) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine) and housed in pathogen-free conditions. AsPC-1 cells (1 × 107) were injected subcutaneously into the flanks of 4- to 6-week-old mice (n = 3/group). Tumors were measured using a caliper and the volume was calculated as (length × width2) × 0.5. Once tumors achieved an average size of ~ 100 mm3 mice were injected via intraperitoneal injection (10 mg/kg, three times weekly) with selected D-peptides. Treatments were continued every three days until day 36. Xenografts were then excised and evaluated by gross inspection.
ELISA assay
Polystyrene microtitre Immulon 2HB plate was coated with freshly prepared (400 ng/50 ml) DCLK1 isoform 4 (50 ml/well) in 0.1 M NaHCO3, pH 8.6, overnight at 4 C. After blocking with 1% gelatin in TTBS at RT, different concentrations of gelsolin or FGF19 were added (2 mg/50ul/well, 1 mg/well, 0.5 mg/well, and 0.25 mg/well at 50 ml/well, proteins diluted in TTBS), and incubated at RT. To study the competitive ability of peptides, we used 20 mg/well of either D-peptide 1, D-peptide 3, D-peptide 5, or the C-terminal peptide antigen for the mAb CBT-15. After incubation at RT, plates were washed with TTBS, and bound gelsolin or FGF19 was detected. Bound plasma gelsolin (ab233671; Abcam, Waltham, MA) was immunodetected with anti-gelsolin Ab (ab11081; Abcam, Waltham, MA) or FGF19 (969-FG-025; Bio-Techne, Minneapolis, MN) immunodetected by anti-FGF19 (sc-390621; Santa Cruz Biotechnology, Dallas, TX). Detection was by alkaline phosphatase-conjugated goat anti-mouse IgG (ab97020; Abcam, Waltham, MA) followed by 1-STEP NBT/BCIP substrate (#34042; ThermoFisher Scientific, Waltham, MA) and quantified at 450 nm on a microplate reader.
Modeling of the peptide binding interaction with DCLK1 NKEBD
We modeled peptide binding interactions with DCLK1 NKEBD using the high-quality partial structure 3VSM_A (100% identity, 0% gaps)20. While this structure provides a representative model for DCLK1 NKEBD, peptide configurations and binding conformations remain undefined and require computational modeling. The D-peptide 1 sequence is present in other proteins with resolved tertiary structures. A PDB search identified several relevant structures, including the X-ray structure of calcium-free human gelsolin (PDB: 3FFN_A)44. We used Autodock45 and ICM-Dock (Molsoft LLC, San Diego, CA) to predict ligand binding affinities, allowing ligand flexibility but assuming a rigid receptor. To address receptor flexibility, we employed docking platforms such as GOLD (The Cambridge Crystallographic Data Centre, Cambridge, UK), CABS-Dock46 and FlexAID47. Candidate conformations were identified through clustering based on pose, configuration, and predicted binding energy, enabling molecular dynamics (MD) simulations. We further applied free energy perturbation (FEP) methods based on MD simulations to estimate ligand binding affinities48.
Statistical analysis
For statistical analyses, we used either a student t-test or analysis of variance (ANOVA) for continuous outcomes or the Kruskal-Wallis test for the number of spheroids using GraphPad Prism, with P values < 0.05 (two-sided) considered statistically significant. Data were transformed as necessary. All experiments were performed independently a minimum of three times. If the overall statistical significance was reached, pairwise comparisons (e.g., DCLK1 D-peptide vs. control D-peptide) were conducted with multiple tests adjusted by Bonferroni’s method.
Data availability
All data are available in the main text or the supplementary materials.
References
Lu, Q. et al. Role of DCLK1 in oncogenic signaling (Review). Int. J. Oncol. 61, 137. https://doi.org/10.3892/ijo.2022.5427 (2022).
Chandrakesan, P. et al. Regulatory roles of Dclk1 in epithelial mesenchymal transition and cancer stem cells. J. Carcinog. Mutagen. https://doi.org/10.4172/2157-2518.1000257 (2016).
Safa, A. R. Epithelial-mesenchymal transition: A hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance. J. Cancer Metastasis Treat. 6, 36 (2020).
Sureban, S. M. et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 71, 2328–2338. https://doi.org/10.1158/0008-5472.CAN-10-2738 (2011).
Ferguson, F. M. et al. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat. Chem. Biol. https://doi.org/10.1038/s41589-020-0506-0 (2020).
Burgess, H. A. & Reiner, O. Alternative splice variants of doublecortin-like kinase are differentially expressed and have different kinase activities. J. Biol. Chem. 277, 17696–17705. https://doi.org/10.1074/jbc.M111981200 (2002).
Ge, Y. et al. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int. J. Cancer. 143, 1162–1175. https://doi.org/10.1002/ijc.31400 (2018).
Ye, L. et al. DCLK1 and its oncogenic functions: A promising therapeutic target for cancers. Life Sci. 336, 122294. https://doi.org/10.1016/j.lfs.2023.122294 (2024).
Patel, O. et al. Structural basis for small molecule targeting of doublecortin like kinase 1 with DCLK1-IN-1. Commun. Biol. 4, 1105. https://doi.org/10.1038/s42003-021-02631-y (2021).
Patel, O. et al. Biochemical and structural insights into doublecortin-like kinase domain 1. Structure 24, 1550–1561. https://doi.org/10.1016/j.str.2016.07.008 (2016).
Cheng, L. et al. DCLK1 autoinhibition and activation in tumorigenesis. Innov. (N Y). 3, 100191. https://doi.org/10.1016/j.xinn.2021.100191 (2022).
Cheng, L. et al. DCLK1 autoinhibition and activation in tumorigenesis. Innov. (N Y). 3, 100191. https://doi.org/10.1016/j.xinn.2021.100191 (2022).
Bailey, J. M. et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 146, 245–256. https://doi.org/10.1053/j.gastro.2013.09.050 (2014).
May, R. et al. Doublecortin and cam kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells. 27, 2571–2579. https://doi.org/10.1002/stem.193 (2009).
Sureban, S. M. et al. DCLK1 monoclonal Antibody-Based CAR-T cells as a novel treatment strategy against human colorectal cancers. Cancers (Basel). https://doi.org/10.3390/cancers12010054 (2019).
Weygant, N. et al. Small molecule kinase inhibitor LRRK2-IN-1 demonstrates potent activity against colorectal and pancreatic cancer through Inhibition of doublecortin-like kinase 1. Mol. Cancer. 13, 103. https://doi.org/10.1186/1476-4598-13-103 (2014).
Carli, A. L. E. et al. Structure-Guided prediction of the functional impact of DCLK1 mutations on tumorigenesis. Biomedicines https://doi.org/10.3390/biomedicines11030990 (2023).
Venkat, A. et al. Mechanistic and evolutionary insights into isoform-specific ‘supercharging’ in DCLK family kinases. eLife 12, RP87958. https://doi.org/10.7554/eLife.87958 (2023).
Jang, D. M. et al. Structural basis of Inhibition of DCLK1 by ruxolitinib. Int. J. Mol. Sci. 22, 8488 (2021).
Kawaguchi, Y. et al. Crystallization and X-ray diffraction analysis of chondroitin lyase from baculovirus: Envelope protein ODV-E66. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 190–192. https://doi.org/10.1107/s1744309111053164 (2012).
Hsieh, C. H. & Wang, Y. C. Emerging roles of plasma gelsolin in tumorigenesis and modulating the tumor microenvironment. Kaohsiung J. Med. Sci. 38, 819–825. https://doi.org/10.1002/kjm2.12578 (2022).
Thompson, C. C. et al. Pancreatic cancer cells overexpress gelsolin family-capping proteins, which contribute to their cell motility. Gut 56, 95–106. https://doi.org/10.1136/gut.2005.083691 (2007).
Hsieh, C. H. & Wang, Y. C. Emerging roles of plasma Gelsolin in tumorigenesis and modulating the tumor microenvironment. Kaohsiung J. Med. Sci. 38, 819–825. https://doi.org/10.1002/kjm2.12578 (2022).
Asare-Werehene, M. et al. Plasma Gelsolin confers chemoresistance in ovarian cancer by resetting the relative abundance and function of macrophage subtypes. Cancers (Basel). https://doi.org/10.3390/cancers14041039 (2022).
Yang, B. et al. Macrophage DCLK1 promotes obesity-induced cardiomyopathy via activating RIP2/TAK1 signaling pathway. Cell. Death Dis. 14, 419. https://doi.org/10.1038/s41419-023-05960-4 (2023).
Huang, Z. et al. Macrophage DCLK1 promotes atherosclerosis via binding to IKKbeta and inducing inflammatory responses. EMBO Mol. Med. 15, e17198. https://doi.org/10.15252/emmm.202217198 (2023).
Undi, R. B. et al. Targeting doublecortin-like kinase 1 (DCLK1)-regulated SARS-CoV-2 pathogenesis in COVID-19. J. Virol. 96, e00967–e00922 (2022). https://doi.org/10.1128/jvi.00967-22
Sorolla, A. et al. Precision medicine by designer interference peptides: Applications in oncology and molecular therapeutics. Oncogene 39, 1167–1184. https://doi.org/10.1038/s41388-019-1056-3 (2020).
Wang, H. et al. Peptide-based inhibitors of protein–protein interactions: Biophysical, structural and cellular consequences of introducing a constraint. Chem. Sci. 12, 5977–5993. https://doi.org/10.1039/D1SC00165E (2021).
Lee, A. C., Harris, J. L., Khanna, K. K. & Hong, J. H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20102383 (2019).
López-Otín, C. & Matrisian, L. M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer. 7, 800–808. https://doi.org/10.1038/nrc2228 (2007).
Uhlig, T. et al. The emergence of peptides in the pharmaceutical business: From exploration to exploitation. EuPA Open. Proteom. 4, 58–69. https://doi.org/10.1016/j.euprot.2014.05.003 (2014). https://doi.org/https://doi.org/
Li, H. et al. Roles of d-Amino acids on the bioactivity of host defense peptides. Int. J. Mol. Sci. https://doi.org/10.3390/ijms17071023 (2016).
Wang, L. et al. Therapeutic peptides: Current applications and future directions. Signal. Transduct. Target. Therapy. 7, 48. https://doi.org/10.1038/s41392-022-00904-4 (2022).
Choi, J. S. & Joo, S. H. Recent trends in cyclic peptides as therapeutic agents and biochemical tools. Biomol. Ther. (Seoul). 28, 18–24. https://doi.org/10.4062/biomolther.2019.082 (2020).
Essler, M. & Ruoslahti, E. Molecular specialization of breast vasculature: A breast-homing phage-displayed peptide binds to aminopeptidase P in breast vasculature. Proc. Natl. Acad. Sci. U S A. 99, 2252–2257. https://doi.org/10.1073/pnas.251687998 (2002).
Myrberg, H., Zhang, L., Mäe, M. & Langel, U. Design of a tumor-homing cell-penetrating peptide. Bioconjug. Chem. 19, 70–75. https://doi.org/10.1021/bc0701139 (2008).
Cordova, A. et al. Aminopeptidase P mediated targeting for breast tissue specific conjugate delivery. Bioconjug. Chem. 27, 1981–1990. https://doi.org/10.1021/acs.bioconjchem.5b00481 (2016).
Lebl, M. et al. One-bead-one-structure combinatorial libraries. Biopolymers 37, 177–198. https://doi.org/10.1002/bip.360370303 (1995).
Chandrakesan, P. et al. DCLK1-Isoform2 alternative splice variant promotes pancreatic tumor immunosuppressive M2-Macrophage polarization. Mol. Cancer Ther. 19, 1539–1549. https://doi.org/10.1158/1535-7163.Mct-19-0776 (2020).
Chandrakesan, P. et al. Dclk1 + small intestinal epithelial tuft cells display the hallmarks of quiescence and self-renewal. Oncotarget 6, 30876–30886. https://doi.org/10.18632/oncotarget.5129 (2015).
Chandrakesan, P. et al. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 5, 9269–9280. https://doi.org/10.18632/oncotarget.2393 (2014).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Nag, S. et al. Ca2 + binding by domain 2 plays a critical role in the activation and stabilization of Gelsolin. Proc. Natl. Acad. Sci. U S A. 106, 13713–13718. https://doi.org/10.1073/pnas.0812374106 (2009).
mgl-admin. AutoDock, https://autodock.scripps.edu.
Kurcinski, M., Jamroz, M., Blaszczyk, M., Kolinski, A. & Kmiecik, S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 43, W419–W424. https://doi.org/10.1093/nar/gkv456 (2015).
Morency, L. P., Gaudreault, F. & Najmanovich, R. Applications of the NRGsuite and the molecular docking software flexaid in computational drug discovery and design. Methods Mol. Biol. 1762, 367–388. https://doi.org/10.1007/978-1-4939-7756-7_18 (2018).
Wang, L., Chambers, J. & Abel, R. Protein-Ligand binding free energy calculations with FEP. Methods Mol. Biol. 2022, 201–232. https://doi.org/10.1007/978-1-4939-9608-7_9 (2019).
Park, I. Y. et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962. https://doi.org/10.1016/j.cell.2016.07.005 (2016).
Lang, L., Shull, A. Y. & Teng, Y. Interrupting the FGF19-FGFR4 axis to therapeutically disrupt cancer progression. Curr. Cancer Drug Targets. 19, 17–25. https://doi.org/10.2174/1568009618666180319091731 (2019).
Yang, Y. et al. FOXM1/DVL2/Snail axis drives metastasis and chemoresistance of colorectal cancer. Aging (Albany NY). 12, 24424–24440. https://doi.org/10.18632/aging.202300 (2020).
Zhang, Y. et al. Gelsolin promotes cancer progression by regulating epithelial-mesenchymal transition in hepatocellular carcinoma and correlates with a poor prognosis. J. Oncol 2020, 1980368. https://doi.org/10.1155/2020/1980368 (2020).
Dufait, I. et al. Perforin and granzyme B expressed by murine Myeloid-Derived suppressor cells: A study on their role in outgrowth of cancer cells. Cancers (Basel). https://doi.org/10.3390/cancers11060808 (2019).
Acknowledgements
The authors wish to thank members of the Houchen lab and Dr. Min Li for comments made during the conception and writing of this work.
Author information
Authors and Affiliations
Contributions
C.W.H., B.E.A., and M.B. conceived the concept. The experiments were carried out by P.C., R.M., K.P., D.Q., and L.L.M. L.L.M. wrote the manuscript and created final figures with editorial assistance from K.P., M.B., and C.W.H. Funding for this work was acquired by C.W.H. All authors have reviewed this manuscript.
Corresponding author
Ethics declarations
Competing interests
Courtney W. Houchen has an ownership interest in COARE Holdings, Inc., and M.L.B. is a co-founder of Trocar Pharmaceuticals and of Shuttle Pharmaceuticals. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moore, L.L., Qu, D., Chandrekesan, P. et al. A novel D-peptide modulates DCLK1 gelsolin interactions, reducing PDAC tumor growth. Sci Rep 15, 35811 (2025). https://doi.org/10.1038/s41598-025-19722-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-19722-z
