
Abstract
This study investigated the role of ubiquitin specific peptidase 42 (USP42) in breast cancer proliferation, focusing on its modulation of apoptosis via the JNK/p38 signaling pathway. USP42 expression levels in breast cancer cell lines were assessed using western blotting and RT-qPCR. In vitro, cell proliferation was evaluated using CCK-8 assay and clonogenic assay assessed, while apoptosis was measured by flow cytometry evaluated. Western blotting was used to analyze the expression of apoptosis-related proteins and those associated with the JNK/p38 pathway. The effect of USP42 knockdown on breast cancer cell proliferation was examined in vivo using a xenograft nude mice model. USP42 protein levels were significantly higher in breast cancer tissues than in normal breast tissues. Moreover, USP42 expression was positively correlated with the advanced T stage, N stage, and pathological stage. USP42 knockdown in MCF7 and MDA-MB-231 cells resulted in decreased proliferation and increased apoptosis rates. USP42 silencing upregulated caspase-3 and Bax expression, while downregulating Bcl-2. Phosphorylation of JNK and p38 increased significantly following USP42 silencing. Treatment with SP600125 (JNK inhibitor) or SB203580 (p38 MAPK inhibitor) effectively recused JNK and p38 activation. Both inhibitors also reduced the apoptotic cell population, which was upregulated by USP42 silencing. These findings highlight USP42 promotes breast cancer progression by reducing JNK and p38 activation and inhibiting apoptosis, suggesting its potential as a therapeutic target in breast cancer treatment.
Similar content being viewed by others


Introduction
According to recent Cancer Statistics, breast cancer accounts for 31% of all female malignancies in the United States in 2023, with 297,790 new cases reported1. Breast cancer is a heterogeneous and complex disease comprising diverse subtypes, each with distinct molecular characteristics2. The prognosis of patients with breast cancer is significantly influenced by both the molecular subtype of the tumor and the patient’s pathological stage3,4. Although advancements in diagnostic techniques and treatments have improved breast cancer cure rates, some patients still experience recurrence or succumb to disease progression. The persistent challenge underscores the urgent need to identify novel molecular targets and to elucidate the complex mechanisms underlying breast cancer development.
Deubiquitinating enzymes (DUBs), a class of proteases, have emerged as crucial regulators of ubiquitination dynamics in various biological processes. Ubiquitination plays a pivotal role in several cellular activities5,6. Ubiquitin-specific peptidase 10 (USP10) modulated hepatic steatosis by inhibiting SIRT6 ubiquitination and degradation in non-alcoholic fatty livers of disease patients7. Similarly, USP1 inhibits osteosarcoma progression by interacting with and degrading TAZ8. Recent studies have highlighted the significant contribution of DUBs in breast cancer development.USP36, for example, suppressed breast cancer progression by regulating PKM29, whereas USP22 affects estrogen receptor stability in ERα-positive breast cancer10. Ubiquitin-specific protease 42 (USP42), a member of the DUB family, has been investigated in other malignant such as acute myelogenous leukemia11 and gastric cancer12. Mechanistically, USP42 has been shown to deubiquitinate and stabilize p53 in response to cellular stress, enhancing p53-dependent gene expression and promoting cell cycle arrest or apoptosis13. Independently of p53, USP42 also deubiquitinates histone H2B, a modification that facilitates RNA polymerase II elongation and promotes transcriptional activation14. Despite extensive functional studies in transcription regulation and stress responses, its role in breast cancer has not been addressed.
In the intricate network of cellular signaling, p38 MAPK (p38 mitogen-activated protein kinase) plays a crucial role in responding to various environmental stressors and inflammatory cytokines. It is believed to promote apoptosis through multiple pathways, including p53 phosphorylation15, Fas/Fasl-mediated apoptosis16, and c-Jun N-terminal kinase (JNK) activation17. Previous studies have demonstrated that the JNK/p38 signaling pathway triggers apoptosis and affects cell proliferation and survival in tumor cells, including in breast cancer18,19,20. The study underscores the upregulated expression of USP42 in both breast cancer cells and tissues, indicating its significant role in promoting proliferation via apoptosis, mediated by the JNK/p38 pathway. This discovery positions USP42 as a promising molecular target, warranting further investigation in the field of breast cancer therapeutics.
Result
USP42 exhibited elevated expression levels and was significantly correlated with the clinicopathological features of breast cancer
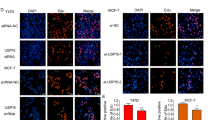
Our initial investigations focused on the gene expression patterns in breast cancer and normal tissues. Analysis of data derived from TCGA tumor tissue and GTEx database revealed a marked increase in USP42 expression in breast tumor tissues compared to that in normal tissues (Fig. 1A). These findings suggested a potential pro-carcinogenic role for USP42 in breast cancer. GEPIA database analysis indicated that patients with high USP42 expression had poorer prognosis (Fig. 1C). To further validate the differential expression of USP42 and its correlation with clinicopathological factors, IHC was performed on tumor and adjacent normal tissues from 77 breast cancer. USP42 expression was higher in tumor tissues than in normal tissues (Figs. 1B, D). Further investigation of the correlation between USP42 expression and clinicopathological features revealed significant associations with tumor size (T stage), the number of lymph node metastases (N stage), and pathological stage (Table 1).

USP42 expression in breast tissues and its prognostic significance. (A) Comparison of USP42 mRNA expression in breast cancer tissues and normal breast tissues from the TCGA and GTEx databases. (B) Differential USP42 protein expression in breast carcinoma tissues compared with paired normal tissues. (C) Kaplan–Meier survival analysis of breast cancer patients stratified with by high and low USP42 expression based on TCGA data analyzed using GEPIA. (D) Representative immunohistochemical staining for USP42 in breast carcinoma tissues and paired normal tissues. Scale bars, 50 μm (magnification, ×200). (***: p < 0.001).
USP42 expression were notably elevated in breast cancer cell lines
To further validate the role of USP42 in breast cancer cells, we examined USP42 expression in MCF10A (normal mammary epithelial cell) and various breast cancer cell lines (MCF-7, BT549, MDA-MB-231). The results demonstrated that the expression of USP42 in breast cancer cells surpassed that in MCF10A cells at both mRNA and protein levels (Figs. 2A, B). These finding highlight the potential significance of USP42 in breast cancer.

USP42 expression in human breast cancer cells. (A, B) RT-qPCR (A) and western bolt (B) analyses of USP42 expression levels in MCF10A, MCF-7, MDA-MB-231, and BT-549 cells. (C, D) RT-qPCR and western bolt analysis of USP42 expression levels in MCF-7(C) and MDA-MB-231(D) cells after transfection with shUSP42-1#,−2#,−3# or the negative control. (Data are presented as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001).
USP42 knockdown suppressed proliferation of breast cancer cells
To further substantiate the functional role of USP42 in breast cancer cell lines, we selected MDA-MB-231 and MCF-7 cells as experimental models was based on the prior analysis of USP42 expression patterns. Stable knockdown of USP42 in these cell lines was achieved through lentiviral-mediated silencing. We selected shRNAs (small hairpin RNAs)−1# and 2# based on the silencing effect of USP42 at the mRNA and protein levels (Figs. 2C, D). Subsequent Cellular Counting Kit-8 (CCK-8) and clonogenic assays confirmed a significant reduction in cell viability following USP42 knockdown (Figs. 3A, B). These findings provide evidence supporting the role of USP42 in promoting both the proliferation.

USP42 downregulation inhibits cell growth. (A, B) Cell viability assessed by CCK-8 (A) and clonogenic assay (B) in MCF-7 and MDA-MB-231 cells transfected with shUSP42-1#,2# or the negative control. (Data are presented as the mean ± SD. ***: p < 0.001).
USP42 knockdown promoted apoptosis in breast cancer cells
Previous studies have indicated the potential impact of apoptosis on proliferation21,22. To investigate the potential alteration in apoptosis in breast cancer cells following USP42 knockdown, we used the Annexin V/PI double staining assay. The results demonstrated a significant increase in apoptosis in both MCF-7 and MDA-MB-231 cells following USP42 knockdown (Figs. 4A, B). Furthermore, examination of apoptosis-related proteins revealed that knockdown of USP42 led to upregulation of cleaved caspase 3 and Bax, along with downregulation of Bcl-2 (Fig. 4C). These results suggested that USP42 promotes breast cancer cell viability by inhibiting apoptosis in breast cancer cells.
USP42 knockdown regulated phosphorylation of JNK and p38 in breast cancer cells
Previous studies highlighted the significance of JNK and p38 in the apoptosis. Western blot analysis was performed to investigate alterations in the protein levels of total JNK, phosphorylated JNK (p-JNK), total p38, and phosphorylated p38 (p-p38) following USP42 knockdown. In MCF-7 and MDA-MB-231 cells with USP42 knockdown, no significant changes were observed in total JNK and p38 protein levels. However, phosphorylation levels of both JNK and p38 increased significantly (Fig. 4D). These findings suggested that USP42 influences breast cancer cells by activating the JNK and p38 signaling pathways.

USP42 downregulation increases apoptosis and regulates JNK and p38 phosphorylation in breast cancer cells. (A, B) Flow cytometric analysis of apoptotic cell numbers following USP42 downregulation in MCF-7 (A) and MDA-MB-231 (B) cells. (C) Western blot analysis of cleaved-caspase3, Bcl-2 and Bax protein levels in MCF-7 and MDA-MB-231 cells. β-actin served as an internal reference. (D) Western blotting analysis of p-p38, p38, JNK and p-JNK protein expression in MCF-7 and MDA-MB-231 cells. β-actin served as an internal reference.
USP42 regulated breast cancer cell proliferation and apoptosis through the JNK/p38 signaling pathway
To further elucidate the role of USP42 in the regulation of breast cancer proliferation and apoptosis, rescue experiments were conducted. These experiments used 40µM SP600125 (JNK inhibitor) or 10µM SB203580 (p38-MAPK inhibitor) to disrupt the JNK/p38 signaling pathway. The results of the CCK-8 assay (Figs. 5A, C) and clonogenic assay (Figs. 5B, D) demonstrated that both SP600125 and SB203580 partially rescued the decrease in cell viability induced by USP42 knockdown. Moreover, the treatment of breast cancer cells with these inhibitors partially attenuated the apoptosis rate induced by USP42 silencing (Fig. 6). These findings suggest a mechanistic link USP42-mediated breast cancer cell progression mediated by USP42.

USP42 downregulation inhibits cell growth via the JNK/p38 signaling pathway. (A-D) Proliferation of MCF-7 and MDA-MB-231 cells transfected with shUSP42-2# or negative control, as assessed by CCK-8 assay (A, C) and clonogenic assay (B, D). The cells were treated with SP600125 (JNK inhibitor), SB203580 (p38 MAPK inhibitor), or DMSO (negative control). (ns: no significance, *: p < 0.05, **: p < 0.01, ***: p < 0.001).

USP42 downregulation increases breast cancer cells apoptosis via the JNK/p38 signaling pathway. (A, D) Flow cytometry analysis of apoptotic MCF-7 and MDA-MB-231 cells transfected with shUSP42-2# or the negative control, treated with SP600125 (A), SB203580 (D) or DMSO. (B, C) Western blot analysis of cleaved caspase 3, Bcl-2, Bax, p-p38 MAPK, p38 MAPK, JNK and p-JNK expression in MCF-7 and MDA-MB-231 cells transfected with shUSP42-2# or negative control, treated with SP600125 (B), SB203580 (C) or DMSO. β-actin served as an internal reference. (*: p < 0.05, **: p < 0.01, ***: p < 0.001).
USP42 knockdown inhibited breast cancer growth in vivo by activating the JNK/p38 signaling pathway
To substantiate the functional role of USP42 in tumor growth, we conducted in vivo experiments using a xenograft mouse model. MDA-MB-231 cells stably transfected with sh-USP42 lentivirus or control vector (four per group) were subcutaneously injected into Balb/c nude mice (Fig. 7A). Subsequent analysis revealed significant suppression of transplanted tumor growth (Figs. 7B-D) following USP42 knockdown. Furthermore, in vivo experiments provided evidence that USP42 regulates breast cancer cells growth via JNK and p38 signaling pathways (Fig. 7E).

USP42 promotes breast cancer tumor growth in vivo. (A) Schematic representation of in vivo experimental design. (B) Subcutaneous administration of MDA-MB-231 cells stably transfected with shUSP42-2# or the control vector into nude mice (n = 4 per group). (C) Representative images of xenograft tumors in nude mice (n = 4 per group). (D) Tumor weights in the shUSP42 and control vector groups (n = 4 per group). (E) Western blot analysis of USP42, p-p38 MAPK, p38 MAPK, JNK and p-JNK protein expression in tumor tissues of xenograft mouse model (n = 4 per group). (Data are presented as mean ± SD. *** p < 0.001).
Discussion
Ubiquitination, as a critical component of post-translational protein modification, plays a multifaceted role beyond protein degradation, and serves as a pivotal regulator of diverse cellular functions. The intricately regulated and reversible biological process are balanced by DUBs, which reverse ubiquitin modifications. Extensive research has underscored the therapeutic potential of DUBs as targets in various cancers, including breast cancer23,24,25. However, the precise mechanism of USP42 in breast cancer remains unclear. To elucidate its role, we performed a comprehensive analysis of raw data from public databases. The results revealed a significant elevation in USP42 mRNA levels in breast tissue samples (Fig. 1A). Immunohistochemical analysis demonstrated significantly elevated protein expression of USP42 in tumor tissues compared to that in healthy mammary tissues (Figs. 1B, C). Moreover, analysis of mRNA and protein levels demonstrated that USP42 exhibited significantly higher expression in breast cancer cells than in MCF10A (Figs. 2A, B). Notably, analysis of clinicopathologic data from 77 breast cancer patients revealed a positive correlation between USP42 expression and the clinicopathologic stage of the patients (Table 1).
Previous studies have demonstrated that USP42 may play diverse roles in various tumors. USP42 disrupted mRNA splicing and predicts adverse outcomes in lung cancer26,27. Additionally, USP42 regulates the malignant proliferation phenotype of colon cancer cells via the paracrine Wnt signaling pathway28. To confirm the role of USP42 in breast cancer, we conducted functional validation experiments by selectively suppressing USP42 expression in two distinct breast cancer cells. These results demonstrated a significant enhancement in proliferation coupled with a concomitant suppression of apoptosis. Finally, the oncogenic role of USP42 in breast cancer was confirmed by establishing a xenograft model in nude mice.
Apoptosis plays a central role in preserving the cellular balance and preventing uncontrolled proliferation. Dysregulation of apoptosis is a hallmark feature in various cancers29,30,31. Our findings showed that the percentage of apoptotic cells increased significantly after USP42 knockdown. Furthermore, changes were noted in key apoptosis-related proteins in breast cancer cells (Fig. 5)32,33,34. These findings suggest that USP42 acts as an oncogene in breast cancer, affecting proliferation by mediating apoptosis.
The mitogen-activated protein kinase (MAPK) signaling pathway plays a crucial role in tumor development35. The p38 and JNK pathways are essential components of MAPK signaling pathway. Numerous studies have confirmed that p38 and JNK signaling pathways are associated with apoptosis and proliferation36,37,38.The p38-JNK pathway is as necessary for apoptosis induction following p53 activation. MAPK (JNK and p38) activation is mediated by p53, which in turn regulates both phosphorylated (p-p53) and total p53 levels39,40. Hock et al. demonstrated that USP42 exerted its biological functions by regulating the stability of p5313.
In the present study, silencing of USP42 resulted in a significant increase in the phosphorylation levels of JNK and p38, which was accompanied by upregulation of pro-apoptotic markers, including cleaved caspase-3 and Bax, as well as downregulation of the anti-apoptotic protein Bcl-2. Notably, pharmacological inhibition of JNK or p38 partially attenuated the apoptosis induced by USP42 knockdown, indicating that these pathways act predominantly in a pro-apoptotic manner within our experimental setting. These findings align with previous reports demonstrating that under conditions of cellular stress, activation of the JNK/p38 axis contributes to apoptosis induction in breast cancer and other malignancies16,17,18,36,37,38. These observations suggested that USP42 silencing activates the JNK/p38 pathway, ultimately accelerating apoptosis in breast cancer cells.
This study provides valuable insights into the functional significance of USP42 in breast cancer cells, and several avenues remain to be explored. Specifically, further investigations are warranted to assess the phenotypic consequences of USP42 overexpression in diverse breast cancer cell lines. In addition, elucidating the direct interactions and downstream targets of USP42 is essential for a comprehensive understanding of its molecular mechanisms. The relationship between USP42 and survival-related events was not assessed in this study because of the limited follow-up period of the included patients. Extending the follow-up duration of patients is crucial for evaluating the role of USP42 in the survival of breast cancer patients.
Conclusion
USP42 is a prominent factor in breast cancer, exerting an influence on proliferation by modulating apoptosis through the JNK/p38-mediated pathway. These findings suggest that targeting USP42 may represent a potential therapeutic intervention strategy for breast cancer therapy. Moreover, this study provides valuable insights into the intricate mechanisms underlying breast cancer progression.
Materials and methods
Tissues
Primary tumor (T) and adjacent non-tumorous (N) tissue samples were obtained from 77 patients at Jiangxi Cancer Hospital. These patients had not receive any adjuvant treatments, such as radiotherapy or chemotherapy, prior to surgery. All patients have written informed consent and all experiments were approved by the Medical Ethics Committee of Jiangxi Cancer Hospital with the ethical code 2023ky182.
Bioinformatics analysis
USP42 expression data, measured by HTSeq-FPKM, was obtained from TCGA cohort pan-cancer database (https://portal.gdc.cancer.gov/) and Genotype-Tissue Expression (GETx) project database (https://www.gtexportal.org). The Wilcoxon rank-sum test was used to analyze disparities between tumor and non-tumorous specimens. Survival curve for the USP42-high and -low groups were generated using the Gene Expression Profiling Interactive Analysis (GEPIA) database(http://gepia.cancer-pku.cn/), with quartile USP42 expression levels as the basis for the analysis.
Cell culture
The human breast cancer cell lines MCF-7, MDA-MB-231, and BT549 were obtained from the Cell Resource Center of Beijing Xiehe, while the mammary gland epithelial cell line MCF10A was obtained from the American Type Culture Collection (ATCC). MCF-7 and BT549 cells were cultured in RPMI-1640(Gibco-BRL, Australia). MDA-MB-231 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco-BRL, Australia). MCF10A cells were cultured in Mammary Epithelial Basal Medium (MEBM) with 100 ng/ml cholera toxin, 1% solution of Pen/Strep, and 20ng/ml EGF. All media were supplemented with 10% fetal bovine serum (FBS, Gibco-BRL, Australia). Cells were cultured under humidified conditions at 37 °C and 5% CO2.
Cell transfection and treatment
The human USP42 shRNA plasmid and negative control shRNA (shNC) were acquired from Invitrogen (Thermo Fisher Scientific, Inc). The targeted sequences were as follows: Human USP42-sh1 (5’-CGAGTTCATCTGTACCTGATA-3’), Human USP42-sh2(5’-CGTCTCTTGTCTTCACTGATA-3’), HumanUSP42-sh3(5’-GCGTCTCTTGTCTTCACTGAT-3’) and shNC (5’-CCGGCAACAAGATGAAGAGCACAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT-3’). Lentiviral vectors carrying USP42 shRNA or shNC were constructed and added to the culture media of MCF-7 and MDA-MB-231 cells. After 48 h later, the stable cell lines were selected using puromycin. In specific experiments, cells were treated with the JNK inhibitor SP600125 or the p38-MAPK inhibitor SB203580(MCE, USA), according to the manufacturer’s instructions.
Western blot analysis
Proteins were extracted from cells or tissues using RIPA lysis buffer. The extracted protein samples were subsequently loaded onto SDS-PAGE gels, where proteins were separated into distinct bands based on their molecular weights via electrophoresis. Following separation, the proteins were transferred from the gel onto a PVDF membrane. Specific antibodies targeting the protein of interest were added to the membrane to allow the formation of antigen-antibody complexes. To minimize background signals, unbound antibodies were removed using multiple washing steps. Protein detection was performed using a chemiluminescent reagent. The resulting protein bands were visualized and analyzed using a Bio-Rad ChemiDoc XRS system to determine the presence and relative abundance of the target protein. The experimental procedure aligned with methodologies described in the literature41. The protein marker used in western blot was the GoldBand Plus 3-color Regular Range Protein Marker (8–180 kDa, Yeasen Biotechnology [Shanghai] Co., Ltd.). Specific information for the antibodies used in the experiment is as follows: USP42 (1:500, sc-390604, Santa Cruz Biotechnology, Inc., USA), JNK (9252, Cell Signaling Technology, 1:1000), p-JNK (9251, Cell Signaling Technology, 1:1000), p38 (9212, Cell Signaling Technology, 1:1000), p-p38 (9211, Cell Signaling Technology, 1:1000), β-actin (sc-47778, Santa Cruz Biotechnology, 1:1000).
Reverse transcription and quantitative real-Time PCR(RT-qPCR)
Total RNA was extracted from samples using TRIzol reagent and subsequently transcribed into complementary DNA (cDNA). The resulting cDNA was combined with PCR primers, SYBR Green fluorescent dye, and PCR reaction components. PCR amplification was performed under specific thermal cycling conditions involving repeated replication of the DNA sequence through controlled temperature fluctuations. During PCR amplification, fluorescence signals were detected, and the signal intensity of each PCR cycle was recorded. Based on the obtained cycle threshold (Ct) values, the relative quantity of target RNA in the samples was analyzed using the relative quantification method with a standard curve. The corresponding primer sequences were as follows: USP42,5’- AATCTTCAGACCCATCAGCCT-3‘(forward) and 5’- AGAACCTGCATCCATGTCTCC-3(reverse); GADPH: 5’-GGTGAAGGTCGGAGTCAACG-3‘(forward) and 5’-CAAAGTTGTCATGGATGHACC − 3‘(reverse). ALL experiments were conducted in triplicates to ensure reproducibility and statistical significance.
Immunohistochemistry (IHC)
Tissue samples were fixed on slides in formalin, embedded in paraffin, and sectioned. Tissue sections were treated for antigen retrieval, including protein dissociation, antigen retrieval, and antigen exposure, to enhance antibody binding efficiency. Non-specific binding was blocked using non-specific proteins, such as bovine serum albumin (BSA), to prevent antibody binding to non-target areas. Tissue samples were incubated with primary antibodies, which allowed them to bind specifically to the target protein. The samples were washed multiple times to remove unbound primary USP42 antibodies (sc-390604, Santa Cruz Biotechnology, Inc.USA), and reduce background signals. Secondary antibodies labeled with fluorophores or enzymes were added, which specifically bound to the primary antibodies to enhance the signal. The samples were washed multiple times to remove unbound secondary antibodies, and to reduce the background signals. The corresponding substrate was added to enzyme-labeled secondary antibodies, and the reaction products were observed. A microscope was used to examine the tissue sections and determine the expression of the target protein based on staining intensity and location. USP42 expression scores in tumor cells were determined by combining the proportion of positively stained tumor cells and the intensity of staining, as previously described42.
Cell proliferation assays
For Cell Counting Kit 8 (CCK-8) assays, breast cancer cells were seeded into 96-well plates at a density of 3,000 cells per well. The CCK-8 reagent (K1080, APExBIO Technology LLC, USA) was added to the wells at 0, 24, 48, 72 h and 96 h. The plates were incubated at 37 °C for 2 h, after which absorbance was measured at 450 nm using a microplate reader. For the colony formation assays, 1500 cells were seeded in each well of 6-well plate containing 3 mL of medium per well. After approximately 14 days of incubation, the resulting cell colonies were fixed with a 4% paraformaldehyde solution for 15 min. Subsequently, the colonies were stained with a 0.2% crystal violet solution (Solarbio, China) and quantified using Image J software. All experiments were performed in triplicates to ensure reliability and consistency.
Flow cytometry assay
The treated cell samples were collected and processed appropriately, including washing and centrifuging, to obtain single-cell suspension. The cell samples were then stained with apoptosis markers, such as Annexin V and Propidium Iodide (PI). The stained cell samples were injected into a flow cytometer (FACSVantage SE; BD, Franklin Lakes, NJ, USA), which utilized lasers to irradiate cells and detect the fluorescence signal intensity within them. Based on the staining characteristics of Annexin-V/PI (FA101-01, TransGen Biotech Co.,Ltd, Beijing, China), the flow cytometer identified and differentiated cells in various states, including live, apoptotic, and necrotic cells, and necrotic cells. The proportion of apoptotic cells and other relevant parameters in the samples were analyzed based on the data obtained from the flow cytometer42.
Orthotopic xenografts analysis
Animal experiments were approved by the Animal Ethics Committee of Kangtai Medical Laboratory Service Hebei Co. on March 14, 2022 (approval number: MDL2022-03-14-02) and conducted with the formal approval of the animal care committees. Nude mice were maintained in sterile conditions, including sterilized food and water, and regular cleaning and disinfection of the cages and the equipment. Each cage contained up to six nude mice. The laboratories maintained strict control over temperature (22–24 °C) and relative humidity (40–60%). The animals were housed in isolation cages with filtered air vents to prevent the entry of pathogen entry. Due to their susceptibility to infections, their health, including activity levels, appetite, and any signs of illness, were regularly monitored. Nude mice were randomly assigned to either the sh-USP42 or control vector group (n = 4 per group). Tumor growth was initiated by subcutaneously injecting MDA-MB-231 cells into 3-week-old female nude mice at a concentration of 1 × 10^6 cells in 200 µl of a 1:1 PBS –Matrigel mixture. Tumor dimensions were evaluated every three days using caliper, and tumor volume was calculated using the formula ((L×W2)/2). At the end of the animal experiments, mice were euthanized by cervical dislocation. Xenografted tumors were surgically excised, and protein levels within the tumors were determined using western blot analysis. All experiments were conducted in accordance with relevant guidelines and regulations. All studies involving live animals is reported in accordance with the ARRIVE guidelines.
Statistical analysis
Statistical analysis was performed using SPSS software (version: 21.0, Chicago, USA). Continuous data were presented as means ± standard deviation (SD), whereas categorical data were expressed as frequency counts and percentages. Student’s t-test was used for comparisons between two groups. For comparisons among multiple groups, one-way analysis of variance (ANOVA) was conducted, followed by the Student-Newman-Keuls test for post-hoc multiple comparisons. The association between USP42 expression and clinicopathologic characteristics was evaluated using the χ2 test or Fisher’s exact test as appropriate. Differences in survival curves were assessed using the log rank test. A p < 0.05 was considered statistically significant.
Data availability
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 73, 17–48. https://doi.org/10.3322/caac.21763 (2023).
Xu, Y. et al. Twist1 promotes breast cancer invasion and metastasis by Silencing Foxa1 expression. Oncogene 36, 1157–1166. https://doi.org/10.1038/onc.2016.286 (2017).
Tekpli, X. et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat. Commun. 10, 5499. https://doi.org/10.1038/s41467-019-13329-5 (2019).
Jugniot, N., Massoud, T. F., Dahl, J. J. & Paulmurugan, R. Biomimetic nanobubbles for triple-negative breast cancer targeted ultrasound molecular imaging. J. Nanobiotechnol. 20, 267. https://doi.org/10.1186/s12951-022-01484-9 (2022).
Hoeller, D., Hecker, C. M. & Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer. 6, 776–788. https://doi.org/10.1038/nrc1994 (2006).
Dikic, I. & Schulman, B. A. An expanded lexicon for the ubiquitin code. Nat. Rev. Mol. Cell. Biol. 24, 273–287. https://doi.org/10.1038/s41580-022-00543-1 (2023).
Luo, P. et al. Ubiquitin-Specific peptidase 10 (USP10) inhibits hepatic Steatosis, insulin Resistance, and inflammation through Sirt6. Hepatology 68, 1786–1803. https://doi.org/10.1002/hep.30062 (2018).
Yuan, P. et al. USP1 Inhibition suppresses the progression of osteosarcoma via destabilizing TAZ. Int. J. Biol. Sci. 18, 3122–3136. https://doi.org/10.7150/ijbs.65428 (2022).
Wu, H. et al. miR-140-3p/usp36 axis mediates ubiquitination to regulate PKM2 and suppressed the malignant biological behavior of breast cancer through Warburg effect. Cell. Cycle. 22, 680–692. https://doi.org/10.1080/15384101.2022.2139554 (2023).
Wang, S. et al. USP22 positively modulates ERα action via its deubiquitinase activity in breast cancer. Cell. Death Differ. 27, 3131–3145. https://doi.org/10.1038/s41418-020-0568-2 (2020).
Paulsson, K. et al. A novel and cytogenetically cryptic t(7;21)(p22;q22) in acute myeloid leukemia results in fusion of RUNX1 with the ubiquitin-specific protease gene USP42. Leukemia 20, 224–229. https://doi.org/10.1038/sj.leu.2404076 (2006).
Hou, K. et al. Overexpression and biological function of Ubiquitin-Specific protease 42 in gastric cancer. PLoS ONE. 11, e0152997. https://doi.org/10.1371/journal.pone.0152997 (2016).
Hock, A. K., Vigneron, A. M., Carter, S., Ludwig, R. L. & Vousden, K. H. Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO J. 30, 4921–4930. https://doi.org/10.1038/emboj.2011.419 (2011).
Hock, A. K., Vigneron, A. M. & Vousden, K. H. Ubiquitin-specific peptidase 42 (USP42) functions to deubiquitylate histones and regulate transcriptional activity. J. Biol. Chem. 289, 34862–34870. https://doi.org/10.1074/jbc.M114.589267 (2014).
Perfettini, J. L. et al. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J. Exp. Med. 201, 279–289. https://doi.org/10.1084/jem.20041502 (2005).
Liu, W. H., Cheng, Y. C. & Chang, L. S. ROS-mediated p38alpha MAPK activation and ERK inactivation responsible for upregulation of Fas and FasL and autocrine Fas-mediated cell death in Taiwan Cobra phospholipase A(2)-treated U937 cells. J. Cell. Physiol. 219, 642–651. https://doi.org/10.1002/jcp.21713 (2009).
Wagner, E. F. & Nebreda, A. R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer. 9, 537–549. https://doi.org/10.1038/nrc2694 (2009).
Chen, J. et al. Microtubule depolymerization and phosphorylation of c-Jun N-terminal kinase-1 and p38 were involved in gambogic acid induced cell cycle arrest and apoptosis in human breast carcinoma MCF-7 cells. Life Sci. 83, 103–109. https://doi.org/10.1016/j.lfs.2008.05.003 (2008).
Guo, Y. et al. CLDN6-induced apoptosis via regulating ASK1-p38/JNK signaling in breast cancer MCF-7 cells. Int. J. Oncol. 48, 2435–2444. https://doi.org/10.3892/ijo.2016.3469 (2016).
Ge, D. et al. Novel effects of sphingosylphosphorylcholine on the apoptosis of breast cancer via autophagy/AKT/p38 and JNK signaling. J. Cell. Physiol. 234, 11451–11462. https://doi.org/10.1002/jcp.27802 (2019).
Liu, Y. et al. Dihydroartemisinin induces apoptosis and inhibits proliferation, migration, and invasion in epithelial ovarian cancer via Inhibition of the Hedgehog signaling pathway. Cancer Med. 7, 5704–5715. https://doi.org/10.1002/cam4.1827 (2018).
Song, S. et al. A novel YAP1 inhibitor targets CSC-Enriched Radiation-Resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol. Cancer Ther. 17, 443–454. https://doi.org/10.1158/1535-7163.Mct-17-0560 (2018).
Liang, Y. et al. Targeting the circBMPR2/miR-553/USP4 axis as a potent therapeutic approach for breast cancer. Mol. Therapy Nucleic Acids. 17, 347–361. https://doi.org/10.1016/j.omtn.2019.05.005 (2019).
Yang, R. et al. Hypoxia-induced circWSB1 promotes breast cancer progression through destabilizing p53 by interacting with USP10. Mol. Cancer. 21, 88. https://doi.org/10.1186/s12943-022-01567-z (2022).
Yoon, J. Y., Seo, S. U., Woo, S. M. & Kwon, T. K. USP41 enhances Epithelial-Mesenchymal transition of breast cancer cells through snail stabilization. Int. J. Mol. Sci. 24 https://doi.org/10.3390/ijms24021693 (2023).
Liu, S. et al. USP42 drives nuclear speckle mRNA splicing via directing dynamic phase separation to promote tumorigenesis. Cell. Death Differ. 28, 2482–2498. https://doi.org/10.1038/s41418-021-00763-6 (2021).
Uriarte, M., Milot, E. & Affar, E. B. USP42 deubiquitinase in the arena of liquid-liquid phase separation and nuclear speckles. Cell. Death Differ. 28, 2022–2024. https://doi.org/10.1038/s41418-021-00794-z (2021).
Giebel, N. et al. USP42 protects ZNRF3/RNF43 from R-spondin-dependent clearance and inhibits Wnt signalling. EMBO Rep. 22, e51415. https://doi.org/10.15252/embr.202051415 (2021).
Fan, Y. & Bergmann, A. Apoptosis-induced compensatory proliferation. The cell is dead. Long live the cell! Trends Cell. Biol. 18, 467–473. https://doi.org/10.1016/j.tcb.2008.08.001 (2008).
Tai, C. J., Hsu, C. H., Shen, S. C., Lee, W. R. & Jiang, M. C. Cellular apoptosis susceptibility (CSE1L/CAS) protein in cancer metastasis and chemotherapeutic drug-induced apoptosis. J. Exp. Clin. Cancer Res. 29, 110. https://doi.org/10.1186/1756-9966-29-110 (2010).
Michie, J., Kearney, C. J., Hawkins, E. D., Silke, J. & Oliaro, J. The Immuno-Modulatory effects of inhibitor of apoptosis protein antagonists in cancer immunotherapy. Cells 9 https://doi.org/10.3390/cells9010207 (2020).
Ahagh, M. H. et al. Synthesis, characterization, anti-proliferative properties and DNA binding of benzochromene derivatives: increased Bax/Bcl-2 ratio and caspase-dependent apoptosis in colorectal cancer cell line. Bioorg. Chem. 93, 103329. https://doi.org/10.1016/j.bioorg.2019.103329 (2019).
Zhang, Y., Yang, X., Ge, X. & Zhang, F. Puerarin attenuates neurological deficits via Bcl-2/Bax/cleaved caspase-3 and Sirt3/SOD2 apoptotic pathways in subarachnoid hemorrhage mice. Biomed. Pharmacother. 109, 726–733. https://doi.org/10.1016/j.biopha.2018.10.161 (2019).
Moradipour, A., Dariushnejad, H., Ahmadizadeh, C. & Lashgarian, H. E. Dietary flavonoid carvacrol triggers the apoptosis of human breast cancer MCF-7 cells via the p53/Bax/Bcl-2 axis. Med. Oncol. 40, 46. https://doi.org/10.1007/s12032-022-01918-2 (2022).
Khavari, T. A., Rinn, J. & Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell. Cycle. 6, 2928–2931. https://doi.org/10.4161/cc.6.23.4998 (2007).
Cui, W. J. et al. Myosin light chain kinase is responsible for high proliferative ability of breast cancer cells via anti-apoptosis involving p38 pathway. Acta Pharmacol. Sin. 31, 725–732. https://doi.org/10.1038/aps.2010.56 (2010).
Guo, J., Qiu, X., Zhang, L. & Wei, R. Smurf1 regulates macrophage proliferation, apoptosis and migration via JNK and p38 MAPK signaling pathways. Mol. Immunol. 97, 20–26. https://doi.org/10.1016/j.molimm.2018.03.005 (2018).
Zhai, Z. et al. JAC1 targets YY1 mediated JWA/p38 MAPK signaling to inhibit proliferation and induce apoptosis in TNBC. Cell. Death Discovery. 8 https://doi.org/10.1038/s41420-022-00992-9 (2022).
Ghosh, J., Das, J., Manna, P. & Sil, P. C. The protective role of Arjunolic acid against doxorubicin induced intracellular ROS dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials 32, 4857–4866. https://doi.org/10.1016/j.biomaterials.2011.03.048 (2011).
Alaaeldin, R. et al. Azilsartan Modulates HMGB1/NF-κB/p38/ERK1/2/JNK and Apoptosis Pathways during Renal Ischemia Reperfusion Injury. Cells 12, (2023). https://doi.org/10.3390/cells12010185
Chen, J. et al. AFF3 is a prognostic biomarker correlated with immune infiltrates in Triple-Negative breast cancer. CEOG 50 https://doi.org/10.31083/j.ceog5008165 (2023).
Luo, H. J. et al. G-protein coupled Estrogen receptor 1 expression in primary breast cancers and its correlation with clinicopathological variables. J. Breast Cancer. 14, 185–190. https://doi.org/10.4048/jbc.2011.14.3.185 (2011).
Acknowledgements
Not applicable.
Funding
This work was supported by Science and Technology Program Project of Health Commission of Jiangxi Province (202510069). Open Research Fund of Jiangxi Cancer Hospital & Institute (Grant Number: KFJJ2023ZD01, KFJJ2023YB06). General Project of Jiangxi Provincial Administration of Traditional Chinese Medicine (2024B0854). Science and Technology Research Project of Jiangxi Provincial Education Department (Grant Number: GJJ2203530). Jiangxi Cancer Hospital Doctoral Start-up Fund (Grant Number: BSQDJ202309).
Author information
Authors and Affiliations
Contributions
GW, QL, TY and XW contributed to study conceptualization and project administration. CH, LY and JC contributed to data curation and formal analysis. WH, LH, JX, JL conceived and designed the experiments; QL contributed validation and visualization; CH, JC and RT contributed to writing-original draft and manuscript revision. LH and JL contributed to investigation and methodology. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the principles of the Declaration of Helsinki. Animal experiments were approved by the Animal Ethics Committee of Kangtai Medical Laboratory Service Hebei Co. (approval number: MDL2022-03-14-02). The study was approved by the Ethics Committee of the Jiangxi Cancer Hospital (Grant Number: 2023ky182), and all participants provided written in-formed consent to participate and for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, C., Chen, J., Tian, R. et al. Deubiquitinating enzyme USP42 promotes breast cancer progression by inhibiting JNK/p38-mediated apoptosis. Sci Rep 15, 36751 (2025). https://doi.org/10.1038/s41598-025-20573-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-20573-x
