
Abstract
Lysosomal autophagy plays a critical role in ensuring the continuity of autophagic flux, however bioinformatics studies investigating biomarkers associated with lysosomal autophagy in cerebral ischemia-reperfusion injury(CIRI) remain scarce. Three datasets (GSE61616, GSE97537, and GSE82146) were retrieved from the GEO database. After batch correction, GSE61616 and GSE97537 were integrated into a combined dataset, while GSE82146 served as the validation set. Lysosomal autophagy-related genes (LRGs) obtained from the GeneCards database were intersected with differentially expressed genes (DEGs) identified between the CIRI group and the control group, resulting in the identification of 36 lysosomal autophagy-related differentially expressed genes (LRDEGs). Functional enrichment analysis using Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene Set Enrichment Analysis (GSEA) revealed that LRDEGs are primarily involved in biological processes such as membrane fusion and SNARE binding and are significantly enriched in inflammation-related pathways. We built a diagnostic model for CIRI and identified the biomarkers Vamp8. The results were further validated by quantitative real-time PCR (qPCR) and Western blotting. Simultaneously, immune infiltration analysis was performed. Finally, the mRNA-miRNA and mRNA-RBP interaction regulatory networks of the key genes revealed that Vamp8 is regulated by miR-325-3p, Elavl1, Zfp36, and Pabpc1.
Similar content being viewed by others


Introduction
Stroke presents a formidable menace to human health and well-being1, which is associated with high recurrence and mortality rates, often resulting in adverse consequences such as limb paralysis, language deficits, and cognitive impairments. These complications significantly increase the burden on families and the broader economy. The rising incidence of stroke in younger populations is particularly concerning2. Stroke constitutes a complex pathological network formed by multiple factors, including abnormal activation of the immune system, chronic inflammatory cascade responses, lipid metabolism imbalance, and the synergistic actions of risk factors.Several studies have demonstrated the simultaneous occurrence of complex immune activation and suppression processes during stroke, involving interactions between central and peripheral immunity3. These processes require the involvement of numerous immune cells and molecules across innate and adaptive immune pathways. Following acute brain ischemia, reperfusion injury induces disruptions in cerebral glucose and oxygen metabolism, potentially resulting in localized tissue edema or hemorrhage upon restoration of blood flow-a phenomenon capable of compounding neurological impairments via intricate cellular cascades and diverse modes of cell demise4.
Autophagy denotes a fundamental cellular mechanism whereby dysfunctional organelles and metabolic remnants are shuttled towards lysosomes for degradation during periods of stress or nutrient deprivation—thereby furnishing energy for sustenance.The successive processes, including autophagy initiation, autophagosome formation and maturation, autophagosome-lysosome fusion, and degradation of autophagic substrates in autolysosomes, are collectively known as autophagic flux5. Evidence indicates that impaired autophagic flux constitutes a critical factor contributing to neuronal injury following ischemic stroke6. Autophagic flux disorders primarily encompass an abnormal accumulation of autophagic substrates, impaired fusion between autophagosomes and lysosomes, and inefficient degradation of autophagic substrates by lysosomes due to lysosomal dysfunction7. Dysregulation at any stage of these processes may result in neuronal autophagic injury or death.An adequate number of lysosomes and preserved lysosomal function are crucial for maintaining autophagic flux8. Investigating the regulatory mechanisms underlying these processes may uncover potential therapeutic targets for ischemic stroke intervention.Prior investigations have primarily concentrated on early-stage autophagy processes, scrutiny regarding the role played by lysosomes in sustaining overall autophagic flux remains scarce.
Therefore, our research takes lysosomal autophagy as the entry point, combining bioinformatics and experimental verification methods, and attempts to deeply explore the pathogenic targets of the autophagy-lysosome pathway in CIRI from the gene level, which aims to open up a new direction for autophagy research after brain injury.
Materials and methods
Data download
The rat cerebral ischemia-reperfusion injury(CIRI) data were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) using the R package GEOquery9. The GSE6161610, GSE97537 and GSE8214611 all contained samples from Rattus norvegicus, with the tissue origin being the brain. The chip platform for GSE61616 and GSE97537 was GPL1355, while the chip platform for GSE82146 was GPL17117. Specific details could be found in Table S1.In GSE61616, 5 samples suffered middle cerebral artery occlusion and reperfusion (MCAO/R), while 5 samples underwent a sham procedure as controls. GSE97537 included 7 MCAO/R samples and 5 sham samples. GSE82146 included 15 MCAO/R samples and 12 sham samples. All these samples were incorporated into the research.
We gathered Lysophagy-Related Genes (LRGs) from the GeneCards database12(https://www.genecards.org/) using “Lysophagy” as a search term and filtering for only “Protein Coding” genes, this process yielded a total of 99 LRGs. After converting these genes to their rat counterparts (Rattus norvegicus, TaxID: 10116) using the R package homologene, we obtained 89 LRGs. We utilized the R package sva13 to remove batch effects from training sets GSE61616 and GSE97537, which comprised 12 samples of MCAO/R and 10 sham samples. Using the R package limma14for standardization, principal component analysis (PCA)15was used to verify whether batch removal was effective. Additionally, we standardized the validation set GSE82146 using the R package limma.
Lysophagy-Related differentially expressed Genes(LRDEGs) analysis
Utilize the limma package to identify differentially expressed genes (DEGs) between the MCAO/R group and the sham group within the training dataset(|log2(FC)| > 1 and adj.P < 0.05), intersecting them with LRGs to obtain LRDEGs. The expression of LRDEGs were verified using the R package pROC.
Functional enrichment analysis
GO(https://www.geneontology.org/)16 and KEGG(https://www.genome.jp/kegg/)17 are effective research tools for studying gene functional enrichment and biological characteristics. We utilized the R package clusterProfiler18 to conduct GO and KEGG enrichment analyses on LRDEGs(adj.P < 0.05, q < 0.05). The p-value was adjusted by Benjamini-Hochberg (BH).
Construction of a diagnostic model for MCAO/R
To develop a MCAO/R diagnostic model for training set, logistic regression analysis was performed on LRDEGs. Subsequently, Support Vector Machine(SVM)19 model was constructed using the LRDEGs identified in the logistic regression analysis. Genes were selected based on the criteria of achieving the highest accuracy and the lowest error rate to further screen the LRDEGs associated with lysosomal autophagy. Thereafter, Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis was conducted on the screened LRDEGs using the glmnet R package20, with parameters set.seed(500) and family="binomial”. LASSO regression extends linear regression by incorporating a penalty term (lambda × absolute value of the slope), which mitigates overfitting and enhances the generalization capability of the model. The resulting LASSO regression analysis yielded the MCAO/R diagnostic model, wherein the included LRDEGs were identified as key genes. The formula for calculating the LASSO risk score is presented below:
Validation of the MCAO/R diagnostic model
In accordance with the findings of logistic regression analysis, the R package rms was employed to construct a nomogram21 for depicting the interconnections among key genes.Based on the LASSO regression analysis, the accuracy and discriminatory power of the MCAO/R diagnostic model were appraised via Calibration analysis. The R package ggDCA was utilized to perform decision curve analysis (DCA) on key genes22.Subsequently, the R package pROC was utilized to plot the ROC and compute the AUC, thereby evaluating the diagnostic performance of the LASSO risk score for MCAO/R. Furthermore, the MCAO/R group was stratified into high risk group and low risk group according to the median value of the LASSO risk score in the MCAO/R diagnostic model.The expression levels of key genes in the validation set GSE82146 were incorporated into the risk coefficients obtained from the LASSO regression analysis in the training set to calculate the risk score of MCAO/R in GSE82146.Ultimately, the R package pROC was applied to plot the ROC of key genes and calculate the AUC, aiming to assess the diagnostic utility of the expression levels of key genes in relation to the occurrence of MCAO/R. The closer the AUC approaches 1, the more optimal the diagnostic performance.
Gene set enrichment analysis (GSEA)
The genes in the MCAO/R group and sham group of the training set were sorted according to the log2(FC), the R package clusterProfiler was used to conduct GSEA23.Then, the genes of the high-risk and low-risk groups in the MCAO/R were also sorted according to the log2(FC), and GSEA was conducted again.The GSEA utilized the following parameters: a seed of 2022, 1000 permutations, gene set size from 10 to 500. The gene set c2.cp.all.v2022.1.Hs.symbols.gmt [All Canonical Pathways] (3050) was sourced from the Molecular Signatures Database (MSigDB)24 for GSEA analysis. Filtering criteria for gene sets in GSEA involved an adj.P < 0.05 and q < 0.05, employing p-value adjustment through the BH approach.
Immune cell infiltration analysis
Initially, R package homologene was used to convert training set IDs from rat (Rattus norvegicus, TaxID: 10116) to human (Homo sapiens, TaxID: 9606). The CIBERSORT25 algorithm was then applied to obtain the pertinent characteristics of immune cells were acquired .Subsequently, the expression variances between the experimental group and the control group were analyzed using R package ggplot22. The Spearman algorithm was employed to calculate the correlations among immune cells, as well as between immune cells and key genes.
Construction of key gene regulatory networks
To explore the connection between biomarkers associated with diagnostic models and miRNAs, relevant microRNAs (miRNAs) were obtained from the StarBase v3.0 database26 (https://rnasysu.com/encori/) based on the condition of phyloP > 2. Subsequently, the Cytoscape software was employed for the visualization of regulatory networks involving mRNA and miRNA. RNA-Binding Proteins (RBPs)27 are pivotal in gene regulation processes, playing critical roles in regulating various life activities such as RNA synthesis, selective splicing, modification, transport, and translation. Utilizing the StarBase v3.0 database, we predicted RBPs targeting key genes associated with diagnostic models under the condition of clipIDnum > 3 and visualized mRNA-RBP regulatory networks using Cytoscape software.
Experimental animals
Male Sprague-Dawley rats, aged 6–8 weeks and weighing approximately 250–300 g, were purchased from Weitong-Lihua Experimental Animal Co. Ltd(Beijing, China). The rats were raised at the Institute of Translational Medicine, the First Hospital of Jilin University, Changchun City, Jilin Province, China. Animals were maintained at a temperature of 22 ± 2 ◦C, humidity of 65 ± 5%, and housed under a 12-h light-dark cycle with ad libitum access to food and water.
Animal models
According to the experimental requirements, the rats were randomly divided into 5 groups, with 6 rats in each group: Sham group, MCAO/ R 24 h, 48 h, 72 h, 7 days group. Rats were subjected to transient MCAO as previously described28. After blocking the blood flow for 1 h, reperfusion was restored. Autophagy exhibits dynamic variations throughout the progression of CIRI, therefore brain tissue samples were obtained at 24 h, 48 h, 72 h and 1 week after reperfusion, respectively. Animals in the sham group underwent the same anesthesia, surgical exposure and vascular isolation steps as those in the model group, but without substantive vascular occlusion.
Brain tissue samples collection
Brain tissue samples were collected at 24 h, 48 h, 72 h, and 7 days after MCAO/R. Rats were euthanized via intraperitoneal injection of 150 mg/kg pentobarbital. Following injection, physiological responses were monitored to ensure the animals were in a euthanized state. The rats were then placed on a surgical table, and the skin was incised along the midline of the thorax with sterile surgical scissors to expose the heart and major blood vessels. A perfusion catheter was inserted into the left ventricle through the cardiac apex, and pre-cooled PBS (4 °C) was injected to ensure catheter patency, maintaining perfusion for approximately 5 to 10 min. After perfusion, the brain was swiftly removed and immediately placed in a -80 °C freezer to preserve RNA and protein integrity.
Ethics statement
Compliance with guidelines
All methods involving live vertebrates were carried out in accordance with relevant guidelines and regulations. The study protocols were approved by the Experimental Animal Research Ethics Committee of Jilin University (No. 20230409).
ARRIVE guidelines
All methods and reporting adhere to the ARRIVE guidelines (https://arriveguidelines.org) to ensure the transparency and reproducibility of the research.
qPCR and RT-qPCR
Total RNA was extracted from the sham and MCAO/R tissue samples using RNeasy Mini kit (Qiagen), and the concentration and purity thereof were determined. cDNA was generated with the First-Strand cDNA Synthesis Kit (Takara, Dalian, China). qPCR was conducted using Universal Blue SYBR Green qPCR Master Mix (Servicebio, Wuhan, China). β-actin was used as an internal control.
Regarding miRNA, miRNAs were isolated through the miRcute miRNA Isolation Kit (Tiangen, Beijing, China) and reverse transcribed with the miRcute Plus miRNA First-Strand cDNA Kit (Tiangen, Beijing, China). RT-qPCR was performed on CFX Connect Real-Time System (Bio-Rad). U6 snRNA was employed as an internal reference.
The relative expression levels of the target genes were calculated by the \(\:{\text{2}}^{\text{−ΔΔ}\text{Ct}}\) method. The primer sequences are as follows: Vamp8, 5’-CTGCCTTGGGTGGAAACAGA-3’ (forward) and 5’-TTGTTTCGGAGGTGGTCCAG-3’ (reverse); β-actin: 5’-GTGACGTTGACATCCGTAAAGA-3‘(forward) and 5’-GTAACAGTCCGCCTAGAAGCAC-3’ (reverse). miR-325-3p,5’-CGGTTTATTGAGCACCTCCTATCAA-3‘(forward) and 5’-TTACTCCTTGGAGGCCATGTA-3‘(reverse); U6:5’-CTCGCTTCGGCAGCACA-3‘(forward) and 5’-AACGCTTCACGAATTTGCGT-3’ (reverse).
Western blotting
Total protein concentration was measured using a bicinchoninic acid (BCA) protein assay kit (Epizyme, China, 23225). Each lane was loaded with an equal amount of protein (20 µg) to ensure quantitative accuracy. Proteins were separated by 4–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% nonfat milk for 90 min at room temperature, followed by incubation with rabbit-derived primary antibodies against Vamp8(1:1000,85400-4-RR, proteintech)and β-actin (1:1000, 81115-1-RR, proteintech) overnight at 4 °C. Afterward, the membranes were washed three times with TBST (Tris-buffered saline, pH 7.4, containing 0.05% Tween 20; Sigma, P1379) and further incubated with horseradish peroxidase-conjugated secondary antibodies, Goat Anti-Rabbit IgG (H&L) (1:10,000, CSA2115, Cohesion Biosciences), for 1 h at room temperature. Finally, immunoblot signals were detected using an enhanced chemiluminescence system.
Statistical analysis
All data processing and analysis were performed using R software (Version 4.3.1). Unless otherwise stated, for comparisons of two continuous variables, the statistical significance of normally distributed variables was assessed using the independent two-sample t-test. Differences between non-normally distributed variables were evaluated using the Mann-Whitney U test (also referred to as the Wilcoxon Rank Sum Test). For comparisons involving three or more groups, the Kruskal-Wallis test was employed. Correlation coefficients between different molecules were calculated using Spearman’s rank correlation analysis. Unless otherwise specified, all statistical p-values were two-tailed, and P < 0.05 was considered statistically significant.
Results
Datasets integration and LRDEGs identification
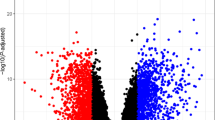
The framework of the article is presented in Fig. 1. Initially, the batch effects among samples of the training sets GSE61616 and GSE97537 were eliminated using the R package sva, yielding a combined set. For the validation set GSE82146, standardization was carried out using the R limma package. The batch effects among samples in both the combined set and the validation set were essentially removed.(Fig. S1).We obtained a total of 589 DEGs between CIRI and control samples(|log2FC| > 1,adj.P < 0.05). Among these DEGs, 519 DEGs were up-regulated (log2FC > 1 and adj.P < 0.05), while 70 DEGs were down-regulated (log2FC < -1 and adj.P < 0.05).The intersection of all the DEGs that met the criteria and the 89 LRGs yielded 36 LRDEGs(Fig. 2), among them, the ROC curves (Fig. 3) of 25 genes:Akap8l, Atp6v1a, Atp6v1d, Atp6v1e1, Atp6v1f, C3, Eef1a1, Faf2, G3bp1, Hnrnpa1, Lamp2, Lamtor3, Napa, Plaa, Ptp4a2, Rpl23, Snap23, Snapin, Stx16, Stx4, Stxbp1, Vamp3, Vamp8, Vim, Ywhaz, showed high accuracy (P<0.001,AUC > 0.9).

Flow chart.
MCAO/R1, Middle Cerebral Artery Occlusion Reperfusion; LRGs2, Lysophagy-Related Genes; DEGs3, Differentially Expressed Genes; LRDEGs4, Lysophagy-Related Differentially Expressed Genes; SVM5, Support Vector Machines; LASSO6, Least Absolute Shrinkage and Selection Operator; GO7, Gene Ontology; KEGG8, Kyoto Encyclopedia of Genes and Genomes; GSEA9, Gene Set Enrichment Analysis; CIRI10, cerebral ischemia-reperfusion injury; RBP11, RNA-Binding Protein.

Identification of differential expression genes(DEGs). (a) Volcano plot of DEGs between cerebral ischemia-reperfusion injury (CIRI) and control groups in combined dataset. (b) Venn diagram of DEGs and lysophagy-related genes(LRGs) in combined dataset. (c) Heatmap of lysophagy-related differentially expressed genes(LRDEGs) in the CIRI and control groups.

Identification of biomarkers from lysophagy-related differentially expressed genes(LRDEGs). (a) Boxplot of LRDEGs in middle cerebral artery occlusion and reperfusion (MCAO/R) group and control group. (b-i) ROC curves of LRDEGs in MCAO/R group and the control group(AUC>0.8).*P < 0.05, **P < 0.01, ***P < 0.001.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG)
The biological processes (BP), cellular components (CC) and molecular functions (MF), associated with the 36 LRDEGs in CIRI were further investigated through GO and KEGG.Utilizing these 36 LRDEGs, GO and KEGG enrichment analyses revealed specific enrichments, as detailed in Table S2. The findings suggested that the LRDEGs show significant enrichment in a range of BP, including membrane fusion, vesicle fusion, organelle membrane fusion, organelle fusion, and exocytic process within CIRI. Additionally, they showed enrichments in cellular components including vacuolar membrane, SNARE complex, proton-transporting V-type ATPase, V1 domain, proton-transporting V-type ATPase complex, proton-transporting two-sector ATPase complex, catalytic domain; molecular functions such as SNARE binding, SNAP receptor activity, molecular adaptor activity, protein-macromolecule adaptor activity, syntaxin binding; and also exhibited enrichments in several biological pathways like SNARE interactions in vesicular transport, Synaptic vesicle cycle, Phagosome, Collecting duct acid secretion, mTOR signaling pathway. Furthermore, the results of GO and KEGG enrichment analyses were visualized using a bubble chart(Fig. 4A). Moreover, a network diagram was constructed to illustrate the relationships between BP, CC, MF and KEGG(Fig. 4B-E). The connections represent the relationships between the corresponding molecules and their associated annotations within the respective entries. Additionally, the size of the nodes is proportional to the number of molecules contained in each entry.

Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis for lysophagy-related differentially expressed genes(LRDEGs). (a) GO and KEGG enrichment bubble chart of LRDEGs; BP, biological process; CC, cellular component; MF, molecular function. (b-e) GO and KEGG enrichment network plot of LRDEGs. The size of the dot was directly proportional to the number of molecules it represents; larger dots indicated a higher number of associated molecules(adj.P < 0.05 and q < 0.05).
Gene set enrichment analysis (GSEA) for combined dataset
GSEA was executed to examine the correlation between the expression of all genes in the combined dataset and the involvement in biological processes, cellular components, and molecular functions (Fig. 5A). The findings were detailed in Table S3. The findings suggested that the combined set showed significant enrichment in a variety of biologically relevant functions and signaling pathways such as REACTOME_INTERLEUKIN_10_SIGNALING (Fig. 5B), REACTOME_INTERLEUKIN_4_AND_INTERLEUKIN_13_SIGNALING (Fig. 5C), REACTOME_NEUTROPHIL_DEGRANULATION (Fig. 5D), WP_OVERVIEW_OF_PROINFLAMMATORY_AND_PROFIBROTIC_MEDIATORS (Fig. 5E), REACTOME_SIGNALING_BY_INTERLEUKINS (Fig. 5F), and WP_IL18_SIGNALING_PATHWAY (Fig. 5G).

Gene set enrichment analysis(GSEA) for combined dataset.
(a) GSEA-based bubble plots representing 6 biological pathways associated with lysophagy-related differentially expressed genes(LRDEGs).(b-h)The 6 biological pathways associated with LRDEGs identified via GSEA(adj.P < 0.05 and q < 0.05, Benjamini-Hochberg).
Identification and determination of diagnostic model for MCAO/R
The results indicated that 22 LRDEGs were statistically significant in the logistic regression model (p < 0.05) (Fig. 6A). Based on these 22 genes, we constructed a SVM model to identify the optimal number of genes that minimized the error rate while maximizing diagnostic accuracy (Fig. 6B-C). The analysis revealed that the SVM model achieved the highest accuracy using a subset of 9 genes: Atp6v1a, Atp6v1f, G3bp1, Plaa, Skp1, Stx16, Vamp8, Vcp, and Ywhaz. Subsequently, we developed a diagnostic model for MCAO/R based on these 9 genes using LASSO regression analysis. Visual inspection of the LASSO variable trajectory plot (Fig. 6D) and the LASSO regression coefficient plot (Fig. 6E) confirmed that the final model retained 2 LRDEGs:G3bp1 and Vamp8, as the most significant predictors.

Diagnostic model of middle cerebral artery occlusion and reperfusion (MCAO/R). (a) Forest Plot of 22 lysophagy-related differentially expressed genes (LRDEGs) included in the logistic regression model in MCAO/R. (b-c) For the Support Vector Machine (SVM) model constructed using LRDEGs, the highest accuracy is achieved when the gene count is set to 9. (d-e) Diagram of variable trajectories and diagnostic model of least absolute shrinkage and selection operator(LASSO) regression model.
Validation of diagnostic model for MCAO/R
The Nomogram demonstrated that the effectiveness of Vamp8 in the diagnostic model was greater than that of G3bp1(Fig. 7A).In the calibration curve of the MCAO/R diagnostic model, although the calibration line indicated by the dotted line deviates from the diagonal line of the ideal model to some extent, the overall performance was close to being consistent. The results of the decision curve analysis (DCA) showed that within a specific range, the performance of this model was significantly better than the All positive and All negative baseline, and the net benefit of the model was relatively high, indicating that the diagnostic model had a good construction effect(Fig. 7B-D). By substituting the expression levels of key genes in the validation set GSE82146 into the analysis, it was found that the expression of G3bp1 and Vamp8 were highly consistent with those in the combined dataset(Fig. 7E). In addition, the expression levels of Vamp8 in the MCAO/R high-risk group and low-risk group showed statistically significant differences (P < 0.05). Vamp8 exhibited higher diagnostic efficacy than G3bp1(0.7 < AUC < 0.9)(Fig. 7F-G). In the results of the WB analysis, we found that with the prolongation of ischemia-reperfusion time in rats, VAMP8 exhibited a trend of initially increasing and then decreasing compared to sham group(P<0.05, Fig. 8A-B). We also utilized qPCR to evaluate the relative expression levels of Vamp8 mRNA in rats at 24 h, 48 h, 72 h, and 7 days following MCAO/R and sham group. The findings revealed that relative expression of Vamp8 mRNA was statistically significantly increased at 24 h and decreased at 7 days compared to sham group, the highest relative expression level was observed in 24 h group(P<0.05, Fig. 8C). However, the relative expression of Vamp8 mRNA at 48 h and 72 h did not show statistical significance compared to the sham group. This might be attributed to the relatively small sample size.

Diagnostic and validation analysis of middle cerebral artery occlusion and reperfusion (MCAO/R). (a) Nomogram of Vamp8 and G3bp1 in the diagnostic model of MCAO/R. (b-c) Calibration Curve plot and decision curve analysis (DCA) plot of the Vamp8 and G3bp1 in MCAO/R diagnostic model. (d) Group comparison of Vamp8 and G3bp1 in the high risk and low risk groups of MCAO/R in combined dataset. (e) Group comparison of Vamp8 and G3bp1 in the high risk and low risk groups of MCAO/R in validation set GSE82146. (f-g) ROC curves of Vamp8 and G3bp1 in MCAO/R.

Cerebral ischemia-reperfusion injury (CIRI) results in dynamic changes in the function of the autophagy-lysosome pathway. (a) Time-dependent changes in VAMP8 expression were observed in the sham group and various middle cerebral artery occlusion and reperfusion(MCAO/R) experimental groups, with β-actin serving as a loading control. Original blots/gels are presented in Figure S2. (b) Quantitative analysis of the immunoblotted proteins was performed using ImageJ. Statistical comparisons were conducted with ANOVA followed by Tukey’s test. Data are presented as mean ± SEM (n = 6 per group). (c) A qPCR assay was performed to assess the relative expression of Vamp8 mRNA. The relative expression levels of Vamp8 mRNA were calculated by the\(\:{\text{}\text{2}}^{\text{−ΔΔ}\text{Ct}}\:\)method(n = 6 per group), with β-actin employed as an internal reference. *P < 0.05.
GSEA for high-risk and low-risk groups of MCAO/R
In order to investigate the impact of gene expression levels on MCAO/R, differential analysis was first conducted on the high-risk and low-risk groups of MCAO/R samples from the combined dataset. Subsequently, GSEA was employed to explore the relationship between the expression of all genes in MCAO/R and the associated biological processes, cellular components, and molecular functions(Fig. 9A). The specific results were detailed in Table S4.The results revealed that all genes in the combined set were significantly enriched in biologically relevant functions and signaling pathways such as REACTOME_CROSSLINKING_OF_COLLAGEN_FIBRILS (Fig. 9B), WP_INFLAMMATORY_RESPONSE_PATHWAY (Fig. 9C), REACTOME_ASSEMBLY_OF_COLLAGEN_FIBRILS_AND_OTHER_MULTIMERIC_STRUCTURES (Fig. 9D), REACTOME_MET_PROMOTES_CELL_MOTILITY (Fig. 9E), REACTOME_INTERLEUKIN_10_SIGNALING (Fig. 9F), and REACTOME_NEUTROPHIL_DEGRANULATION (Fig. 9G).

Gene set enrichment analysis (GSEA) for high-risk and low-risk groups of middle cerebral artery occlusion and reperfusion (MCAO/R). (a)The GSEA bubble plots depicted 6 biological pathways associated with lysophagy - related differentially expressed genes (LRDEGs) in the high risk and low risk groups of MCAO/R. (b-j) The 6 biological pathways associated with LRDEGs in the high risk and low risk groups of MCAO/R(adj.P < 0.05 and q < 0.05, adjusted by Benjamini-Hochberg).
Immune-related analysis of MCAO/R
The infiltration abundance of 22 types of immune cells in combined dataset was obtained through the CIBERSORT algorithm(Fig. 10A). In Fig. 10B, we found the results indicated significant differences in the levels of expression for 9 types of immune cells, including B_cells_memory, Plasma_cells, T_cells_CD8, T_cells_CD4_naive, T_cells_regulatory_Tregs, Monocytes, Macrophages_M0, Dendritic_cells_resting, and Neutrophils. These variances were observed in the two groups within the combined dataset (P < 0.05) .Furthermore, a correlation heatmap was used to display results indicating that monocytes exhibited maximal positive correlation with resting dendritic cells (r = 0.88,Fig. 10C), while also demonstrating maximal negative correlation with naive CD4 + T cells (r = -0.81).Finally, a bubble plot depicted the associations between important genes and the abundance of immune cell infiltration in combined dataset(Fig. 10D). The results indicated significant positive correlations between G3bp1 and resting dendritic cells as well as Vamp8 and resting dendritic cells (r > 0.8; p < 0.05). In contrast, G3bp1 exhibited significant negative correlations with regulatory T-cells and naive CD4 + T-cells (r < -0.8; P < 0.05).

Immune infiltration analysis. (a) The relative percentages of various immune cell types in the control group and the middle cerebral artery occlusion and reperfusion (MCAO/R) group (b) Box plots showing the expression of various immune cells in the control and MCAO/R group. (c-d) Correlation between biomarkers(Vamp8,G3bp1) and immune cells. Negative correlations are shown in blue and positive correlations in red.
Construction of key gene regulatory networks
We extracted miRNAs and RNA-binding proteins(RBPs) related to G3bp1 and Vamp8 from the StarBase database and respectively constructed mRNA-miRNA and mRNA-RBP regulatory networks, consisting of 42 miRNAs and 48 RBPs and visualized it using Cytoscape software. We found that Vamp8 was regulated by miR-325-3p(Fig. 11A) as well as Elavl1, Zfp36 and Pabpc1(Fig. 11B). Specific information was provided in Figure S3.

Regulatory network analysis of biomarkers. (a) mRNA-miRNA regulatory network of Vamp8 and G3bp1.(b) mRNA-RBP regulatory network of Vamp8 and G3bp1.
Discussion
Ischemic stroke is a serious and potentially life-threatening disease resulting from the blockage of blood flow in the vessels of the brain, which leading to a range of neurological deficits. Within the therapeutic time window, current treatment modalities primarily include intravenous thrombolysis and mechanical thrombectomy. However, some patients may experience secondary injury known as CIRI. Research indicates that upregulation of autophagy induced by cerebral ischemia leads to dysfunction in lysosomal storage within neurons and related synaptic damage29. The majority of studies have focused exclusively on the initiation phase of autophagy, while overlooking the stability of membrane fusion during autolysosome formation and the critical role of lysosomal autophagy in sustaining autophagic flux. Therefore, we focus on lysosomal autophagy as the entry point to explore potential regulatory targets within the autophagy/lysosome pathway following CIRI.
In this study, we initially identified 36 differentially expressed genes associated with lysosomal autophagy in the context of cerebral ischemia-reperfusion injury. Using GO and KEGG enrichment analyses, these genes were found to be predominantly localized in cellular components such as the SNARE complex and proton-transporting V-type ATPase. They are involved in biological processes including organelle membrane fusion and exocytosis, while exhibiting molecular functions such as SNARE binding, SNAP receptor activity, and syntaxin binding. Furthermore, these genes were significantly enriched in pathways related to vesicular transport (SNARE interactions), phagosome formation, and mTOR signaling. GSEA analysis revealed that these genes were also highly enriched in inflammation-related pathways. To identify precise regulatory targets, we constructed a diagnostic model for cerebral ischemia-reperfusion injury and validated it through multiple approaches. Ultimately, Vamp8 and G3bp1 were identified as key genes within this diagnostic model, with Vamp8 demonstrating superior efficacy. Immune infiltration analysis indicated a positive correlation between Vamp8 expression and resting dendritic cells. Subsequently, by constructing mRNA-miRNA and mRNA-RBP regulatory networks, we determined that Vamp8 is regulated by miR-325-3p, Elavl1, Zfp36, and Pabpc1. Through qPCR and WB experiments, we confirmed that Vamp8 expression levels in MCAO/R groups were significantly higher than those in the sham group, peaking at 24 h and gradually decreasing at 48 h, 72 h, and 7 days. We hypothesize that during the early stages of ischemia (24 h), lysosomal function remains intact, and the upregulation of Vamp8 enhances the fusion of autophagosomes and lysosomes, thereby facilitating the clearance of damaged organelles. Between 48 and 72 h post-ischemia, lysosomal dysfunction occurs in the ischemic penumbra, leading to reduced acidification capacity, downregulation of Vamp8, decreased efficiency of autophagosome-lysosome fusion, and impaired autophagic flux. By day 7, the lysosomal system collapses, driving cells into an irreversible death phase. The expression of Vamp8 may be suppressed via transcriptional inhibition or epigenetic silencing mechanisms.
Vamp8 is responsible for encoding v-SNARE and is primarily located on the lysosomal membrane30.It participates in the assembly of a SNARE complex with synaptosomal associated protein 29 and SNARE protein syntaxin 17, playing a crucial role in mediating the membrane fusion process of autophagosomes and lysosomes31,32. Under normal circumstances, the phosphorylation of Vamp8 is regulated by upstream mTORC133. It is dynamically distributed on the endosomal, lysosomal membrane, and plasma membrane. When cells are under starvation conditions, autophagy is upregulated. In a synergistic action with Ras-related protein 21(RAB21) and myotubularin-related protein 13 (MTMR13), VAMP8 migrates to the lysosomal membrane, promoting autophagosome formation34. The SNARE interactions in vesicular transport pathways fulfill a pivotal function during the process of cerebral ischemia-reperfusion. Ischemia damages the structure and function of cell membranes and organelle membranes, affecting the formation of SNARE complexes, thereby hindering membrane fusion. This disruption leads to impaired neurotransmitter release and affects inter-neuronal signaling. By regulating the expression and activity of SNARE proteins and their regulatory factors, it is possible to restore membrane structure, facilitate intracellular material transport, and protect neuronal function. Regulating the expression and activity of SNARE proteins, SNARE regulatory factors, and genes related to the autophagosome-lysosome pathway can contribute to the restoration of neurotransmitter vesicle cycling and neuronal function. Within this pathway, there are regulatory relationships among these aforementioned genes. Proteins such as STX4, STX16, SNAP23, and VAMP3/8 in the SNARE complex directly participate in this pathway35by modulating the fusion process between organelle membranes and vesicles. Snapin acts as a regulator of SNARE complexes36, influencing the activity and localization of SNARE proteins. Meanwhile, STXBP1 serves as a ligand for SNARE proteins by binding to and activating SNARE complexes to promote membrane fusion37.Therefore, we propose that Vamp8 modulates lysosomal autophagy by regulating the function of downstream SNARE proteins.
Furthermore, we found the GSEA analysis of the high-risk CIRI group revealed significant enrichment in inflammation-related pathways, coinciding with an upregulation in Vamp8.The immune pathway is critical in the progression of stroke. The inflammatory response mechanism of atherosclerosis involves both innate and adaptive immunity38. Dendritic cells serve as professional antigen-presenting cells, and our research has revealed significant positive correlations between dendritic cells and Vamp8. Previous studies have proved the substantial dendritic cell aggregation during the formation of atherosclerosis39. This may be attributed to the ability of dendritic cells to activate macrophages, promote Th1 expression, and subsequently release interferon and other cell factors that contribute to atherogenesis40,41.
miRNAs are a class of small non-coding RNA molecules, typically 20–24 nucleotides in length, that play a critical role in the regulation of cellular processes42. miR-325-3p is expressed in mice, rats, and humans, playing a crucial role in regulating gene expression, cell proliferation, and apoptosis. Research has shown that miR-325-3p protects rat neurons from ischemia-reperfusion injury by inhibiting RIP343. A recent study confirmed that hsa-miR-325-3p plays an important role in reversing the capillarization of liver sinusoidal endothelial cells44.
This study utilized multiple RNA-seq datasets (GSE61616, GSE97537 and GSE82146) and achieved high-quality data integration through advanced batch effect removal methods (R package sva). Compared to previous analyses relying on single datasets, this approach provides a more reliable and comprehensive gene expression profile.Through an in-depth analysis of lysosomal autophagy-related genes, we successfully identified 36 LRDEGs. This screening method, relating LRDEGs to CIRI, reveals the potential mechanisms of autophagy during CIRI process for the first time in related research. We demonstrated the intersection between LRDEGs and overall DEGs through Venn diagram analysis, highlighting the unique role of lysosomal autophagy in CIRI. This finding lays the foundation for future explorations of autophagy’s role in CIRI and related diseases. By integrating logistic regression and SVM algorithms, we developed a novel diagnostic model for CIRI, specifically optimizing model specificity through LASSO regression analysis. This multi-model ensemble approach not only enhances predictive accuracy but also provides a new tool for clinical diagnosis. However, this study inevitably has certain limitations, such as featuring a sample size that is relatively small and has undergone limited analysis for clinical validation. Future research could further assess the potential application value of lysosomal autophagy regulatory factors in diagnosing and predicting prognosis in cases of stroke. Furthermore, although mice and rats exhibit a high degree of genomic conservation, significant differences in gene regulatory mechanisms may exist between species, potentially affecting the expression and function of Vamp8 and its related factors. Therefore, we recommend that future research incorporate mechanistic experiments and systematic analyses of other species to achieve a more comprehensive understanding of the interaction networks involving Vamp8 and its biological significance across different organisms. This will provide important insights into the potential role of Vamp8 in disease processes.
Data availability
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplemental information.GEO database (https://www.ncbi.nlm.nih.gov/geo/); Genecards database(https://www.ncbi.nlm.nih.gov/gds); MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp).
References
Lo, E. H., Dalkara, T. & Moskowitz, M. A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 4 (5), 399–415. https://doi.org/10.1038/nrn1106 (2003).
Ekker, M. S. et al. Stroke incidence in young adults according to age, subtype, sex, and time, trends. Neurology 92, e2444–e2454. https://doi.org/10.1212/WNL.0000000000007533 (2019).
Iadecola, C., Buckwalter, M. S. & Anrather, J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. J. Clin. Invest. 130, 2777–2788. https://doi.org/10.1172/JCI135530 (2020).
Zhang, M. et al. Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets. Signal. Transduct. Target. Ther. 9 (1), 12. https://doi.org/10.1038/s41392-023-01688-x (2024).
Liu, S. Z., Yao SJ,Yang, H. & Autophagy Regulator of cell death. Cell. Death Dis. 14 (10), 648. https://doi.org/10.1038/s41419-023-06154-8 (2023).
Mo, Y., Sun, Y. Y. & Liu, K. Y. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 15 (8), 1388–1396. https://doi.org/10.4103/1673-5374.274331 (2020).
Zhang, Y. et al. Weakened interaction of ATG14 and the SNARE complex blocks autophagosome-lysosome fusion contributes to fluoride-induced developmental neurotoxicity. Ecotoxicol. Environ. Saf. 230, 113108 (2021).
Ling, D. et al. Attenuation of histone H4 lysine 16 acetylation (H4K16ac) elicits a neuroprotection against ischemic stroke by alleviating the autophagic/lysosomal dysfunction in neurons at the penumbra. Brain Res. Bull. 184, 24–33 (2022).
Davis, S. Meltzer PS.GEOquery: a Bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23 (14), 1846–1847. https://doi.org/10.1093/bioinformatics/btm254 (2007).
Wang, L., Yu, Y., Yang, J., Zhao, X. & Li, Z. Dissecting xuesaitong’s mechanisms on preventing stroke based on the microarray and connectivity map. Mol. Biosyst. 11 (11), 3033–3039. https://doi.org/10.1039/c5mb00379b (2015).
Wang, H., Anggraini, F. T., Chen, X. & DeGracia, D. J. Embryonic lethal abnormal vision proteins and adenine and uridine-rich element mRNAs after global cerebral ischemia and reperfusion in the rat. J. Cereb. Blood Flow. Metab. 37 (4), 1494–1507. https://doi.org/10.1177/0271678X16657572 (2017).
Stelzer, G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinf. ;54:1.30.1–1.30.33.doi: https://doi.org/10.1002/cpbi.5. (2016).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28 (6), 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47. https://doi.org/10.1093/nar/gkv007 (2015).
Salem, K. B. & Abdelaziz, A. B. Principal component analysis (PCA). Tunis Med. 99 (4), 383–389 (2021).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47 (D1), 419–426. https://doi.org/10.1093/nar/gky1038 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16 (5), 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19 (1), 432. https://doi.org/10.1186/s12859-018-2451-4 (2018).
Engebretsen, S. & Bohlin, J. Statistical predictions with Glmnet. Clin. Epigenetics. 11 (1), 123. https://doi.org/10.1186/s13148-019-0730-1 (2019).
Wu, J. et al. A nomogram for predicting overall survival in patients with low-grade endometrial stromal sarcoma: A population-based analysis. Cancer Commun. (Lond). 40 (7), 301–312. https://doi.org/10.1002/cac2.12067 (2020).
Calster, B. V. et al. Reporting and interpreting decision curve analysis: A guide for investigators. Eur. Urol. 74 (6), 796–804. https://doi.org/10.1016/j.eururo.2018.08.038 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102 (43), 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27 (12) 1739-40. (2011). https://doi.org/10.1093/bioinformatics/btr260
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12 (5), 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42 (Database issue), D92–D97. https://doi.org/10.1093/nar/gkt1248 (2014).
Singh, A. RNA-binding protein kinetics. Nat. Methods. 18 (4), 335. https://doi.org/10.1038/s41592-021-01122-6 (2021).
Hu, X. et al. Microglia/ macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia, Stroke.2012(43)11;3063–3070. https://doi.org/10.1161/STROKEAHA.112.659656
Zhang, X. et al. Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons.Autophagy. 17(6):1519–1542. (2021). https://doi.org/10.1080/15548627.2020.1840796
Tian, X. ,Teng J,Chen J.New insights regarding SNARE proteins in autophagosome-lysosome fusion.Autophagy. 10.1080/15548627.2020.1823124. (2021).
Chen, Q. et al. Prefused lysosomes cluster on autophagosomes regulated by Vamp8. Cell. Death Dis. 12 (10), 939. https://doi.org/10.1038/s41419-021-04243-0 (2021).
Itakura, E., Kishi-Itakura, C. & Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151 (6), 1256–1269. https://doi.org/10.1016/j.cell.2012.11.001 (2012).
Huang, H. et al. mTOR-mediated phosphorylation of Vamp8 and SCFD1 regulates autophagosome maturation. Nat. Commun. 12 (1), 6622. https://doi.org/10.1038/s41467-021-26824-5 (2021).
Zhao, Y. G., Codogno, P. & Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell. Biol. 22 (11), 733–750. https://doi.org/10.1038/s41580-021-00392-4 (2021).
Mohseni, R. ArabSadeghabadi Z,Ziamajidi N,Abbasalipourkabir R,RezaeiFarimani A.Oral administration of Resveratrol-Loaded solid lipid nanoparticle improves insulin resistance through targeting expression of SNARE proteins in adipose and muscle tissue in rats with type 2 diabetes. Nanoscale Res. Lett. 14 (1), 227. https://doi.org/10.1186/s11671-019-3042-7 (2019).
Chheda, M. G., Ashery, U., Thakur, P., Rettig, J. & Sheng, Z. H. Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat. Cell. Biol. 3 (4), 331–338. https://doi.org/10.1038/35070000 (2001).
Verhage, M. et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287 (5454), 864–869. https://doi.org/10.1126/science.287.5454.864 (2000).
Fani, L. et al. The association of innate and adaptive immunity, subclinical atherosclerosis, and cardiovascular disease in the Rotterdam study: a prospective cohort study. PLoS Med. 17 (5), e1003115. https://doi.org/10.1371/journa.pmed.1003115 (2020).
Worbs, T. Hammerschmidt SI,Förster R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 17 (1), 30–48. https://doi.org/10.1038/nri.2016.116 (2017).
Gil-Pulido, J. & Zernecke, A. Antigen-presenting dendritic cells in atherosclerosis. Eur. J. Pharmacol. 816, 25–31. https://doi.org/10.1016/j.ejphar.2017.08.016 (2017).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127 (3), 402–426. https://doi.org/10.1161/CIRCRESAHA.120.316903 (2020).
Cai, Y. & Yu X,Hu S,Yu, J. A. brief review on the mechanisms of miRNA regulation.Genomics Proteomics Bioinformatics. 7 (4) 147 – 54. (2009). https://doi.org/10.1016/S1672-0229(08)60044-3
Yi, S. et al. miR-325-3p protects neurons from oxygen-glucose deprivation and reoxygenation injury via inhibition of RIP3. Dev Neurosci. 42 (2–4) 83–93. (2020). https://doi.org/10.1159/000509108
Liu, W. et al. Nucleic acid spheres for treating capillarisation of liver sinusoidal endothelial cells in liver fibrosis. Nat. Commun. 16 (1), 4517. https://doi.org/10.1038/s41467-025-59885-x (2025).
Acknowledgements
We would like to acknowledge the support of the National Natural Science Foundation of China for this research project.
Funding
This work was supported by the National Natural Science Foundation of China [grant number 82071351, 82301538 and 82371371].
Author information
Authors and Affiliations
Contributions
FY-X: Conceptualization, Methodology, Software, Data curation, Writing- Original draft preparation. CJ: Visualization, Investigation. YX and LY: Supervision. ST-F: Software, Validation. CL: Writing- Reviewing and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, Yx., Cao, J., Yin, X. et al. Vamp8 modulates cerebral ischemia-reperfusion injury via the autophagy-lysosome pathway. Sci Rep 15, 34654 (2025). https://doi.org/10.1038/s41598-025-20879-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-20879-w
