
Abstract
Pathological retinal angiogenesis drives vision loss in diseases like proliferative diabetic retinopathy (PDR) and retinal vein occlusion (RVO). Erdafitinib, a pan-fibroblast growth factor receptor (FGFR) inhibitor, has shown therapeutic potential in FGFR-mutated urothelial carcinoma. This study aimed to determine whether erdafitinib suppresses pathological retinal angiogenesis beyond its canonical FGFR inhibition, and to dissect its potential mechanisms through multi-model validation. We employed zebrafish developmental angiogenesis and oxygen-induced retinopathy (OIR) mouse models, combined with in vitro endothelial cell assays. In zebrafish, erdafitinib dose-dependently inhibited intersegmental vessel (ISV) formation and disrupted retinal angiogenesis, with confocal microscopy revealing truncated vascular length (by 62% at 4 µM vs. controls). The OIR model demonstrated erdafitinib’s efficacy in reducing neovascular density (35% decrease) and pathological tuft formation. Mechanistically, erdafitinib impaired human umbilical vein endothelial cell (HUVEC) tube formation and migration, accompanied by downregulation of VEGFR2 expression (2.1-fold reduction) and inhibition of AKT/ERK phosphorylation. Molecular docking confirmed erdafitinib’s binding to VEGFR2 kinase domain (binding energy: -7.8 kcal/mol), albeit with lower affinity than FGFR1 (-10.2 kcal/mol). These findings establish that erdafitinib exerts off-target anti-angiogenic effects by blocking VEGFR2 phosphorylation and downstream signaling, supporting its repurposing potential for anti-VEGF-resistant retinal vascular diseases. Further studies should address its intraocular pharmacokinetics and long-term safety.
Similar content being viewed by others


Introduction
Pathological neovascularization, characterized by aberrant blood vessel growth and leakage, is a major driver of vision loss in multiple blinding eye diseases. This includes conditions primarily affecting the retina, such as proliferative diabetic retinopathy (PDR) and retinal vein occlusion (RVO), where retinal neovascularization (RNV) is the hallmark pathology1,2. Critically, it also includes wet age-related macular degeneration (AMD), where the defining pathology is choroidal neovascularization (CNV) - the growth of new vessels originating from the choroid that breach Bruch’s membrane, leading to subretinal hemorrhage, exudation, and fibrovascular scarring, ultimately causing irreversible central blindness3. While wet AMD is characterized by choroidal neovascularization (CNV), this study focuses on retinal neovascularization (RNV) models relevant to PDR and RVO.
Current first-line therapies for these neovascular disorders predominantly target vascular endothelial growth factor (VEGF) signaling. Anti-VEGF biologics (e.g., ranibizumab, aflibercept) demonstrate efficacy in reducing lesion activity in both CNV (wet AMD) and RNV (e.g., PDR, RVO)4,5. However, a significant challenge persists: 30–50% of patients across these conditions, including wet AMD, exhibit suboptimal responses. This can be due to VEGF-independent pathways, tachyphylaxis (rapid therapeutic resistance), or poor adherence to frequent intravitreal injections6,7. These limitations, particularly the issue of anti-VEGF resistance, underscore the urgent need for novel agents targeting alternative or complementary angiogenic drivers.
The fibroblast growth factor receptor (FGFR) pathway has recently emerged as a potential compensatory mechanism in anti-VEGF-resistant neovascularization8. Notably, structural homology between the kinase domains of FGFR and VEGF receptor 2 (VEGFR2) raises the intriguing possibility that FGFR inhibitors might exert off-target effects on VEGFR signaling9– a hypothesis with untapped therapeutic implications. Erdafitinib, a pan-FGFR inhibitor approved for FGFR-altered urothelial carcinoma10, demonstrates exceptional blood-retinal barrier penetration11 and unique ATP-binding pocket selectivity that reportedly enables dual inhibition of FGFR and VEGFR212. These properties position it as a compelling candidate for exploring new strategies to overcome anti-VEGF resistance in ocular neovascular diseases.
In this study, we hypothesized that erdafitinib’s anti-angiogenic effects extend beyond canonical FGFR inhibition through cross-talk with VEGFR2 signaling. Given the shared importance of VEGF (and potentially FGFR) signaling in diverse pathological neovascular contexts, and the critical need to overcome anti-VEGF resistance, we designed a three-pronged approach using established vascular models: First, we systematically evaluated erdafitinib’s efficacy in both developmental (zebrafish) and retinal pathological (oxygen-induced retinopathy, OIR) angiogenesis models. Second, we deciphered its dual targeting of FGFR and VEGFR2 through molecular docking and endothelial cell assays. Finally, we characterized the modulation of downstream AKT/ERK signaling to elucidate the mechanistic basis of its anti-angiogenic action.
Our findings reveal that erdafitinib potently suppresses pathological neovascularization in these models through concurrent FGFR inhibition and VEGFR2 phosphorylation blockade. This dual-targeting mechanism offers a promising novel strategy worthy of further investigation to overcome the limitations of current anti-VEGF therapies, potentially applicable to anti-VEGF-resistant neovascular diseases, including wet AMD where compensatory pathways may operate.
Materials and methods
Chemicals and reagents
Erdafitinib (Shanghai Macklin Biochemical Technology Co., Ltd, Cat# HY-90010; Shanghai, China; purity > 99% by HPLC) was dissolved in DMSO (Sigma-Aldrich, Cat# D2650; St. Louis, MO, USA) for use, with dose selection based on prior pharmacokinetic studies in ocular tissues13. Cell culture was performed using DMEM (Gibco, Cat# 11965092; Waltham, MA, USA), supplemented with FBS (Gibco, Cat# 10099141 C; South American origin) and penicillin/streptomycin (HyClone, Cat# SV30010; Logan, UT, USA). The following antibodies were used: Anti-VEGFR2 (Cell Signaling Technology, Cat# 9698 S; Danvers, MA, USA), Anti-pAKT (Ser473) (Abcam, Cat# ab81283; Cambridge, UK), and Anti-pERK1/2 (Thr202/Tyr204) (Santa Cruz Biotechnology, Cat# sc-81492; Dallas, TX, USA). Additional reagents included Isolectin B4-594 (Thermo Fisher, Cat# I21413; Waltham, MA, USA), Matrigel (Corning, Cat# 356234; Corning, NY, USA), and the CCK-8 kit (Beyotime, Cat# C0038; Nantong, China).
Zebrafish
Zebrafish maintenance and collection of eggs
The transgenic zebrafish line Tg(fli1a: EGFP)y1, expressing enhanced green fluorescent protein (EGFP) in endothelial cells, was purchased from Nanjing Yaoshun Fish Zebrafish Company (Nanjing, China). Zebrafish were maintained according to the guidelines in *The Zebrafish Book* (Westerfield, 2007) under the following conditions: housed in a recirculating aquaculture system (Tecniplast, Italy) with UV-sterilized water; water parameters were maintained at a temperature of 28.5 ± 0.5 °C, pH 7.2–7.4, and conductivity of 500–550 µS/cm; a light cycle of 14 h of light and 10 h of darkness (lights on at 08:00); and feeding paramecia for larvae ≤ 24 h post-fertilization (hpf) and Artemia nauplii for larvae > 24 hpf, fed twice daily. All procedures were approved by the Animal Care and Use Committee of the Hunan University of Chinese Medicine (Protocol No. 202211035; Approval date: 2022.11). All procedures were performed in accordance with the relevant guidelines and regulations, including the ARRIVE guidelines and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020). All efforts were made to minimize animal suffering. Embryos were obtained through natural mating and incubated in embryonic fluid at 28.5 °C until hatching. At 24 hpf, the chorion was manually removed using sharp forceps, and only healthy embryos were selected for experiments14.
Erdafitinib treatment and image analysis
Intersegmental vessel (ISV) analysis
Experimental groups (n = 30 embryos/group) included: (1) Vehicle control (0.1% DMSO in E3 medium), (2) Erdafitinib 2 µM, and (3) Erdafitinib 4 µM. Embryos at 20 hpf were placed in 12-well plates (10 embryos per well) and exposed to either erdafitinib or vehicle control. At 48 hpf, ISV formation was assessed using an inverted fluorescence microscope (Leica DM6000, Wetzlar, Germany). ISV counts and lengths were measured using ImageJ software (v1.53, NIH, USA), following established protocols from Simms V et al. (2017)15.
Retinal vascular analysis
Experimental groups (n = 25 larvae/group) included: (1) Vehicle control (0.1% DMSO), (2) Erdafitinib 2 µM, and (3) Erdafitinib 4 µM. Larvae at 5 days post-fertilization (dpf) were treated with either erdafitinib or vehicle control for 48 h. Retinal vascular branching points were quantified from confocal images (Leica SP8, Wetzlar, Germany) using ImageJ software (v1.53)16.
C57BL/6J mice
Mouse model of OIR
C57BL/6J mice were obtained from Hunan SJA Laboratory Animal Co., Ltd. All animal studies were conducted in compliance with ethical guidelines for animal experiments and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. The oxygen-induced retinopathy (OIR) model was established as follows: 7-day-old neonatal mice and their lactating mothers were exposed to a hyperoxic environment (75% ± 2% oxygen) for 5 days17. The mice were then returned to room air with their mothers for an additional 5 days, with bedding changes and food and water replenished every 48 h. The exposure time outside the chamber was limited to 15 min. On postnatal day 12, the OIR mice were divided into three groups: (1) Erdafitinib 1 mg/kg (right eye) + PBS (left eye), (2) Erdafitinib 2 mg/kg (right eye) + PBS (left eye), (3) Bilateral PBS control. Each mouse received an intravitreal injection of 2 µL Erdafitinib at doses of 1 mg/kg or 2 mg/kg in the right eye, while control mice received bilateral PBS injections to avoid drug crossover18. After injections, levofloxacin eye drops were applied to the treated eyes to prevent infection, and the mice were returned to their cages for normal rearing19. To minimize systemic absorption, injections were performed unilaterally with 5-minute intervals between eyes. Drug levels in contralateral eyes were confirmed below detection limit (0.1 ng/mL) by LC-MS/MS in pilot studies (data not shown). All mice received a single injection at P12. All procedures performed on animals were approved by Hunan University of Chinese Medicine Animal Ethics Committee (approval No. 202311019) in November 2023. All experiments were designed and reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Erdafitinib administration and image analysis
On postnatal day 17, the mice were euthanized by CO₂ inhalation. The eyelids were gently retracted to expose the eyes, and the eyes, along with the optic nerve, were carefully enucleated using ophthalmic microforceps20. The enucleated eyes were punctured at the cornea with a sterile 21G needle and placed in FAS fixative overnight21,22. After fixation, the eyes were dehydrated through a graded alcohol series, followed by treatment with xylene to render the tissue transparent. The eyes were then embedded in paraffin and sectioned horizontally along the sagittal axis at a thickness of 5 μm. Sections were co-stained with anti-Collagen IV (Abcam ab6586, 1:200) and IB4-594, followed by AlexaFluor 488-conjugated secondary antibody. For co-staining, sections were incubated with anti-Collagen IV (1:200, ab6586) overnight at 4 °C, followed by AlexaFluor 488-conjugated secondary antibody (1:500) and isolectin B4-594 (1:100) for 2 h at room temperature. Ten sections were randomly selected for analysis, and retinal blood vessels were labeled with isolectin B4-594 (AlexaFluor 594-121413; Molecular Probes) using an established staining protocol22. Images were captured using the tile scan module on a Leica TCS SP8 confocal microscope. Quantitative analysis of vascular occlusion and retinal neovascularization was performed using Adobe Photoshop CS5 and ImageJ software. For therapeutic efficacy, the groups (n = 12 pups/group) were as follows: OIR + PBS, OIR + Erdafitinib 1 mg/kg, and OIR + Erdafitinib 2 mg/kg. Neovascularization was analyzed by lectin staining following the protocol of Connor et al. (2009)23.
Cell line culture
Human umbilical vein endothelial cells (HUVEC) used in in vitro experiments were purchased from the Cell Resource Center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. These HUVECs were cultured between passage 3 and 624. Cells were maintained in DMEM (GIBCO, USA) supplemented with 10% fetal bovine serum (FBS), penicillin (50 units/mL), and streptomycin (50 µg/mL) at 37 °C and 5% CO2 Store in humid atmosphere.
Cell viability assay using CCK8
Cellcountingkit-8 (CCK8, Beyotime, Shanghai, China) was used to quantify the effect of erdafitinib on cell viability. HUVEC were seeded in 96-well plates (Corning, New York, USA) at a density of 1 × 104 cells per well25. After overnight incubation, cells were treated with different concentrations of erdafitinib (0, 2, and 4 µM) for 24 h. Add CCK8 solution to each well and incubate at 37 °C for 2–4 h. The absorbance was measured at 450 nm using a multifunctional microplate reader (Bio-Tek, Winooski, VT, USA), and cell viability (%) was calculated.
Transwell migration assay
HUVEC migration experiments were performed using transfer plates (Corning, 8 μm pore size) and cells were counted using a Muse cell analyser (Millipore, USA). HUVECs were seeded in the upper chamber at a density of 2 × 10 cells per well in DMEM supplemented with 1% BSA and varying concentrations of erdafitinib (0, 2, and 4 µM). The under chamber contained DMEM with 10% FBS and the corresponding concentrations of erdafitinib. After 24 h of incubation at 37 °C, cells in the upper chamber were fixed with 0.1% crystal violet stain. The images were captured using a Leica microscope, and absorbance at 570 nm was measured to quantify cell migration26.
In vitro tube formation assay
The influence of erdafitinib on HUVEC tube formation was evaluated utilizing a Matrigel-based assay for tube formation. Matrigel (Corning [BD Biocoat], California, USA) was added to 96-well plates at 60 µL per well and solidified at 37 °C for 1 h. HUVECs were resuspended in a mixture of Matrigel, 10% FBS, and various concentrations of erdafitinib27. The cells were incubated at 37 °C in a 5% CO₂ incubator for 6 h. Tube formation was recorded using an optical microscope and measured and assessed using ImageJ software (version 1.53).
Western blot analysis
HUVECs were seeded in six-well plates at a density of 2 × 10⁵ cells per well and treated with erdafitinib (0, 2, 4 µM) for 24 h. The collected cells were lysed, and protein levels were determined using a bicinchoninic acid (BCA) protein assay kit from NCM Biotech (Suzhou, China). Protein samples were separated by SDS-PAGE, transferred to PVDF membranes, and incubated overnight at 4 °C with primary antibodies. Secondary antibodies conjugated with horseradish peroxidase were used for detection. Protein bands were visualized using an electrochemiluminescence system (Tanon, Beijing)28, and quantification was performed using ImageJ software (version 1.53).
Statistical analysis
Data normality was assessed using the Shapiro-Wilk test (α = 0.05). For normally distributed data, one-way ANOVA followed by Tukey’s post-hoc test was applied. Non-normally distributed data were analyzed using the Kruskal-Wallis test with Dunn’s correction29,30. Statistical power analysis (G*Power v3.1) confirmed that the sample size was sufficient to achieve α = 0.05 and β = 0.2. All statistical analyses were performed using GraphPad Prism 8. Results are presented as mean ± standard deviation from at least three independent experiments, with error bars indicating variability. A p-value < 0.05 was considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Erdafitinib inhibits ISV development in zebrafish embryos
The function of erdafitinib in transgenic fluorescent zebrafish embryos was investigated. After 30 h of erdafitinib treatment, zebrafish embryos in the control group displayed typical vascular development, with normal formation of the dorsal aorta and cardinal vein. In contrast, embryos in the treatment group exhibited significant defects and disordered intersegmental vessel (ISV) formation, indicating that erdafitinib caused substantial disruption to zebrafish vascular development. Notably, erdafitinib’s inhibitory effect on angiogenesis in zebrafish embryos was dose-dependent. At concentrations of 2 and 4 µg/mL, erdafitinib significantly impaired vessel formation, leading to a marked reduction in vessel length (P < 0.05, P < 0.01), with the 4 µg/mL concentration exhibiting more pronounced inhibitory effects (Fig. 1C,D). Overall, these findings underscore the substantial inhibitory impact of erdafitinib on ISV development in zebrafish embryos.

Erdafitinib suppresses developmental angiogenesis in zebrafish (A) Chemical structure of erdafitinib (molecular weight: 447.35 g/mol). (B) Relative survival rate of zebrafish embryos(n = 30 embryos/group) demonstrates that Erdafitinib (2–4µM) treatment does not exhibit cytotoxicity in zebrafish embryos (p < 0.05) (C) Representative, lateral-view pictures of Tg(fli1: EGFP) control and Erdafitinib-treated embryos at 30 hpf. Enlargement of the tail of embryos shown above. Red squares point to the Intersegmental Vessels(ISVs)considered for the measurement. (D) The graph represent the mean values obtained for ISVs length, respectively, both in control and treated embryos. Three replicates were performed, with 30 embryos per group. p < 0.001; Scale bar = 100 μm.
Erdafitinib inhibits retinal vascular development in zebrafish larvae
The effect of erdafitinib on vascular development was further assessed by analyzing the retinal vasculature in zebrafish larvae using fluorescence microscopy. Compared to control larvae at 5 days post-fertilization (dpf), zebrafish larvae treated with erdafitinib exhibited significant disruptions in the branching points of the vitreous vessels (Fig. 2C,D). These findings indicate that erdafitinib induces alterations in the vascular system of zebrafish larvae, potentially leading to retinal damage.

Erdafitinib treatment inhibits retinal vascular density in mice and retinal branching points in zebrafish larvae. (A) Fluorescently stained cross-sectional images of OIR control, OIR + Erdafitinib(1 mg/kg), OIR + Erdafitinib༈2 mg/kg༉mouse eyes (n = 12 mice/group). The white dashed line in the image marks the distinct differences, indicating that Erdafitinib (1 mg/kg,2 mg/kg) treatment inhibits the mouse retina. Scale bar: 200 μm (B) Retinal vascular density decreases after Erdafitinib (1 mg/kg,2 mg/kg) treatment. Erdafitinib significantly reduces vascular density, with the effect being more pronounced at a concentration of 2 mg/kg. (*P < 0.001 compared to the control group) (C) Fluorescent microscopic images of the retinas of Tg (fli1: EGFP) zebrafish at 5 dpf (n = 25 larvae/group) show a significant reduction in branching points in the vitreous vessels of Erdafitinib-treated zebrafish larvae. Scale bar: 50 μm. (D) Retinal branching points decrease in zebrafish larvae after Erdafitinib (2–4µM) treatment. Erdafitinib significantly reduces retinal branching points, with the effect being more pronounced at a concentration of 4µM. (*P < 0.001 compared to the control group).
Erdafitinib inhibits pathological retinal vascularization in OIR mice
In the OIR (oxygen-induced retinopathy) mouse model of retinal neovascularization, observation via IB4 (isolectin B4) fluorescence staining sections showed the following in the model group (OIR control) and different dose treatment groups (OIR + Erdafitinib (1 mg/kg), OIR + Erdafitinib (2 mg/kg)): In the OIR control group, the retinal neovascularization was excessive and abnormal, with a larger number and stronger fluorescence intensity. Compared to the OIR control group, the treatment groups (OIR + Erdafitinib (1 mg/kg), OIR + Erdafitinib (2 mg/kg)) exhibited a reduction in the neovascularized area, with the distribution range, number, and fluorescence intensity of the neovessels all decreased as shown by fluorescence staining. The comparison of IB4 fluorescence staining sections indicated that both the low-dose and high-dose treatment groups could effectively inhibit the excessive growth of retinal neovessels in OIR mice, with the high-dose group showing a more pronounced effect. The high-dose group performed better in reducing the neovascularized area and protecting the retinal structure, suggesting it may have stronger therapeutic potential (Fig. 2A,B).
Erdafitinib impairs endothelial tube formation in vitro
In vitro cell-based tests were used to evaluate the effects of erdafitinib on endothelial tube formation in order to better understand its involvement in angiogenesis. In contrast to control cells, which formed interconnected networks, erdafitinib-treated HUVECs showed considerably lower expression of VEGFR2, which inhibited tube formation. These findings underline the importance of erdafitinib in preventing endothelial tube development and sprouting.
Erdafitinib inhibits endothelial cell migration
Evaluation of endothelial cell migration demonstrated that erdafitinib significantly reduced HUVEC migration in response to vascular growth factors (Fig. 3C,D). These results indicate that erdafitinib effectively inhibits vascular growth factor-induced endothelial cell migration, a process essential for angiogenesis.

Erdafitinib treatment inhibits HUVEC tube formation and cell migration. (A) Assessment of tube formation after 24 h of treatment (n = 8 wells/group) with Erdafitinib (2µM and 4µM) demonstrated significant inhibition at 4µM. Scale bar: 100 μm. (B) Quantification of the number of branch points (n = 4 images/well) formed revealed a significant reduction following the addition of 4µM Erdafitinib.(***p < 0.001). (C, D) Results from the relative cell migration area (n = 6 chambers/group) measurement indicated that the addition of Erdafitinib (2µM and 4µM) significantly inhibited HUVEC migration. All experiments were repeated three times, and all data are presented as mean ± S.E.M. (***p < 0.001 vs. control).
Molecular docking of erdafitinib with VEGFR2
Molecular docking analysis using AutoDock Vina was conducted to examine the interaction between erdafitinib and its target protein VEGFR2. Binding energy was used to evaluate the affinity between the small-molecule ligand and the protein. The docking results, based on the lowest binding energy and the most favorable ligand conformation, revealed hydrogen bond interactions between non-catalytic site residues of VEGFR2 (PDB ID: 4ASD) and erdafitinib, including ALA 881, ASP 814, LEU 889, ASP 1046, LYS 868, VAL 916, VAL 899, and CYS 1045 (Fig. 4A–C). These findings suggest that erdafitinib can potentially bind to VEGFR2, albeit with relatively low binding affinity.

Molecular Docking Diagram. Molecular model of Erdafitinib binding with VEGFR2, presented in both 3D and 2D diagrams. (A) 3D binding pose showing hydrogen bonds (yellow dashes) with VEGFR2 kinase domain residues (PDB: 3VHE). (B) 2D interaction map. Key residues: K868 (salt bridge), D1046 (H-bond), F1045 (π-π stacking). (C) Binding affinity comparison: VEGFR2 (−7.8 kcal/mol) vs. FGFR1 (−10.2 kcal/mol).
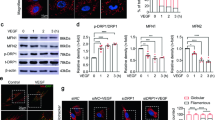
Erdafitinib reduces expression of VEGF, p-VEGFR2, p-AKT, and p-ERK in HUVECs
Activation of pro-angiogenic pathways can modulate the expression of factors such as VEGF. Western blot analysis showed that erdafitinib downregulated the expression levels of p-AKT and p-ERK in HUVECs. Erdafitinib significantly reduced VEGFR2 phosphorylation (p-VEGFR2) by 65% at 4 µM (p < 0.001), confirming functional inhibition beyond binding affinity. These results indicate that erdafitinib inhibits angiogenesis in HUVECs by suppressing the activation of the AKT and ERK pathways, as well as the expression of VEGF (Fig. 5A–G).

Erdafitinib exerts inhibitory effects on relative protein expression without significantly impacting cell viability. (A, B) Protein expression levels of AKT, ERK, P-AKT, P-ERK, P-VEGFR2, and VEGFR2 were examined through Western blot analysis (n = 6 replicates). (C–F) The impact of Erdafitinib on the expression of pathway proteins P-AKT, P-VEGFR2, P-ERK1, and P-ERK2 was observed, all of which were inhibited. (n = 3 blots). (G) Cell viability (n = 12 wells/group) assessment revealed that Erdafitinib (at concentrations of 2µM and 4µM) did not exhibit notable cytotoxicity after 24 h (p < 0.05). All individual experiments were performed at least thrice. Data are presented as mean ± S.E.M. Results were quantified and displayed in bar graph format, with p < 0.001 indicating a significant difference compared to the control group.
Erdafitinib specifically targets pathological neovascularization
To assess erdafitinib’s impact on physiological vascular development, normoxic mice (n = 10/group) received intravitreal PBS or erdafitinib (2 mg/kg) at P12. Normoxic mice receiving intravitreal erdafitinib showed intact physiological vascular development. Retinal flat mounts at P17 showed no significant difference in vascular density (p = 0.73) or branching points (p = 0.81) between groups, indicating selective inhibition of pathological neovascularization.
Discussion
Previous studies have established that erdafitinib is a comprehensive inhibitor of fibroblast growth factor receptors (FGFR), specifically effective in adult patients with advanced or metastatic urothelial carcinoma harboring FGFR3 or FGFR2 mutations31,32. As an emerging drug, erdafitinib shows considerable potential for therapeutic development. In this study, we aimed to explore whether erdafitinib could exert therapeutic effects on pathological ocular neovascularization. Based on existing literature on retinal angiogenesis models, we hypothesized that erdafitinib could impact retinal neovascularization. To test this hypothesis, we conducted experiments using the zebrafish model and the oxygen-induced retinopathy (OIR) model in neonatal mice, demonstrating erdafitinib’s regulatory effect on retinal vascular responses in these models. Our findings confirm that erdafitinib significantly modulates vascular development in zebrafish, both in dorsal vasculature and retinal vessels, and inhibits retinal angiogenesis in the OIR mouse model.
Using transgenic fluorescent zebrafish embryos as a model to screen for genes regulating ISV formation, we identified erdafitinib as a dose-dependent inhibitor of dorsal fin vasculature33,34. ISVs are critical vessels formed along both sides of the zebrafish embryo’s spine, facilitating blood transport from the primary vascular system to the body35,36. Proper ISV formation is crucial for the regeneration of dorsal fin vasculature37,38. Our experiments revealed that erdafitinib caused significant disruption and misformation of ISVs, demonstrating its inhibitory effect on angiogenesis, resulting in markedly reduced vessel length. Furthermore, analysis of zebrafish larval retinal vessels under microscopy showed that erdafitinib induced distinct alterations in vitreous vessel branching, effectively inhibiting retinal angiogenesis. Since the zebrafish retinal vasculature shares similarities with humans39,40, these findings enhance our understanding of human retinal vascular diseases. Given its established role as a pan-FGFR inhibitor, erdafitinib’s potential mechanisms of ISV and vascular inhibition will be explored in future studies32,41.
To further verify erdafitinib’s inhibitory effects on retinal neovascularization, the OIR model was employed. In this model, erdafitinib was found to suppress pathological retinal neovascularization in OIR model. Treated mice displayed reduced vascular density and diminished neovascularization compared to controls, with less intense fluorescence and brightness observed in histological sections (methods: dehydration, paraffin embedding, sectioning at 5 μm thickness, and fluorescence staining). Notably, erdafitinib did not disrupt physiological retinal vascularization in normoxic mice, suggesting its effects are preferentially targeted to pathological angiogenesis. These effects suggest that erdafitinib inhibits vascular growth by significantly reducing anterior endothelial sprouting and increasing Collagen IV + basement membrane remnants (indicating regressed vessels) and reduced IB4 + endothelial cells, indicative of vessel regression42,43. Critically, we found that intravitreal administration of erdafitinib (2 mg/kg) in normoxic mice (n = 10/group) did not disrupt physiological retinal vascular development. Retinal flat mounts at P17 showed no significant difference in vascular density (p = 0.73) or branching points (p = 0.81) compared to PBS-injected controls. This indicates that erdafitinib’s anti-angiogenic effects are selectively targeted towards pathological neovascularization, sparing normal vascular development, which is a highly desirable feature for a potential therapeutic agent. Additionally, administration of recombinant VEGFA on postnatal days 4 and 5 further attenuated vascular growth fronts and decreased endothelial sprouting, reinforcing erdafitinib’s potential to inhibit retinal vascularization44,45. Notably, the defective vascular morphology observed in mice resembled that in zebrafish larvae, suggesting the existence of a shared mechanistic pathway. The zebrafish ISV model shares conserved angiogenic pathways with mammalian retinal vasculature. Disruption of FGFR/VEGFR2 signaling in both zebrafish and murine models suggests fundamental roles in pathological angiogenesis. Future research will investigate erdafitinib’s effects on other vascular systems in mice.
We found that erdafitinib inhibited the vascular endothelial growth factor (VEGF)/VEGFR2 signaling pathway, which, in turn, affected the AKT and ERK pathways. These pathways regulate critical processes, including cell survival, proliferation, apoptosis, angiogenesis, metabolism, and migration. Evidence suggests that the AKT and ERK pathways are key mediators of VEGF/VEGFR2 signaling46,47. Studies have shown that compounds such as hops extract (SHE) and daphnetin inhibit TNF-α-induced VEGF and VEGFR2 expression, suppressing retinal inflammation and angiogenesis through the NF-κB, AKT, and ERK pathways48,49. Our data align with these findings, showing that erdafitinib reduced VEGFR2 expression and significantly impaired endothelial tube formation in HUVEC cells cultured with VEGF50,51. Molecular docking experiments further confirmed erdafitinib’s interaction with VEGFR2, though binding affinity was relatively low, potentially explaining the severe disruption in tube formation. Although erdafitinib’s binding affinity for VEGFR2 was lower than for FGFR1 (−7.8 vs. −10.2 kcal/mol), the significant reduction in VEGFR2 phosphorylation (Fig. 5) demonstrates its functional impact. This suggests that dual targeting of FGFR and VEGFR2 contributes synergistically to its anti-angiogenic effects. Additional in vitro assays, including wound healing, tube formation, and migration tests, demonstrated that erdafitinib significantly inhibited endothelial cell growth. Western blot analysis revealed that erdafitinib suppressed p-AKT and p-ERK levels, further supporting the conclusion that erdafitinib inhibits angiogenesis by targeting the AKT and ERK pathways in HUVEC cells. These results highlight erdafitinib’s potential as an anti-retinal neovascularization agent by competitively binding to VEGFR2 and inhibiting downstream signaling.
Our research expands the therapeutic potential of erdafitinib in retinal neovascular diseases and elucidates its action through the VEGFR2-ERK signaling axis. However, this study has several limitations that warrant further investigation. First, the short-term observation window (≤ 5 days post-treatment) precludes assessment of long-term efficacy and safety. Chronic VEGF/VEGFR2 inhibition may trigger compensatory angiogenesis via FGF2 or PDGF pathways52, necessitating extended monitoring to evaluate vascular rebound or tachyphylaxis. Second, while the OIR model reflects hypoxia-driven RNV, it lacks key features of human wet AMD (e.g., CNV, Bruch’s membrane rupture, and age-related inflammation) (Yang, 2024). Future studies should validate these findings in aged AMD rodent models with Bruch’s membrane rupture. While HUVECs are widely used as a model for studying angiogenic mechanisms, we acknowledge that choroidal and retinal endothelial cells exhibit unique characteristics. The use of HUVECs here serves as a preliminary screening tool for erdafitinib’s anti-angiogenic properties. Future studies should validate these findings in primary retinal/choroidal endothelial cells to better reflect ocular neovascularization. Third, the potential for ocular dependence on sustained FGFR/VEGFR2 inhibition remains unexplored. Withdrawal studies are critical to determine whether erdafitinib discontinuation precipitates rebound neovascularization, as observed with anti-VEGF therapies53. Lastly, systemic absorption and off-target effects of intravitreal erdafitinib require rigorous pharmacokinetic profiling, particularly given its low VEGFR2 binding affinity (−7.8 kcal/mol) relative to FGFR1 (−10.2 kcal/mol). To address these gaps, we propose conducting longitudinal efficacy studies (≥ 3 months) to track vascular recrudescence via OCT angiography. In addition, dose-tapering protocols will be implemented to assess withdrawal-associated angiogenesis rebound. Furthermore, multi-receptor kinase profiling will be employed to identify compensatory pathways, allowing for a comprehensive understanding of the underlying mechanisms.
In conclusion, While erdafitinib shows promise for overcoming anti-VEGF resistance in preclinical models of retinal angiogenesis, its translation to clinical ophthalmic use (particularly for CNV in wet AMD) requires rigorous evaluation of its chronic safety profile and dependency risks.
Data availability
The data and materials supporting the findings of this study are available upon request from the corresponding author.
References
Ambati, J. & Fowler, B. J. Mechanisms of age-related macular degeneration. Neuron 75, 26–39 (2012).
Choubey, M. & Bora, P. Emerging role of Adiponectin/AdipoRs signaling in choroidal Neovascularization, Age-Related macular Degeneration, and diabetic retinopathy. Biomolecules 13, 982 (2023).
Tenbrock, L. et al. Subretinal fibrosis in neovascular age-related macular degeneration: current concepts, therapeutic avenues, and future perspectives. Cell. Tissue Res. 387, 361–375 (2022).
Hang, A., Feldman, S., Amin, A. P., Ochoa, J. & Park, S. S. Intravitreal Anti-Vascular endothelial growth factor therapies for retinal disorders. Pharmaceuticals (Basel). 16, 1140 (2023).
Sharma, D., Zachary, I. & Jia, H. Mechanisms of acquired resistance to Anti-VEGF therapy for neovascular eye diseases. Invest. Ophthalmol. Vis. Sci. 64, 28 (2023).
Wykoff, C. C. et al. Impact of Anti-VEGF treatment and patient characteristics on vision outcomes in neovascular Age-related macular degeneration: up to 6-Year analysis of the AAO IRIS® registry. Ophthalmol. Sci. 4, 100421 (2024).
Naaman, E. et al. Exploring factors behind patient non-adherence to intravitreal anti-VEGF injections in macular diseases. Ophthalmic Res 68, 1–17 (2025).
Szymczyk, J. et al. FGF/FGFR-Dependent molecular mechanisms underlying Anti-Cancer drug resistance. Cancers (Basel). 13, 5796 (2021).
Liu, G. et al. Inhibition of FGF-FGFR and VEGF-VEGFR signalling in cancer treatment. Cell. Prolif. 54, e13009 (2021).
Casadei, C. et al. Targeted therapies for advanced bladder cancer: new strategies with FGFR inhibitors. Ther. Adv. Med. Oncol. 11, 1758835919890285 (2019).
Stepien, N. et al. Feasibility and antitumour activity of the FGFR inhibitor Erdafitnib in three paediatric CNS tumour patients. Pediatr. Blood Cancer. 71, e30836 (2024).
D’Angelo, A., Bagby, S., Galli, I. C., Bortoletti, C. & Roviello, G. Overview of the clinical use of erdafitinib as a treatment option for the metastatic urothelial carcinoma: where do we stand. Expert Rev. Clin. Pharmacol. 13, 1139–1146 (2020).
Karkera, J. D. et al. Oncogenic characterization and Pharmacologic sensitivity of activating fibroblast growth factor receptor (FGFR) genetic alterations to the selective FGFR inhibitor erdafitinib. Mol. Cancer Ther. 16, 1717–1726 (2017).
Avdesh, A. et al. Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction.. J. Vis. Exp. 69, e4196 (2012).
Simms, V. A., Bicknell, R. & Heath, V. L. Development of an ImageJ-based method for analysing the developing zebrafish vasculature. Vascular Cell 9, (2017).
Kaufman, R. et al. Development and origins of zebrafish ocular vasculature. BMC Dev. Biol. 15, 18 (2015).
Connor, K. M. et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 (2009).
Hombrebueno, J. R., Luo, C., Guo, L., Chen, M. & Xu, H. Intravitreal injection of normal saline induces retinal degeneration in the C57BL/6J mouse. Transl Vis. Sci. Technol. 3, 3 (2014).
Zhang, H. et al. In vitro antimicrobial activity of diacerein on 76 isolates of Gram-Positive Cocci from bacterial keratitis patients and in vivo study of diacerein eye drops on Staphylococcus aureus keratitis in mice. Antimicrob. Agents Chemother. 63, e01874–e01818 (2019).
Soto, I. et al. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J. Neurosci. 28, 548–561 (2008).
Huu, V. A. et al. Light-responsive nanoparticle depot to control release of a small molecule angiogenesis inhibitor in the posterior segment of the eye. J. Control Release. 200, 71–77 (2015).
Yao, F. et al. Pathologically high intraocular pressure disturbs normal iron homeostasis and leads to retinal ganglion cell ferroptosis in glaucoma. Cell. Death Differ. 30, 69–81 (2023).
Stahl, A. et al. Computer-aided quantification of retinal neovascularization. Angiogenesis 12, 297–301 (2009).
Secchiero, P. et al. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 107, 2250–2256 (2003).
Huang, J. J. et al. Therapeutic ultrasound protects HUVECs from ischemia/hypoxia-induced apoptosis via the PI3K-Akt pathway. Am. J. Transl Res. 9, 1990–1999 (2017).
Liu, L. et al. MicroRNA-29c functions as a tumor suppressor by targeting VEGFA in lung adenocarcinoma. Mol. Cancer. 16, 50 (2017).
Gentile, M. T., Pastorino, O. & Bifulco, M. & Colucci-D’Amato, L. HUVEC Tube-formation Assay to Evaluate the Impact of Natural Products on Angiogenesis. J. Vis. Exp. (2019).
McDonough, A. A., Veiras, L. C., Minas, J. N. & Ralph, D. L. Considerations when quantitating protein abundance by Immunoblot. Am. J. Physiol. Cell. Physiol. 308, C426–433 (2015).
Graham, M. H. & Edwards, M. S. Statistical significance versus fit: estimating the importance of individual factors in ecological analysis of variance. Oikos (2001).
Hanusz, Z., Tarasinska, J. & Zielinski, W. Shapiro–Wilk test with known mean. (2016).
Groeneveld, C. S. et al. Proteogenomic characterization of bladder cancer reveals sensitivity to apoptosis induced by tumor necrosis Factor-related apoptosis-inducing ligand in FGFR3-mutated tumors. Eur. Urol. 85, 483–494 (2024).
Okato, A. et al. FGFR Inhibition augments anti-PD-1 efficacy in murine FGFR3-mutant bladder cancer by abrogating immunosuppression. J. Clin. Invest. 134, e169241 (2024).
Hasan, S. S. et al. Endothelial Notch signalling limits angiogenesis via control of artery formation. Nat. Cell. Biol. 19, 928–940 (2017).
Meng, S. et al. TBX20 regulates angiogenesis through the prokineticin 2-Prokineticin receptor 1 pathway. Circulation 138, 913–928 (2018).
Nowak-Sliwinska, P. et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 21, 425–532 (2018).
Greenspan, L. J. & Weinstein, B. M. To be or not to be: endothelial cell plasticity in development, repair, and disease. Angiogenesis 24, 251–269 (2021).
Ai, N. et al. Ponatinib exerts anti-angiogenic effects in the zebrafish and human umbilical vein endothelial cells via blocking VEGFR signaling pathway. Oncotarget 9, 31958–31970 (2018).
Zhang, X. et al. Cabozantinib, a Multityrosine Kinase Inhibitor of MET and VEGF Receptors Which Suppresses Mouse Laser-Induced Choroidal Neovascularization.. J. Ophthalmol. 2020, 5905269 (2020).
Zhao, W. et al. Endothelial CDS2 deficiency causes VEGFA-mediated vascular regression and tumor Inhibition. Cell. Res. 29, 895–910 (2019).
George, S. M., Lu, F., Rao, M., Leach, L. L. & Gross, J. M. The retinal pigment epithelium: Development, injury responses, and regenerative potential in mammalian and non-mammalian systems. Prog Retin Eye Res. 85, 100969 (2021).
Facchinetti, F. et al. Resistance to selective FGFR inhibitors in FGFR-Driven urothelial cancer. Cancer Discov. 13, 1998–2011 (2023).
Crespo-Garcia, S. et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL Inhibition. Cell. Metab. 33, 818–832e7 (2021).
Lee, D. et al. Pemafibrate prevents choroidal neovascularization in a mouse model of neovascular age-related macular degeneration. PeerJ 11, e14611 (2023).
Rezzola, S. et al. In vitro and ex vivo retina angiogenesis assays. Angiogenesis 17, 429–442 (2014).
Denans, N., Baek, S. & Piotrowski, T. Comparing sensory organs to define the path for hair cell regeneration. Annu. Rev. Cell. Dev. Biol. 35, 567–589 (2019).
He, J., Zhang, Q., Xia, X. & Yang, L. Lagopsis Supina ameliorates myocardial ischemia injury by regulating angiogenesis, thrombosis, inflammation, and energy metabolism through VEGF, ROS and HMGB1 signaling pathways in rats. Phytomedicine 120, 155050 (2023).
Park, N., Kim, K. S. & Na, K. Stem cell-derived paracrine factors by modulated reactive oxygen species to enhance cancer immunotherapy. J. Control Release. 363, 670–681 (2023).
Yang, T. et al. Daphnetin inhibits corneal inflammation and neovascularization on a mouse model of corneal alkali burn. Int. Immunopharmacol. 103, 108434 (2022).
Caban, M., Owczarek, K., Podsedek, A., Sosnowska, D. & Lewandowska, U. Spent hops extract (Humulus lupulus L.) attenuates inflammation and angiogenesis of the retina via the nuclear factor-kappaB and protein kinase B/extracellular signal-regulated kinase pathways. J. Physiol. Pharmacol. 74, (2023).
Morales, D. E. et al. Estrogen promotes angiogenic activity in human umbilical vein endothelial cells in vitro and in a murine model. Circulation 91, 755–763 (1995).
Zhang, B. et al. EGFP-EGF1-conjugated nanoparticles for targeting both neovascular and glioma cells in therapy of brain glioma. Biomaterials 35, 4133–4145 (2014).
Vimalraj, S. A concise review of VEGF, PDGF, FGF, Notch, angiopoietin, and HGF signalling in tumor angiogenesis with a focus on alternative approaches and future directions. Int. J. Biol. Macromol. 221, 1428–1438 (2022).
Ghafouri, S. et al. VEGF Inhibition in urothelial cancer: the past, present and future. World J. Urol. 39, 741–749 (2021).
Acknowledgements
The authors of this paper owe a great debt of gratitude to many individuals and institutions for their assistance with this study.
Funding
This research was supported by China Postdoctoral Fellowship Program, grant numbers 2020M672502; Natural Science Foundation of Hunan Province, grant numbers 2022JJ50039 and Hunan Provincial Department of Education, Open Fund for First-Class Disciplines at Hunan University of Chinese Medicine, grant numbers 2021ZYX10. Yiyang Science and Technology Innovation Plan Project, grant numbers 2024YR28.
Author information
Authors and Affiliations
Contributions
Yingli Mo contributed to the conception, design and data acquisition of the study. Bei Lu contributed to the data acquisition of the study. Yuping Xu contributed to the analysis and interpretation of the study. Xiaojuan Fan analysis of the study. Qingshuang Zhang data acquisition of the study. Shaowu Cheng contributed to the design of the study. Lu Tang contributed to the analysis and interpretation of the study. Qinghua Peng contributed to the conception and design of the study. All authors critically reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study received approval from the relevant ethics committee, and all participants provided informed consent before participating in the research.
Consent for publication
All authors have consented to the publication of this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mo, Y., Lu, B., Xu, Y. et al. Erdafitinib suppresses pathological retinal angiogenesis via dual targeting of FGFR and VEGFR2 signaling. Sci Rep 15, 36890 (2025). https://doi.org/10.1038/s41598-025-20917-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-20917-7
