
Abstract
To identify genetic markers that can predict the prognosis of bladder carcinoma (BC) patients using single-cell RNA sequencing (scRNA-seq). The RNA-seq data for BC were obtained from the GSA-Human database (HRA000212). Quality control and normalization were performed using the Seurat package, followed by identification of highly variable genes. Cell clustering and annotation were based on these genes. Functional enrichment analyses (GO and KEGG) were conducted, along with cell-cell communication and trajectory analysis. Prognostic modeling was performed using LASSO-Cox regression, with Receiver Operating Characteristic (ROC) analysis for risk group evaluation. Gene expression validation was carried out via RT-qPCR and western blotting in normal (SV-HUC-1) and BC cell lines (T24, J82, EJ, UM-UC-3, 5637, RT112). A total of 473 upregulated genes and 106 downregulated genes were identified in BC samples. These differentially expressed genes (DEGs) were significantly enriched in apoptosis-related signaling pathways and IL-17 signaling pathway. Cell-cell communication analysis suggested that the CXCL2/MIF-CXCR2 signaling pathway may mediate interactions between epithelial cells and fibroblasts. By integrating bulk RNA sequencing data, we identified 49 genes that are associated with the prognosis of BC. Using the LASSO-Cox regression model, 17 prognostic genes were selected. Stratification of patients into high- and low-risk groups based on these genes allowed for effective survival prediction, supported by the ROC curve analysis. Univariate and multivariate Cox regression analyses further confirmed that the risk score was an independent predictor of overall survival. Furthermore, IGFBP5, KRT14 and SERPINF1 were found to be linked to poor survival outcomes in BC, with their expression levels notably elevated in BC cell lines compared to a normal bladder cell line. This study identified potential prognostic marker genes for BC, offering valuable insights into the prediction of patient survival outcomes.
Similar content being viewed by others

Introduction
Bladder carcinoma (BC), the ninth most common cancer globally and one of the most common malignancy of the urinary system, had nearly 600,000 new cases worldwide in 20201,2. Approximately 75% of BC patients have non-muscle-invasive bladder cancer (NMIBC), 20% have muscle-invasive bladder cancer (MIBC), and another 5% have metastatic BC3,4. Despite the high treatment costs, the clinical prognosis of BC remains poor5. After standard treatment with transurethral resection of bladder tumor (TURBT) and adjuvant chemotherapy, 50–70% of NMIBC cases will recur within five years, with 10% to 30% progressing to MIBC or metastatic BC6. Compared to NMIBC, MIBC progresses and metastasizes more rapidly, with a worse overall prognosis7. The standard treatment for MIBC is radical cystectomy combined with lymph node dissection, typically accompanied by neoadjuvant chemotherapy8. However, the five-year survival rate after surgery is only around 60%9. The high mortality and recurrence rates of BC may be attributed to the high proliferative and invasive nature of cancer cells, as well as malignant biological behaviors such as immune system dysregulation10,11.
Tumor heterogeneity presents significant challenges in identifying potential diagnostic and therapeutic biomarkers through traditional tissue-based transcriptomic sequencing (bulk RNA-seq)5,12. Single-cell RNA sequencing (scRNA-seq) greatly overcomes the limitations of traditional bulk RNA-seq and has emerged in recent years as a powerful method for unveiling tumor heterogeneity13. For example, single-cell transcriptomic profiling of urothelial carcinoma revealed rare epithelial subtypes with epithelial-to-mesenchymal transition and cancer stem cell-like features, as well as distinct immunosuppressive microenvironments associated with tumor progression14. In addition, a recent review highlighted how integrating scRNA-seq with spatial transcriptomics has provided novel insights into bladder cancer pathogenesis and treatment resistance, underscoring the transformative potential of these technologies for precision oncology15. In this research, we employed scRNA-seq in conjunction with data from the TCGA and GTEx databases to identify and categorize various cell types, followed by cluster analysis. Further investigations were carried out using techniques such as Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, pseudotime analysis, and CellChat. Additionally, a risk model was developed through LASSO-Cox regression, which showed considerable promise as a prognostic biomarker and demonstrated strong predictive accuracy for immunotherapeutic responses in BC patients.
Materials and methods
Data source, clustering, and cell annotation
The scRNA-seq data for BC were employed from the GSA-Human database (https://ngdc.cncb.ac.cn/gsa-human/), under the accession code HRA000212. In the quality control process, we retained cells with nFeature_RNA > 200 and < 5000, and percentage of mitochondrial genes (percent.mt) < 5%, to exclude low-quality or dying cells and potential doublets with abnormally high gene counts. To further remove ambient RNA contamination and improve data purity, we utilized the decontX package (version 1.0.0). This method estimates and subtracts contamination from the expression profile of each cell. All downstream analyses were performed using the Seurat package (version 4.4.0), and batch effect correction was conducted using the Harmony package (version 1.2.0). To facilitate clustering, UMAP cluster analysis was carried out using highly variable genes, and cell type annotation was subsequently performed for the identified cell clusters.
To further identify genes associated with prognosis in BC, we utilized publicly available bulk RNA-seq datasets. Gene expression data for normal bladder tissues were obtained from the Genotype-Tissue Expression (GTEx) database, while tumor transcriptomic data were derived from the bladder urothelial carcinoma (BLCA) cohort of The Cancer Genome Atlas (TCGA). Both datasets, along with corresponding clinical information, were downloaded from the UCSC Xena platform (https://xenabrowser.net/datapages/). All analyses were performed using these publicly available data.
Copy number variations (CNVs) estimation
The CNVs in BC epithelial cells were analyzed using the InferCNV package with default settings (https://github.com/broadinstitute/infercnv) to identify highly malignant epithelial cells. Immune and stromal cells were used as reference controls. The CNV score for each cell was defined as the total number of CNVs calculated across all genes within that cell.
Differentially expressed genes (DEGs), GO enrichment analysis and KEGG pathway enrichment analysis
DEGs were identified using the “FindAllMarkers” function in the Seurat package, applying a threshold of |log2FoldChange| > 1 and P adjust < 0.01. Following DEG identification, GO and KEGG pathway enrichment analyses were performed to explore the functional significance of upregulated and downregulated genes16,17. Terms with a P adjust < 0.05 were considered significantly enriched.
Cell-cell communication analysis
Cell-cell communication is a crucial mechanism through which cells coordinate and cooperate to perform various biological functions. To investigate the interactions between different cell types, we constructed a cellular communication network using the CellChat package (Version 1.6.1). It incorporates known ligand-receptor complex compositions and, through a series of calculations and analyses, unveils the intercellular communication network and the interactions between cells. In addition, the NicheNet (v2.2.0) package was used to investigate differential gene expression among cells in BC tissues that may be driven by intercellular communication. NicheNet specifically predicts the prioritization of target genes in sender cells and their corresponding receiver cells, as well as changes in their expression levels during tumorigenesis18.
LASSO-Cox regression analysis
To identify genes associated with prognosis, we employed the LASSO-Cox regression model using the Glmnet package (Version 4.1.8) to establish a risk coefficient model, using survival time and status as dependent variables. The risk index for individuals with different survival outcomes was then calculated, with the median risk score serving as the cut-off point. Based on this median score, patients with BC were classified into low-risk and high-risk groups. Survival analysis and Receiver Operating Characteristic (ROC) curve assessments were subsequently performed to evaluate the predictive accuracy and survival outcomes of these two groups.
Univariate and multivariate Cox proportional hazards regression analyses
To further support the clinical relevance and prognostic value of the identified riskscore, univariate and multivariate Cox proportional hazards regression analyses were performed. Variables included in the analyses comprised the risk score, demographic characteristics (age, sex, race), and clinicopathological parameters (tumor stage, T stage, N stage, and M stage). Hazard ratios (HRs) with 95% confidence intervals (CIs) were calculated to estimate the relative risk of overall survival associated with each variable.Variables with P < 0.05 in univariate analysis were further included in the multivariate Cox model to identify independent prognostic factors.
Pseudotime analysis
Pseudotime analysis is a technique used to infer the developmental or differentiation trajectories of cells from scRNA-seq data, aiming to model the gradual changes that occur as cells progress through biological processes. Pseudotime analysis of BC cells was conducted using the Monocle package (Version 2.30.1). To explore differentiation trajectories and their association with prognosis, cells were ordered based on their pseudotime using the “plot_cell_trajectory” function. The “differentialGeneTest” function was then applied to identify genes responsible for cell branching and differentiation. The results were visualized using the “plot_pseudotime_heatmap” function, providing insights into the dynamic processes of cell differentiation within the BC context.
Cell culture
The immortalized human urothelial cell line SV-HUC-1, along with several human bladder cancer cell lines, including T24, J82, EJ, UMUC-3, 5637, and RT-112, were sourced from the American Type Culture Collection (ATCC). SV-HUC-1 cells were cultured in F-12 K medium, while the bladder cancer cell lines (T24, J82, EJ, UMUC-3, 5637, and RT112) were maintained in RPMI-1640 medium. Both culture media were supplemented with 10% fetal bovine serum and 1% penicillin G-streptomycin. The cells were incubated at 37 ℃ in a CO2 incubator, ensuring a stable environment with 5% CO2.
Reverse transcription quantitative polymerase chain reaction (RT‑qPCR)
Total RNA was isolated from the cell lines using TRIzol reagent. Following extraction, the RNA underwent reverse transcription at 42 ℃ for 15 min using a reverse transcription kit, after which it was incubated at 95 ℃ for 3 min to synthesize complementary DNA (cDNA). RT-qPCR analysis was performed on the Agilent Technologies AriaMx real-time PCR System (Agilent, USA). Amplification was carried out using the TransStart® Top Green qPCR SuperMix (+ Dye II) kit. The primer sequences used in this analysis are provided in Supplementary Table S1.
Western blot
A cocktail of protease and phosphatase inhibitors, along with phenylmethylsulphonyl fluoride (PMSF), was added to a chilled 1% NP-40 solution and combined with the protein extracts from the cells. The protein concentration was determined using the BCA assay kit, and the samples were heated at 95 ℃ for 10 min. The proteins were then resolved by 8–12% SDS-PAGE, followed by transfer onto a PVDF membrane. The PVDF membranes were blocked with Protein-Free Rapid Blocking Buffer on a shaker at room temperature for 15 min, then incubated overnight with the primary antibody at 4℃. On the following day, the membranes were incubated with the appropriate secondary antibody at room temperature for 1 h. The immunoblot bands were then detected using the ECL Plus chemiluminescent substrate. Details on the antibodies and their concentrations are provided in Supplementary Table S2.
Statistical analysis
Data processing was conducted using R software (version 4.3.3). Statistical significance was assessed using the Student’s t-test. Kaplan Meier survival curves, along with log-rank tests, were employed to evaluate the relationship between the expression of prognostic genes and BC prognosis. P value of less than 0.05 was considered statistically significant.
Results
Cell annotation and cluster analysis
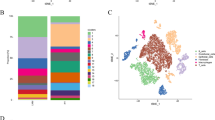
Transcriptomic data from 8 BC samples and 3 para-tumor samples were analyzed using scRNA-seq from the HRA000212 dataset in GSA-Human. After performing quality control and correcting for batch effects, a total of 56,403 single cells were clustered into eight major groups, including fibroblasts, T and NK, endothelial, myeloid, epithelial, mast, plasma and B cells, which were visualized using the UMAP 2D map (Fig. 1A-B). Cluster-specific genes were used to annotate cell types, employing classic markers referenced in previous studies (Fig. 1C)4,19,20,21. The distribution of cell types in both para-tumor and BC tissues is shown (Fig. 1D). Epithelial cells were significantly enriched in BC tissues, consistent with tumor cell expansion.

Cell annotation and cluster analysis. A-B, Transcriptomic data from 8 BC samples and 3 adjacent normal tissue samples were subjected to quality control and batch effect correction. scRNA-seq analysis clustered 56,403 single cells into 8 major populations. C, The dot plot shows cell type annotation, which was performed based on population-specific genes and classical marker genes. D, The distribution of cell types revealed differences in the composition and proportion of cell populations across different tissue samples.(Fib: fibroblast cells, T/NK: T and NK cells, Endo: endothelial cells, Mye: myeloid cells, Epi: epithelial cells, Mast: mast cells, Plasma: plasma cells, B: B cells.).
InferCNV, DEGs and function enrichment analyses
The CNVs in epithelial cells were inferred using the inferCNV method, allowing the malignancy of cancer cells to be assessed(Fig. 2A). The CNV scores were calculated for epithelial cells(Fig. 2B), and a threshold was defined as one standard deviation below the median CNV value. Cells with CNV scores below this threshold were classified as low-malignancy epithelial cells, whereas those above the threshold were classified as high-malignancy epithelial cells22(Fig. 2C).

CNVs in epithelial cells, DEGs and function enrichment analyses. A, CNVs in epithelial cells were inferred using the inferCNV method, revealing the presence of copy number alterations across the epithelial cell population. B, CNV scores were calculated for each epithelial cell. C, Epithelial cells were classified into low- and high-malignancy groups. D, DEG analysis of epithelial cells identified 473 upregulated genes and 106 downregulated genes. E-F, GO and KEGG enrichment analyses revealed significant enrichment in apoptosis-related signaling pathways and the IL-17 signaling pathway. The significance and functional annotations of the relevant pathways are shown in the figure.
A total of 16,726 high-malignancy epithelial cells were isolated and re-clustered. To investigate the pathogenesis of BC, DEG analysis was performed on the epithelial cells. Using a threshold of |log2FC| > 1 and P value < 0.01, 473 upregulated genes and 106 downregulated genes were identified (Fig. 2D). To gain further insights into the potential biological processes and pathways involved, GO and KEGG enrichment analyses were conducted, revealing significant enrichment in apoptosis-related signaling pathways and the IL-17 signaling pathway (Fig. 2E-F).
Cell-cell communication analysis
Ligand-receptor interactions between different cell types were modeled using CellChat functions to construct cell-cell communication networks (Fig. 3A-B). The signaling pathway analysis of bladder epithelial cells revealed that the MIF-CD74/CXCR4 interaction had the highest signaling intensity. The expression levels of genes involved in the MIF-CD74/CXCR4 signaling pathway were subsequently evaluated across various cell types (Fig. 3C-D).

Cell-cell communication analysis. A-B, The CellChat function was used to simulate ligand-receptor interactions between different cell types, constructing the intercellular communication network. The figure illustrates the ligand-receptor interactions and their network structure between various cell types. C-D, For bladder epithelial cells, the signal strength of MIF-CD74/CXCR2/CXCR4 interaction was analyzed, and the expression levels of genes involved in this signaling pathway across different cell types were evaluated. E, Ligand-target interaction network highlighting CXCL2 as a key ligand in bladder cancer.
We subsequently used NicheNet to explore intercellular interactions between epithelial cells and other cell types, aiming to investigate how cell-cell communication influences epithelial cells. All cell types were designated as sender cells, while epithelial cells were defined as receiver cells, and a BC-specific cell interaction network was constructed. We found that ligand-target interactions were predominantly centered on the ligand CXCL2, which was primarily expressed in fibroblasts (Fig. 3E). This suggests that the CXCL2/MIF-CXCR2 signaling pathway may play a critical role in mediating interactions between epithelial cells and other cell types, particularly fibroblasts.
Prediction and validation of prognostic gene signatures in BC cells
To further identify genes associated with prognosis in BC, differential gene expression analysis was performed using data from the GTEx database and BLCA from TCGA. A total of 116 upregulated genes and 252 downregulated genes were identified with a threshold of |log2FC| > 1 and P value < 0.01 (Fig. 4A). These differential genes in BLCA were intersected with the DEGs identified in bladder epithelial cells from scRNA-seq, resulting in 49 overlapping genes (Fig. 4B).

Prediction and validation of prognostic gene signatures in BC cells. A, Volcano plot of differential gene expression analysis from bulk RNA-seq. A total of 116 upregulated genes and 252 downregulated genes were identified. B, Intersection analysis of differential genes from BLCA and DEGs in bladder epithelial cells. C-D, Prognostic gene selection. The LASSO-Cox regression model was used to model the risk coefficients of 49 overlapping genes, resulting in the selection of 17 genes associated with BC prognosis. E, The risk scores for patients were calculated based on the risk coefficients of the 17 genes. Patients were divided into high-risk and low-risk groups based on the median risk score. F, Based on the grouping results from the median risk score, the Kaplan Meier survival curve showed that the prognosis of patients in the high-risk group was significantly worse. G, The ROC curve was evaluated based on the risk score model, further confirming the model’s good performance in predicting overall survival in patients. (***P < 0.001)
To identify prognosis-related genes, risk coefficients for these 49 genes were modeled using Constructing Cox model based on LASSO regression screening variables, with survival time and status as the dependent variables (Fig. 4C-D). Seventeen genes were selected, including CST1, CLIC3, CXCL10, EEF1G, EGR1, GJB2, HMGA1, IGFBP5, KRT14, KRT18, KRT7, RPL17, RPS10, SERINC2, SERPINF1, TACSTD2, TMSB10. The riskscore for each patient was calculated using the following formula:
Based on the median risk score, patients were categorized into high-risk and low-risk groups. Survival analysis and ROC curve evaluation were conducted for these two groups, using the median risk index as the cutoff point (Fig. 4E). Kaplan Meier survival curves demonstrated that the high-risk group had significantly poorer prognosis compared to the low-risk group (Fig. 4F). Afterwards, we evaluated the value of the prognostic signature in the TCGA and GTEx cohort, and the results showed a good prognostic value in BLCA (Fig. 4G).
Expressions and survival analysis of screened genes of BC cells in bulk RNA-seq
The expression levels of the 17 screened genes were analyzed in the training set, revealing that all genes showed significantly different expression between the BC group and BN group (Fig. 5A). Further analysis was conducted to investigate the impact of high and low expression levels of these genes on patient survival. The results indicated that elevated expressions of IGFBP5, KRT14, and SERPINF1 were significantly associated with worse survival outcomes and higher stage in BC patients (Fig. 5B-F).

Expression analysis of 17 selected genes in bulk RNA-seq in BC. A, The results showed significant differences in the expression of these genes between the BC and control groups. B-C, Expression of selected genes and survival analysis. High expression of IGFBP5, KRT14, and SERPINF1 was significantly associated with poorer survival outcomes in BC patients. E-F, High expression of IGFBP5 was significantly associated with worse stage and T classification in patients. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = no significance)
The expressions analysis of screened genes of BC cells in scRNA-seq
We further examined the expression of the screened prognostic genes using BC scRNA-seq. The heatmap displays the expression levels of the 17 screened genes in both BC and adjacent normal tissues, revealing distinct expression patterns between cancerous and adjacent normal tissues (Fig. 6A). Finally, the distribution of gene expression and the proportion of cells expressing each gene are visualized, with statistically significant differences observed across individual cells (Fig. 6B).

Expression analysis of 17 selected genes in scRNA-seq in BC. A, A heatmap showing the expression levels of 17 selected genes in BC and adjacent normal tissues, revealing significant expression differences between cancerous and adjacent normal tissues. B, Expression distribution and cell proportion analysis of selected genes. It showed significant statistical differences in gene expression levels across different cell types. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = no significance)
Pseudotime analysis of BC cells in scRNA-seq
In the present study, we focused on bladder epithelial cells to illustrate their developmental pathways. The trajectory diagram visualizes the temporal progression of bladder epithelial cell development (Fig. 7A). The epithelial cells undergo three distinct differentiation states during their development (Fig. 7B).Additionally, the distribution of both BC and para-tumor epithelial cells across the 8 identified clusters throughout development is shown (Fig. 7C-D). Seventeen screened genes that are strongly associated with cell differentiation and developmental trajectories are highlighted (Fig. 7E).

Pseudotime analysis of BC cells in scRNA-seq. A: The pseudotime analysis of bladder epithelial cells reveals the temporal progression of cell development. B: Differentiation states of bladder epithelial cells. It illustrates three distinct differentiation states that bladder epithelial cells undergo during development, showing the possible differentiation stages throughout the developmental process. C-D: Distribution of developmental trajectories of BC and adjacent epithelial cells. It shows the distribution of BC and adjacent epithelial cells across 8 identified clusters during pseudotime development, revealing distribution differences of cells from different groups along the differentiation trajectory. E: Expression of 17 selected genes along the developmental trajectory of bladder epithelial cells, demonstrating the crucial roles these genes play in cell differentiation and development.
Expressions of screened genes in BC cell lines
We conducted additional analyses to evaluate the expression of IGFBP5, KRT14, and SERPINF1 in the normal bladder cell line (SV-HUC-1) and various BC cell lines (T24, J82, EJ, UM-UC-3, 5637, and RT112) through RT-qPCR and western blot techniques. The mRNA and protein levels of these genes were markedly elevated in the BC cell lines compared to the normal bladder cells, indicating their potential prognostic markers for BC (Fig. 8A-B). Original blots are presented in Supplementary Fig. 1.

Expressions of screened genes in BC cell lines. A. RT-qPCR results showed that the mRNA levels of IGFBP5 and SERPINF1 were significantly upregulated in BC cell lines compared to normal bladder cells. B: Western blot results demonstrated that the protein levels of IGFBP5, KRT14, and SERPINF1 were markedly increased in BC cell lines compared to normal bladder cells. (*P < 0.05, **P < 0.01, ***P < 0.001, ns = no significance)
Notably, KRT14 mRNA levels were higher in the normal bladder epithelial cell line SV-HUC-1 than in bladder cancer cell lines, whereas its protein levels were markedly lower. This discordance may result from post-transcriptional or post-translational regulation, highlighting the complexity of gene expression beyond transcription.
Cox regression analysis of prognostic factors
To further evaluate the clinical relevance and prognostic value of the risk signature, we performed univariate and multivariate Cox proportional hazards regression analyses. In univariate analysis, the riskscore (HR = 2.718, 95% CI: 2.159–3.421, P < 0.0001), age at diagnosis (HR = 1.034, 95% CI: 1.018–1.050, P < 0.0001) and M0 status (HR = 0.224, 95% CI: 0.055–0.919, P = 0.038) were significantly associated with overall survival, whereas most demographic and clinicopathological variables, including sex, race, tumor stage, T stage, and N stage did not reach statistical significance (Table 1).
Variables with P < 0.05 in univariate analysis were incorporated into the multivariate model. The riskscore remained an independent predictor of poor prognosis (HR = 2.578, 95% CI: 2.037–3.263, P < 0.0001), as did age (HR = 1.029, 95% CI: 1.013–1.044, P < 0.0001), while M0 status exhibited a modest protective effect (HR = 0.737, 95% CI: 0.547–0.994, P = 0.045) (Table 1). These results indicate that the risk signature provides independent prognostic information beyond conventional clinicopathological factors and may serve as a robust predictor of patient survival.
Discussion
Due to the extensive reprogramming of the tumor microenvironment (TME) and significant transcriptional heterogeneity, studying different cellular subtypes using traditional RNA sequencing techniques has posed challenges. In 2009, scRNA-seq technology emerged, enabling us not only to investigate cancer cell characteristics at the single-cell level but also to comprehensively describe the functional phenotypes of other cellular components within the TME23. In recent years, scRNA-seq has made significant progress in exploring biomarkers associated with BC progression and TME. In 2019, Yu et al.24 were the first to create a single-cell transcriptomic map of human bladder cells using scRNA-seq. Chen et al.4 established a TME map for BC and identified the role of inflammatory cancer-associated fibroblasts (iCAFs) in BC, revealing potential therapeutic targets. Tian et al.25 revealed that YWHAG expression was significantly elevated in tumor cells compared to normal urothelial cells, as determined by density distribution analysis based on scRNA-seq data. Lu et al.26 confirmed through scRNA-seq data that CXCR6 is a key biomarker for predicting the prognosis of MIBC and the response to immunotherapy. By integrating scRNA-seq with bulk transcriptomic data, our study bridges these complementary approaches to generate a comprehensive understanding of the cellular and molecular determinants of BC progression.
Our single-cell transcriptomic analysis identified a pivotal role for the MIF-CD74/CXCR2/CXCR4 signaling axis in orchestrating crosstalk between epithelial tumor cells and immune/stromal populations within the BC microenvironment. MIF, a pleiotropic inflammatory cytokine, binds the type II transmembrane receptor CD74 as well as chemokine receptors CXCR2 and CXCR4, promoting tumorigenic processes including macrophage recruitment, myeloid-derived suppressor cell (MDSC) expansion, angiogenesis, and immune evasion27,28,29,30. Specifically, CXCL2/MIF-CXCR2 signaling has been shown to drive MDSC recruitment and suppress T cell proliferation, contributing to an immunosuppressive niche associated with poor prognosis in BC patients28. Mechanistic studies demonstrate that MIF1 and its homolog DDT (MIF2) act redundantly through CD74 to sustain pro-tumorigenic signaling, with dual inhibition of MIF1/MIF2 achieving more effective suppression of tumor proliferation and progression compared to single-agent blockade in preclinical models30. Moreover, CD74 itself appears to be differentially expressed in muscle-invasive versus non-muscle-invasive BC, and its knockdown reduces proliferation, invasion, and angiogenesis, underscoring its functional importance as a therapeutic target31. Our scRNA-seq data further suggest that epithelial tumor cells actively exploit this signaling axis to modulate surrounding immune and stromal compartments, especially fibroblasts, consistent with previous reports of MIF-(CD74 + CXCR4) and MIF-(CD74 + CD44) interactions in BC32. Collectively, these findings support a model in which aberrant MIF-CD74/CXCR2/CXCR4 signaling establishes a pro-tumorigenic microenvironment by integrating tumor-intrinsic and stromal/immune cues.From a translational perspective, targeting components of this axis—through receptor blockade, neutralizing antibodies, or dual MIF homolog inhibition—represents a promising strategy to enhance antitumor immunity and improve patient outcomes in BC.
Our study identified IGFBP5, KRT14, and SERPINF1 as key drivers within the prognostic 17-gene signature in bladder cancer, highlighting their emerging roles in tumor progression. IGFBP5, a binding protein in the insulin-like growth factor (IGF) signaling pathway, promotes tumor cell proliferation, migration, and invasiveness via both IGF-dependent and independent mechanisms33. Its regulation downstream of the L-type amino acid transporter 1 (LAT1) underscores its functional relevance, as LAT1 inhibition by JPH203 reduced IGFBP5 expression and suppressed MAPK/ERK and AKT signaling, thereby attenuating bladder cancer cell proliferation and invasion34. Fibulin-3 further enhances invasive potential through upregulation of IGFBP5, with restoration of IGFBP5 rescuing invasion after fibulin-3 knockdown, positioning IGFBP5 as a central mediator of aggressive tumor behavior35. Beyond its pro-proliferative function, IGFBP5 may serve as a surrogate marker for therapeutic responsiveness: high expression inversely correlates with IGF1R activation and sensitivity to IGF1R inhibitors36, and it has been repeatedly identified as a hub gene linking metabolic dysregulation, such as type 2 diabetes, to bladder cancer progression37. Clinically, IGFBP5 is incorporated into molecular diagnostic assays such as Cxbladder®, emphasizing its prognostic and stratification value38.
KRT14, a basal keratin, marks the aggressive basal/squamous molecular subtype in MIBC39. Multiregional immunohistochemical analyses demonstrate that KRT14 expression is enriched in basal/squamous tumors and correlates with advanced tumor stage, particularly pT4 lesions40. Transcriptomic and immunohistochemical profiling further identifies KRT14 among core basal markers co-expressed with KRT5 and CD44, defining the basal/squamous consensus subtype41. High KRT14 levels are associated with high-grade, muscle-invasive tumors and shorter disease-specific survival, reflecting its linkage to squamous differentiation and invasive phenotypes42. Mechanistically, basal keratin expression can be modulated by peroxisome proliferator-activated receptor gamma (PPARγ) signaling and proliferative cues; activation of PPARγ combined with proliferation inhibition downregulates KRT14 and other basal keratins, suggesting potential strategies for reprogramming basal-like tumors toward less aggressive luminal states43.
SERPINF1, encoding pigment epithelium-derived factor (PEDF), functions as a stage-associated hub gene in bladder cancer, with elevated expression linked to tumor cell proliferation and prognostic significance44. SERPINF1 contributes to immune evasion by promoting resistance to PD-1/PD-L1 blockade through enhanced fatty acid production and suppression of CD8⁺ T cell activation; conversely, knockdown or inhibition of fatty acid synthesis restores immunotherapy sensitivity45. These findings indicate that SERPINF1 actively modulates the tumor immune microenvironment, reinforcing aggressive phenotypes.
Beyond their individual roles, IGFBP5, KRT14, and SERPINF1 may operate within a coordinated “proliferation-invasion-immune escape” network. IGFBP5 establishes a proliferative foundation through IGF and LAT1 signaling, KRT14 marks basal-like invasive states, and SERPINF1 promotes immune evasion, collectively enabling tumor clones to expand and invade while escaping host immunity. Potential interactions among these genes may involve shared transcriptional regulators, metabolic pathways, or signaling axes; for example, metabolic rewiring downstream of IGFBP5 or SERPINF1 could influence basal programs marked by KRT14, creating feedback loops that reinforce aggressive tumor behavior.
Translationally, integrating these three genes into a combined biomarker panel could improve prognostic precision by capturing complementary aspects of tumor biology. Therapeutically, a multi-pronged approach emerges: targeting IGF/IGFBP5 signaling to inhibit proliferation, reprogramming KRT14-positive basal-like cells to reduce invasiveness, and modulating SERPINF1-mediated immune suppression to restore anti-tumor immunity. Such combination strategies may enhance patient stratification, optimize immunotherapy efficacy, and mitigate bladder cancer progression by concurrently addressing proliferative, invasive, and immunosuppressive mechanisms.
However, this study has some limitations. First, the limited sample size of scRNA- seq data may restrict the generalizability of our findings. Although scRNA-seq enables high-resolution analysis of cellular heterogeneity within the tumor microenvironment, the number of patients included in the dataset is relatively small. As a result, the identified cell clusters and gene expression patterns may not fully represent the biological diversity observed across a broader population of BC patients. Further studies incorporating larger, multi-center single-cell datasets are necessary to validate the robustness and reproducibility of our results. Second, functional validation of the identified prognostic genes remains preliminary. The biological functions and mechanistic roles of these genes in BC progression have not yet been experimentally explored. In-depth functional assays, such as gene knockdown or overexpression experiments, along with in vivo studies, are required to elucidate the precise molecular pathways through which these genes contribute to tumor behavior and patient prognosis.
Conclusion
In this study, we integrated TCGA and scRNA-seq data to explore the role of BC cell trajectories and intercellular communication, and identified several BC prognostic genes, enabling us to predict patient survival.
Data availability
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249 (2021).
Cañizo, C. G. et al. Characterisation of the tumour microenvironment and PD-L1 granularity reveals the prognostic value of cancer-associated myofibroblasts in non-invasive bladder cancer. Oncoimmunology 14 (1), 2438291 (2025).
Lopez-Beltran, A., Cookson, M. S., Guercio, B. J. & Cheng, L. Advances in diagnosis and treatment of bladder cancer. Bmj 384, e076743 (2024).
Gontero, P. et al. European association of urology guidelines on Non-muscle-invasive bladder cancer (TaT1 and carcinoma in Situ)-A summary of the 2024 guidelines update. Eur. Urol. 86 (6), 531–549 (2024).
Ali, W., Xiao, W., Jacobs, D. & Kajdacsy-Balla, A. Survival and enrichment analysis of Epithelial-Mesenchymal transition genes in bladder urothelial carcinoma. Genes (Basel) 14, 10 (2023).
Leblond, M. M., Zdimerova, H., Desponds, E. & Verdeil, G. Tumor-Associated macrophages in bladder cancer: biological Role, impact on therapeutic response and perspectives for immunotherapy. Cancers (Basel). 13, 18 (2021).
Taylor, J. I. et al. Long-term outcomes of bladder-sparing therapy vs radical cystectomy in BCG-unresponsive non-muscle-invasive bladder cancer. BJU Int. 135 (2), 260–268 (2025).
Lerner, S. P. et al. Standard or extended lymphadenectomy for Muscle-Invasive bladder cancer. N Engl. J. Med. 391 (13), 1206–1216 (2024).
Witjes, J. A. et al. European association of urology guidelines on Muscle-invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur. Urol. 79 (1), 82–104. https://doi.org/10.1016/j.eururo.2020.03.055 (2021).
Dyrskjøt, L. et al. Bladder cancer. Nat. Rev. Dis. Primers. 9 (1), 58. https://doi.org/10.1038/s41572-023-00468-9 (2023).
Chen, Z. et al. Prognosis risk model based on Necroptosis-Related signature for bladder cancer. Genes (Basel) 13, 11 (2022).
Fang, Z., Li, J., Cao, F. & Li, F. Integration of scRNA-Seq and bulk RNA-Seq reveals molecular characterization of the immune microenvironment in acute pancreatitis. Biomolecules 13, 1. https://doi.org/10.3390/biom13010078 (2022).
Huang, D. et al. Advances in single-cell RNA sequencing and its applications in cancer research. J. Hematol. Oncol. 16 (1), 98 (2023).
Lyu, T. et al. Single-Cell RNA sequencing reveals the tumor heterogeneity and immunosuppressive microenvironment in urothelial carcinoma. Cancer Sci. 116 (3), 710–723. https://doi.org/10.1111/cas.16436 (2025).
Yoshihara, K., Ito, K., Kimura, T., Yamamoto, Y. & Urabe, F. Single-cell RNA sequencing and Spatial transcriptome analysis in bladder cancer: current status and future perspectives. Bladder Cancer. 11 (1), 23523735251322017. https://doi.org/10.1177/23523735251322017 (2025).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–D462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 17 (2), 159–162. https://doi.org/10.1038/s41592-019-0667-5 (2020).
Li, J. et al. Epithelial cell diversity and immune remodeling in bladder cancer progression: insights from single-cell transcriptomics. J. Transl Med. 23 (1), 135. https://doi.org/10.1186/s12967-025-06138-6 (2025).
Li, J. et al. Single-cell and bulk RNA-sequence identified fibroblasts signature and CD8 + T-cell - fibroblast subtype predicting prognosis and immune therapeutic response of bladder cancer, based on machine learning: bioinformatics multi-omics study. Int. J. Surg. 110 (8), 4911–4931. https://doi.org/10.1097/JS9.0000000000001516 (2024).
Chen, Z. et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 11 (1), 5077. https://doi.org/10.1038/s41467-020-18916-5 (2020).
Peng, J. et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell. Res. 29 (9), 725–738. https://doi.org/10.1038/s41422-019-0212-1 (2019).
Aran, D. Single-Cell RNA sequencing for studying human cancers. Annu. Rev. Biomed. Data Sci. ;6, 1–22. https://doi.org/10.1146/annurev-biodatasci-020722-091857 (2023).
Yu, Z. et al. Single-Cell transcriptomic map of the human and mouse bladders. J. Am. Soc. Nephrol. 30 (11), 2159–2176. https://doi.org/10.1681/ASN.2019040335 (2019).
Tian, T. et al. YWHAG promotes bladder cancer metastasis by regulating TMOD3 to activate ERK1/2 and JNK phosphorylation in the MAPK pathway. J. Transl Med. 22 (1), 1159. https://doi.org/10.1186/s12967-024-06003-y (2024).
Lu, X. et al. CXCR6 expression predicts prognosis and immunotherapeutic benefit in muscle-invasive bladder cancer. Front. Oncol. 14, 1498579. https://doi.org/10.3389/fonc.2024.1498579 (2024).
Penticuff, J. C., Woolbright, B. L., Sielecki, T. M., Weir, S. J. & Taylor, J. A. MIF family proteins in genitourinary cancer: tumorigenic roles and therapeutic potential. Nat. Rev. Urol. 16 (5), 318–328. https://doi.org/10.1038/s41585-019-0171-9 (2019).
Zhang, H. et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 36 (15), 2095–2104. https://doi.org/10.1038/onc.2016.367 (2017).
Guda, M. R. et al. Pleiotropic role of macrophage migration inhibitory factor in cancer. Am. J. Cancer Res. 9 (12), 2760–2773 (2019).
Woolbright, B. L. et al. Role of MIF1/MIF2/CD74 interactions in bladder cancer. J. Pathol. 259 (1), 46–55. https://doi.org/10.1002/path.6018 (2023).
Gai, J. W. et al. Expression of CD74 in bladder cancer and its suppression in association with cancer proliferation, invasion and angiogenesis in HT-1376 cells. Oncol. Lett. 15 (5), 7631–7638. https://doi.org/10.3892/ol.2018.8309 (2018).
Zhao, Z., Jia, H., Sun, Z., Li, Y. & Liu, L. A new perspective on macrophage-targeted drug research: the potential of KDELR2 in bladder cancer immunotherapy. Front. Immunol. 15, 1485109. https://doi.org/10.3389/fimmu.2024.1485109 (2024).
Waters, J. A., Urbano, I., Robinson, M. & House, C. D. Insulin-like growth factor binding protein 5: diverse roles in cancer. Front. Oncol. 12, 1052457. https://doi.org/10.3389/fonc.2022.1052457 (2022).
Maimaiti, M. et al. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci. Rep. 10 (1), 1292 (2020).
Han, A. L. et al. Fibulin-3 promotes muscle-invasive bladder cancer. Oncogene 36 (37), 5243–5251 (2017).
Neuzillet, Y. et al. IGF1R activation and the in vitro antiproliferative efficacy of IGF1R inhibitor are inversely correlated with IGFBP5 expression in bladder cancer. BMC Cancer. 17 (1), 636. https://doi.org/10.1186/s12885-017-3618-5 (2017).
Nayan, S. I. et al. Network based approach to identify interactions between type 2 diabetes and cancer comorbidities. Life Sci. 335, 122244. https://doi.org/10.1016/j.lfs.2023.122244 (2023).
Harvey, J. C. et al. Analytical Validation of Cxbladder(®) Detect, Triage, and Monitor: Assays for Detection and Management of Urothelial Carcinoma. Diagn. (Basel) 14 (18). https://doi.org/10.3390/diagnostics14182061 (2024).
Koll, F. J. et al. Overexpression of KMT9α is associated with aggressive Basal-like Muscle-Invasive bladder cancer. Cells 12, 4. https://doi.org/10.3390/cells12040589 (2023).
Schallenberg, S. et al. Intratumoral heterogeneity of molecular subtypes in Muscle-invasive bladder Cancer-An extensive multiregional immunohistochemical analysis. Eur. Urol. Focus. 9 (5), 788–798. https://doi.org/10.1016/j.euf.2023.03.012 (2023).
Angeloni, M. et al. Robust consensus molecular subtyping of Muscle-Invasive bladder cancer via 3’ RNA sequencing of Formalin-Fixed Paraffin-Embedded tissues: potential impact for clinical and trial settings. Lab. Invest. 105 (9), 104191. https://doi.org/10.1016/j.labinv.2025.104191 (2025).
Ingenwerth, M., Nyirády, P., Hadaschik, B., Szarvas, T. & Reis, H. The prognostic value of cytokeratin and extracellular collagen expression in urinary bladder cancer. Curr. Mol. Med. 22 (10), 941–949 (2022).
Mehus, A. A. et al. Activation of 37PPARγ and Inhibition of cell proliferation reduces key proteins associated with the basal subtype of bladder cancer in As3+-transformed UROtsa cells. PLoS One. 15 (8), e0237976. https://doi.org/10.1371/journal.pone.0237976 (2020).
Feng, F. et al. Identifying stage-associated hub genes in bladder cancer via weighted gene co-expression network and robust rank aggregation analyses. Med. (Baltim). 101 (51), e32318. https://doi.org/10.1097/MD.0000000000032318 (2022).
Denis, M. et al. In vivo syngeneic tumor models with acquired resistance to Anti-PD-1/PD-L1 therapies. Cancer Immunol. Res. 10 (8), 1013–1027. https://doi.org/10.1158/2326-6066.CIR-21-0802 (2022).
Acknowledgements
The single-cell RNA sequencing analysis in this study was supported by High-performance Computing Platform of Peking University.
Funding
2025 Government-Funded Program for the Training of Outstanding Clinical Medicine Talents(ZF2025133), S&T Program of Hebei: Central Government Guiding Local Science and Technology Development Fund Project (236Z7710G).
Author information
Authors and Affiliations
Contributions
SMZ: Writing-original draft, Formal analysis.RL: Data curation, Writing-review & editing, Validation.TT: Formal analysis, Methodology, Visualization.JLZ: Investigation, Resources.YL: Software, Validation.WZG: Data curation, Methodology, Supervision.YZ: Project administration.YQJ: Formal analysis, Funding acquisition, Visualization.SMH: Resources, Project administration.XSL: Investigation, Writing-review & editing.YQG: Conceptualization, Software, Supervision.YXG: Supervision, Funding acquisition, Investigation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, S., Li, R., Tian, T. et al. Single-cell RNA sequencing identifies potential critical prognostic biomarkers in bladder carcinoma. Sci Rep 15, 39105 (2025). https://doi.org/10.1038/s41598-025-23975-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-23975-z
