
Abstract
Thyroid cancer, the most prevalent endocrine malignancy, frequently presents with lymph node metastasis (LNM), a critical prognostic factor in influencing recurrence and survival. Even with progress in diagnostic and therapeutic strategies, the mechanisms underlying LNM, especially the role of microbial dysbiosis and metabolic reprogramming, remain incompletely understood. This study aimed to elucidate the interplay between gut microbiota, host metabolism, and LNM in thyroid cancer through a multi-tiered analytical framework to address this gap. We selected 90 patients with differentiated thyroid carcinoma (DTC) who underwent surgery. Before iodine-131 treatment, fecal samples were collected, and microbial diversity and metabolite profiles were analyzed using 16 S rRNA sequencing and LC-MS-based metabolomics. Patients were stratified based on lymph node metastasis status (LNM vs. no metastasis), metastatic site (central vs. lateral neck metastasis), and metastatic burden (≤ 5, 5–10, and > 10 metastatic nodes). Our findings revealed notable alterations in the composition of gut microbiota and metabolite profiles associated with LNM. At the phylum level, Proteobacteria was elevated in LNM + patients, while Bacteroidota and Fusobacteriota were depleted. At the genus level, Faecalibacterium and Escherichia_Shigella were enriched in LNM + patients, while Bacteroides was depleted. Metabolomics analysis identified several potential biomarkers, including osmanthuside A, niacin, and alpha-tocopherol, which could distinguish patients with and without LNM. Additionally, specific metabolites such as leukotriene-F4 and 3-Methoxytyramine showed marked differences between central and lateral neck metastasis groups. Our study underscores the potential influence of gut microbiota and metabolites on the advancement and metastasis of thyroid cancer progression. These findings offer a foundation for developing non-invasive biomarkers and therapeutic strategies targeting the gut-thyroid axis.
Similar content being viewed by others


Introduction
Thyroid cancer (TC), especially papillary thyroid carcinoma (PTC), is the most prevalent endocrine malignancy and exhibits a high propensity for lymph node metastasis (LNM). Thyroid cancer is a critical prognostic factor influencing recurrence and survival, with reported LNM rates exceeding 60% even in early-stage cases1,2,3. The lymph node metastasis of thyroid cancer usually first occurs in the central compartment, and then may spread to the lateral cervical region4. In addition, thyroid cancer can also metastasize to mediastinal lymph nodes, but relevant studies are relatively limited. Despite advancements in diagnostic and therapeutic strategies, the mechanisms underlying LNM—particularly the role of microbial dysbiosis and metabolic reprogramming—remain incompletely understood.
Previous studies indicated gut microbiota dysbiosis and metabolic reprogramming in TC pathogenesis and progression5,6. The gut-thyroid axis, mediated by microbial metabolites (such as short-chain fatty acids), may modulate immune responses and tumor microenvironment, influencing metastatic potential7,8. Recent metabolomic studies further reveal distinct serum and tissue metabolic profiles in PTC patients with LNM, suggesting metabolic pathways as potential drivers of metastasis9.
Meanwhile, emerging evidence suggests that the gut and intratumoral microbiota, along with their metabolic byproducts, may modulate tumor progression and metastatic behavior in thyroid cancer10. However, systematic microbial diversity and metabolomic profile comparisons across distinct LNM patterns (e.g., central vs. lateral metastasis) and metastatic burdens (e.g., low vs. high nodal involvement) are lacking.
This study aims to elucidate the interplay between microbiota, host metabolism, and LNM in thyroid cancer through a multi-tiered analytical framework. First, we compare fecal microbial diversity and metabolomic signatures between patients with LNM (Y2) and those without metastasis (W2), building on prior observations that gut microbiota alterations correlate with thyroid cancer pathogenesis10. Second, we stratify LNM patients into central (CLNM) and lateral neck metastasis (LLNM) subgroups, given their divergent clinical implications11. CLNM is more common but often manageable, whereas LLNM is associated with higher recurrence and poorer outcomes12. Recent studies suggest that intratumoral bacteria may differentially influence metastatic routes13, yet whether fecal microbiota and systemic metabolism reflect these distinctions remains unexplored. Finally, we evaluate microbial and metabolic variations across gradients of metastatic burden (≤ 5, 5–10, and > 10 metastatic nodes), a factor increasingly recognized as a predictor of disease aggressiveness2. While metabolomic studies have identified tumor tissue and serum biomarkers associated with LNM, the potential of fecal metabolites as non-invasive indicators of metastatic extent is uncharted. Our integrative approach leverages 16 S rRNA sequencing and LC-MS-based metabolomics to uncover novel biomarkers and mechanistic insights, addressing gaps in current research13. This study not only advances our understanding of the microbiota-metabolite axis in thyroid cancer metastasis but also provides a foundation for developing microbiome- or metabolite-based stratification tools.
Materials and methods
Patients and experimental design
A total of 90 patients undergoing differentiated thyroid carcinoma (DTC) surgery and subsequent iodine-131 treatment at our hospital were enrolled in this study. Postoperative histopathological biopsy confirmed the diagnosis of DTC. The study included 22 men and 68 women, aged between 18 and 66 years, with an average age of 34.5 years. Patients were staged according to the 8th edition of the American Joint Committee on Cancer (AJCC) tumor staging system for thyroid carcinoma. Detailed clinical characteristics, such as the number of lesions, maximum tumor diameter, lymphatic metastasis count, and distant metastasis status, are listed in Supp Table 1. Importantly, microbial diversity and metabolome analyses were conducted at three levels: The initial stage considers the presence of lymph node metastasis, comparing two groups, non-lymph node metastasis (W2) and lymph node metastasis (Y2). The second level, concerning lymph node location, consists of three categories: non-lymph node metastasis (N), central lymph node metastasis (A1), and lateral cervical lymph node metastasis (B1). The third level, number of lymph node metastases, includes three groups, with number of lymph node metastases ≤ 5 (F1), number of lymph node metastases > 5 and < 10 (T1), and number of lymph node metastases > 10 (T2) (Supp Fig. 1).

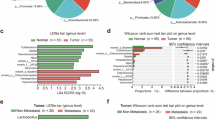
The changes in the gut microbiota of patients with thyroid cancer lymph node metastasis. The number of common and unique OTUs in Y2 and W2 (A). The relative ratio of top abundant taxa in Y2 and W2 at the phylum level (B). The relative ratio of top abundant taxa in Y2 and W2 at the genus level (C). Correlation analysis of top abundant taxa at the phylum level in Y2 and W2 (D). Significantly differential taxa between Y2 and W2 at the phylum level (E). Significantly differential taxa between Y2 and W2 at the genus level (G). Linear discriminant analysis (LDA) effect size (LEfSe) analysis of fecal microbial gneus in Y2 and W2 (F). Y2: lymph node metastasis, W2: non- lymph node metastasis.
Inclusion criteria
Participants were aged 18 to 60 years, residing and working in the Sichuan Basin for an extended period, with a body mass index (BMI) between 18.5 and 23.9. Patients in the research group were diagnosed with differentiated thyroid cancer through postoperative pathology.
Exclusion criteria
We excluded pregnant individuals, patients with previously diagnosed autoimmune thyroid diseases (e.g., Graves’ disease, Hashimoto’s thyroiditis), intestinal diseases (e.g., irritable bowel syndrome, ulcerative colitis, inflammatory bowel disease), a history of smoking, drinking, or drug dependence, other autoimmune diseases, endocrine disorders, severe systemic diseases, malignancy history, postmenopausal women, corticosteroid or immunosuppressant use within six months, antibiotic or probiotic use within one month, and patients with clinically significant unSupp Table diseases or conditions that would hinder their participation in the study.
Sample collection
Fecal samples of the patients were collected after the operation and before the treatment with Iodin-131. Fresh fecal samples were collected from study groups after an overnight fast (≥ 8 h). Gloves were worn to preserve hygiene during the collection. A sterile toothpick or stool sampler was used to obtain the inner midsection of the sample, avoiding surface layer contamination and bacterial DNA degradation. Each sample was divided into 2–3 sterile EP tubes (2 mL each, containing 0.5–2 g of feces) and stored in liquid nitrogen before being transferred to -80 °C for long-term preservation.
Microbial DNA extraction
Bacterial DNA was extracted from fecal samples using the E.Z.N.A.® Stool DNA Kit (Omega, USA) following the manufacturer’s protocols. DNA integrity and fragment sizes were assessed via 1% agarose gel electrophoresis, while DNA quantification was performed using the NanoDrop 2000 (Boston, USA).
High-throughput 16 S ribosomal RNA gene sequencing
Sequencing was conducted at Biomarker Technologies Co., Ltd. (Beijing, China). The V3-V4 region of the 16S rRNA gene was amplified using primers F: ACTCCTACGGGAGGCAGCA and R: GGACTACHVGGGTWTCTAAT, with Phusion® Hot Start Flex 2X Master Mix (New England Biolabs, USA). Unique barcodes were added to the 5’ ends of each sample’s primers. Amplicon purification and quantification were carried out using AMPure XT beads (Beckman Coulter Genomics, USA) and Qubit (Invitrogen, USA). The Agilent 2100 Bioanalyzer (Agilent, USA) was used to assess amplicon size and quantity. The amplicon library was quantified using the Quantification Kit for Illumina (Kapa Biosciences, USA) and sequenced on the Illumina Novaseq platform (Illumina, USA).
Data processing
The DADA2 algorithm within QIIME2 (version 2020.6) was employed to denoise post-quality control data. The default threshold for filtering Amplicon Sequence Variants (ASVs) was set at 0.005% of total sequence reads. Raw reads were filtered using Trimmomatic v0.33, and primer sequences were removed using cutadapt 1.9.1. Clean reads were spliced using Usearch v10, and sequences were aligned against an internal control database using BLASTN v2.9.0 + with a similarity threshold of 0.97, coverage threshold of 0.9, and e-value threshold of 0.001. Denoising and chimera removal were performed using the dada2 method in QIIME2 2020.6. Taxonomic annotation was conducted using the Naive Bayes classifier with the SILVA reference database. Community composition was analyzed at various taxonomic levels (phylum to species), and species abundance matrices were generated using QIIME and visualized with R tools. Alpha diversity was assessed using ACE, Chao1, and Shannon indices, while beta diversity was evaluated using principal coordinate analysis (PCoA). Bacterial abundance and diversity were compared using the Wilcoxon rank-sum test and Welch’s t-test. Linear discriminant analysis (LDA) effect size (LEfSe) was applied to identify differentially abundant taxa.
Nontargeted metabolomics profiling
Fifty milligrams of sample were weighed and mixed with 1000 µL of extraction solvent containing an internal standard (methanol: acetonitrile: water, 2:2:1, with an internal standard concentration of 20 mg/L). The mixture was vortexed for 30 s, processed with stainless steel beads in a bead mill at 45 Hz for 10 min, and ultrasonicated for 10 min in an ice-water bath. Metabolite extraction was analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) with an ACQUITY UPLC HSS T3 column (Waters, Milford, USA). The column temperature was set at 40 °C, with an injection volume of 2 µL and a helium carrier gas flow rate of 0.4 mL/min. The MS scanning range was m/z 70-1050. Raw data were processed using Progenesis QI software for peak extraction and alignment. Metabolites were identified based on the METLIN database and Biomark’s library, with theoretical fragment identification and mass deviation within 100 ppm. Normalized peak area data were used for subsequent analysis. Orthogonal partial least squares-discriminant analysis (OPLS-DA) was conducted to evaluate the clustering of samples. Metabolites with VIP > 1, p-value < 0.05, and fold change (FC) > 2 were considered significantly different between groups. The False Discovery Rate (FDR) correction method accounted for multiple hypothesis testing. Metabolic pathway analysis was performed to identify enriched pathways based on altered metabolites, using the Human Metabolome Database (HMDB) and the KEGG database (https://www.kegg.jp/kegg/kegg1.html)14. Pathway enrichment analysis was conducted using the Python package SciPy.
Statistical analysis
Sequencing data were deposited in the SRA database (accession number: PRJNA1049645). The Mann-Whitney test in GraphPad Prism version 9.1.0 (GraphPad Software, San Diego, CA, USA) analyzed bacterial relative abundance and alpha diversity. Spearman’s rank correlation analysis was performed using the Scipy package in Python to examine the relationships between differentially enriched genus and metabolites. Continuous variables are presented as mean ± standard deviation (SD). Two groups were compared using Student’s t-test or a separate variance estimation t-test for independent samples. Differences between categorical variables were assessed using the chi-square test. All analyses were performed using SPSS version 22.0 (IBM, USA).
Results
The changes in the gut microbiota of patients with thyroid cancer lymph node metastasis
A total of 13,151 OTUs were identified across both groups (Y2 and W2) (Supp Table 2). Among these, 393 OTUs were shared between two groups, while 12,302 and 456 OTUs were unique to patients with lymph node metastasis (LNM+) and without metastasis (LNM-), respectively (Fig. 1A). At the phylum level, Firmicutes accounted for 65.50% (LNM+) vs. 64.92% (LNM-), Bacteroidota for 21.20% vs. 24.94%, Proteobacteria for 7.65% vs. 4.59%, Actinobacteriota for 3.35% vs. 2.80%, and Fusobacteriota for 0.57% vs. 1.94% (Fig. 1B). At the genus level, Faecalibacterium constituted 14.44% (LNM+) vs. 8.42% (LNM-), Bacteroides 8.43% vs. 10.90%, Escherichia_Shigella 4.28% vs. 3.07%, and Lachnospiraceae 4.25% vs. 3.70% (Fig. 1C). Correlation analysis highlighted UCG_005, Christensenellaceae_R_7_group, Alistipes_shahii, NK4A214_group, Ruminococcaceae, and Barnesiella as strongly associated with other genus, suggesting their potential functional roles (Fig. 1D). At the phylum level, significantly different taxa between groups were Sumerlaeota, Spirochaetota, and Bdellovibrionota (Fig. 1E), while at the genus level, they included bacterium_adhufec311, Erysipelatoclostridiaceae, Bacillus, durum, and Alistipes_sp._CHKCI003 (Fig. 1F). Notably, we identified the microorganisms with the greatest difference in abundance at the phylum and genus levels. Roseburia (highest fold-change in LNM+) and Negativicutes (highest in LNM-) exhibited the most divergent abundances (Fig. 1G).
The influence of lymph node metastasis quantity in thyroid cancer on gut microbiota
Among all three groups, 521 OTUs were common, whereas 8,447, 1,858, and 1,779 OTUs were exclusive to the F1, T1, and T2 groups, respectively (Fig. 2A). At the phylum level, we observed that Firmicutes accounted for 65.12%, 65.35% and 67.37% in F1, T1, and T2 groups, respectively. The proportions of Bacteroidota were 21.56%, 20.17% and 22.00% across the three groups. Proteobacteria represented 7.65%, 8.08% and 6.05%, while Actinobacteriota constituted 3.06%, 4.92% and 2.58% in the three groups (Fig. 2B).

The influence of lymph node metastasis quantity in thyroid cancer on gut microbiota. The number of common and unique OTUs in F1, T1, and T2 (A). The relative ratio of top abundant taxa in F1, T1, and T2 at the phylum level (B). The relative ratio of top abundant taxa in F1, T1, and T2 at the genus level (C). Significantly differential taxa in F1, T1, and T2 at the phylum level (D). Linear discriminant analysis (LDA) effect size (LEfSe) analysis of fecal microbial genus in F1, T1, and T2 (E). F1: Number of lymph node metastases is less than 5, T1: Number of lymph node metastases is between 5 and 10, T2: Number of lymph node metastases exceeds 10.
At the genus level, Faecalibacterium accounted for 12.36%, 14.36% and 15.55% in F1, T1, and T2 groups, respectively. The proportions of Bacteroides were 7.87%, 8.37% and 10.29%, while Lachnospiraceae represented 6.49%, 3.63% and 4.22% across the three groups (Fig. 2C).
Meanwhile, results demonstrated that Lachnospiraceae_bacterium_GAM79 showed significant differences across groups, with the highest relative abundance observed in patients with thyroid cancer having > 10 lymph node metastases (T2 group) (Fig. 2D). Additional screening for microbiota with the most significant fold changes revealed that Bifidobacterium, Bifidobacteriaceae, Bifidobacteriales, and Actinobacteriota exhibited substantially higher relative abundance in the T1 group compared to other groups (Fig. 2E).
Gut microbiota analysis of patients with central and lateral cervical lymph node metastasis
Results indicated a total of 331 OTUs were found to be shared in three groups, while 456, 7201, and 4322 OTUs were unique to N, A1, and B1 groups, respectively (Fig. 3A). At the phylum level, the relative abundance of several phyla, including Firmicutes, Bacteroidota, Proteobacteria, Actinobacteriota, Fusobacteriota, Desulfobacterota, and Verrucomicrobiota, was relatively high across the three groups. Specifically, the relative abundance of Firmicutes was 64.91% in group N, 63.87% in group A1, and 68.84% in group B1. Bacteroidota accounted for 24.95%, 21.33%, and 20.94% of the three groups, respectively. Proteobacteria exhibited relative abundances of 4.59%, 8.86%, and 5.19% in N, A1, and B1, respectively (Fig. 3B). At the genus level, Bacteroides had relative abundances of 10.90%, 8.87%, and 7.54% in N, A1, and B1, respectively. Faecalibacterium accounted for 8.42%, 13.60%, and 16.15%, while Phascolarctobacterium was present at 7.73%, 2.05%, and 2.47% in the three groups, respectively (Fig. 3C). We further screened for microbial taxa with significant differences between groups at both the phylum and genus levels. Significant differences between the groups were observed at the phylum level for Sumerlaeota, Spirochaetota, and Bdellovibrionota (Fig. 3D). At the genus level, bacterium_adhufec311, Erysipelatoclostridiaceae, Bacillus, durum, and Alistipes_sp._CHKCI003 exhibited significant differences in abundance between the groups (Fig. 3E). Notably, we identified the microorganisms with the greatest difference in abundance at the phylum, class, and genus levels. The relative abundances of Gammaproteobacteria, Roseburia, and Proteobacteria were significantly higher in patients with central lymph node metastasis compared to the other two groups (Fig. 3F).

Gut microbiota analysis of patients with central and lateral cervical lymph node metastasis. The number of common and unique OTUs in N, A1, and B1 (A). The relative ratio of top abundant taxa in N, A1, and B1 at the phylum level (B). The relative ratio of top abundant taxa in N, A1, and B1 at the genus level (C). Significantly differential taxa in N, A1, and B1 at the phylum level (D). Significantly differential taxa in N, A1, and B1 at the genus level (E). Linear discriminant analysis (LDA) effect size (LEfSe) analysis of fecal microbial genus in N, A1, and B1 (F). N: Location without lymph node metastasis, A1: Central lymph node metastasis, B1: Lateral lymph node metastasis.
Metabolite changes in patients with thyroid cancer lymph node metastasis
OPLS-DA analysis indicated a clear distinction among the groups of the samples (Supp Fig. 2). Metabolomics analysis between Y2 and W2 identified 3255 metabolites (STable 3), with 318, 267, and 261 categorized as Carboxylic acids and derivatives, Prenol lipids, and Fatty Acyls, respectively (Fig. 4A). Compared with patients with lymph node metastasis, 166 metabolites were up-regulated and three metabolites were down-regulated in patients without lymph node metastasis (Fig. 4B). Further functional annotation of the differentially expressed metabolites revealed that they were mainly enriched in pathways such as Tyrosine metabolism, Biosynthesis of vancomycin group antibiotics, Lysine biosynthesis, and Starch and sucrose metabolism (Fig. 4C). The metabolites with the largest fold changes between the two groups included Shikimate, dTMP, and Ganoderic acid F, among others (Fig. 4D). Subsequent ROC curve analysis identified five metabolites that could distinguish between the two groups of patients, namely Osmanthuside A, Niacin (Nicotinic acid), 4 − Megastigmene − 6a,9R − diol 9 − apiosyl−(1−>6) − glucoside, Toluene − cis − dihydrodiol, and alpha − Tocopherol (Fig. 4E).

Metabolite changes in patients with thyroid cancer lymph node metastasis. Metabolite composition classification in Y2 and W2 (A). Volcano map of differentially expressed metabolites in Y2 and W2 (B). KEGG enrichment analysis and related DEMs (C). Differentially expressed metabolite bar graph in Y2 and W2. Horizontal coordinates are log2FC of differential metabolites and vertical coordinates are differential metabolites (D). ROC curve analysis of the candidate biomarkers shows the abundance of the five metabolites and high AUCs for the five metabolites (E). Y2: lymph node metastasis, W2: non- lymph node metastasis.
Metabolite composition analysis of patients with central and lateral cervical lymph node metastasis
Through differential metabolite analysis of different comparison groups, a total of 534 differentially expressed metabolites (DEMs) were identified, with 271, 189, and 176 DEMs detected in the A1vs B1, N vs. A1, and N vs. B1 comparison groups, respectively (Fig. 5A). Compared with patients with central lymph node metastasis, 62 DEMs were up-regulated and 209 DEMs were down-regulated in patients with lateral cervical lymph node metastasis (Fig. 5B). Further functional annotation of the differentially expressed metabolites revealed that they were mainly enriched in pathways such as Bile secretion, Secondary bile acid biosynthesis, and Pentose phosphate pathway (Fig. 5C). The metabolites with the most considerable fold changes between the two groups included Oxyphencyclimine, N(6) − Methyllysine, O − methoxycatechol − O−sulphate, and leukotriene − f4 (Fig. 5D). Subsequent ROC curve analysis identified three metabolites that could distinguish between patients with central and lateral cervical lymph node metastasis, namely − Gingero (AUC: 0.75), Hexazinone (AUC: 0.74), and 5beta − 1,3,7(11) − Eudesmatrien − 8−one (AUC: 0.75) (Fig. 5E).

Metabolite composition analysis of patients with central and lateral cervical lymph node metastasis. Metabolite composition classification in N, A1, and B1 (A). Volcano map of differentially expressed metabolites in A1 and B1 (B). KEGG enrichment analysis and related DEMs in A1 and B1 (C). Differentially expressed metabolite bar graph in A1 and B1. Horizontal coordinates are log2FC of differential metabolites and vertical coordinates are differential metabolites (D). ROC curve analysis of the candidate biomarkers shows the abundance of the three metabolites and the high AUCs for the three metabolites (E). N: Location without lymph node metastasis, A1: Central lymph node metastasis, B1: Lateral lymph node metastasis.
Impact of the number of thyroid cancer lymph node metastases on gut metabolite composition
Differential metabolite analysis identified a total of 390 differentially expressed metabolites, with 254, 110, and 68 DEMs detected in the F1vs T1, F1 vs T2, and T1 vs T2 comparison groups, respectively (Fig. 6A). Compared with the F1 group, 40 DEMs were up-regulated and 214 DEMs were down-regulated in the T1 group (Fig. 6B). Further functional annotation of the differentially expressed metabolites revealed that they were mainly enriched in pathways such as Porphyrin metabolism, Retinol metabolism, and Arachidonic acid metabolism (Fig. 6C). The metabolites with the most significant fold changes between the two groups included cis − Hydroxy Perhexiline, Pratosartan, and − hydroxy − 3−(3 − hydroxyphenyl) propanoic acid − O−sulphate (Fig. 6D). Subsequent ROC curve analysis identified five metabolites that could distinguish between F1 and T1 groups, namely Thapsigargin (AUC = 0.80), trans-4-Aminocyclohexanecarboxylic acid (AUC = 0.77), N- (2R)-2-Hydroxy-2-[(8S,11S)-8-isopropyl-6,9-dioxo-2-oxa-7,10-diazabicyclo[11.2.2]heptadeca-1(15),13,16-trien-11-YL]ethyl-N-isopentylbenzenesulfonamide (AUC = 0.77), Met Val Trp Arg (AUC = 0.76), and 5-Oxoavermectin “1a’’ aglycone (AUC = 0.76) (Fig. 6E). Compared with the F1 group, 4 DEMs were up-regulated and 106 DEMs were down-regulated in the T2 group (Fig. 7A). Further functional annotation of the metabolites with differential expression revealed that they were mainly enriched in pathways such as the regulation of TRP channels by inflammatory mediators, the contraction of vascular smooth muscle, and the biosynthesis of unsaturated fatty acids (Fig. 7B). The 20 metabolites with the most considerable fold changes between the two groups included Conhydrinone, 3 − Methoxytyramine, and Cathasterone (Fig. 7C). Subsequent ROC curve analysis identified five metabolites that could distinguish between groups F1 and T2, namely Ganoderiol I (AUC = 0.77) and 3-Methoxytyramine (AUC = 0.75) (Fig. 7D).

Impact of the number of thyroid cancer lymph node metastases on gut metabolite composition. Metabolite composition classification in F1 and T1 (A). Volcano map of differentially expressed metabolites in F1 and T1 (B). KEGG enrichment analysis and related DEMs in F1 and T1 (C). Differentially expressed metabolite bar graph in F1 and T1. Horizontal coordinates are log2FC of differential metabolites and vertical coordinates are differential metabolites (D). ROC curve analysis of the candidate biomarkers shows the five metabolites’ abundance and the high AUCs for the five metabolites (E). F1: Number of lymph node metastases is less than 5, T2: Number of lymph node metastases exceeds 10.

Metabolite composition analysis of patients with central and lateral Cervical lymph node metastasis. Metabolite composition classification in F1 and T2 (A). Volcano map of differentially expressed metabolites in F1 and T2 (B). KEGG enrichment analysis and related DEMs in F1 and T2 (C). Differentially expressed metabolite bar graph in F1 and T2. Horizontal coordinates are log2FC of differential metabolites, and vertical coordinates are differential metabolites (D). ROC curve analysis of the candidate biomarkers shows the abundance of three metabolites and the high AUCs for the three metabolites (E). F1: Number of lymph node metastases is less than 5, T2: Number of lymph node metastases exceeds 10.
Correlations of gut microbiota and metabolites in thyroid cancer lymph node metastasis
Additionally, we performed a combined study of metabolomics and microbial diversity, examining the relationship between differentially expressed metabolites and microorganisms. In the A1 and B1 groups, correlations were found between 4 differentially expressed metabolites and four differentially expressed microorganisms. Specifically, a strong positive correlation was observed between Actinobacillus and Butyl (S)-3-hydroxybutyrate glucoside. 13-cis-Retinol showed significant positive correlations with Catenisphaera and Papillibacter. Synergistes had a strong positive correlation with Enterodiol. Tepidimonas exhibited a relatively high correlation with 3α,7α,12α-Trihydroxy-5β-cholestan-26-al (Fig. 8).

Integrated correlation of gut microbial genus and fecal metabolites. Heatmap of Spearman’s rank correlation coefficients between differentially abundant genus and metabolites in A1 vs. B1 and F1 vs. T2. The red or blue color in each square represents positive or negative relationships, respectively. Statistically significant correlations were marked with asterisks (*p < 0.05, **p < 0.01, and ***p < 0.001). A1: Central lymph node metastasis, B1: Lateral lymph node metastasis, F1: Number of lymph node metastases is less than 5, T2: Number of lymph node metastases exceeds 10.
In the F1 and T1 groups, correlation analysis between differentially expressed metabolites and microorganisms revealed that Lactonifactor had positive correlations with 3-Oxo-alpha-ionol and Caffeinol. Atopobiaceae showed a strong positive correlation with Caffeinol. A significant positive correlation was observed between Thermoanaerobacterales_bacterium and 3-Oxo-alpha-ionol (Fig. 8).
Discussion
The focus of our research was to investigate the relationship between gut microbiota, metabolites, and lymph node metastasis in thyroid cancer. Through a comprehensive analysis of fecal microbiota and metabolites, we aimed to uncover potential biomarkers and mechanisms underlying the metastatic behavior of thyroid cancer. Our study reveals significant alterations in the gut microbiota composition in the three comparison systems. Identified distinct microbial profiles, functional pathways, and differential taxa imply that gut microbiota may play a role in influencing thyroid cancer progression and its metastatic behavior Microbial Diversity and Metastasis-Associated Shifts The marked disparity in unique OTUs between LNM+ (12,302) and LNM− (456) groups underscores a profound dysbiosis linked to metastasis. Meanwhile, the unique OTU distribution (456, 7201, and 4322 in N, A1, and B1) suggests that lymph node metastasis status is associated with specific microbial community structures. The shared core microbiota (521 OTUs) suggests a conserved gut ecological niche in thyroid cancer, while group-specific OTUs (e.g., 8,447 in F1 vs. ≤1,779 in metastatic groups) indicate that the differences in the gut microbiota may be related to the number of nodules. The dominance of Firmicutes and Bacteroidota in all groups aligns with prior reports linking Firmicutes/Bacteroidota (F/B) ratio shifts to thyroid cancer aggressiveness.
At the phylum level, the elevated Proteobacteria (7.65% vs. 4.59%) in LNM + patients is notable, as this phylum includes opportunistic pathogens implicated in chronic inflammation and cancer progression15,16. Conversely, the lower Bacteroidota (21.20% vs. 24.94%) and Fusobacteriota (0.57% vs. 1.94%) in LNM + may reflect a loss of commensal taxa with anti-inflammatory properties, consistent with studies linking Bacteroides to immune regulation in thyroid cancer5,17. Meanwhile, the elevated Proteobacteria in A1 (8.86%), a phylum implicated in inflammation and cancer progression, may reflect its potential role in central lymph node metastasis18,19.
At the genus level, the enrichment of Faecalibacterium (14.44% vs. 8.42%) in LNM + patients contrasts with its typical role as a butyrate producer with anti-tumor effects20,21. Faecalibacterium is a key SCFA-producing bacterium, particularly butyrate, which has anti-inflammatory and immunomodulatory effects22,23. previous researches indicated SCFAs may indirectly influence tumor metastasis by modulating the immune microenvironment. For example, SCFAs can recruit myeloid-derived suppressor cells (MDSCs) and promote the formation of a pro-inflammatory pre-metastatic niche (PMN), thereby accelerating gastric cancer (GC) metastasis24. Additionally, SCFAs may affect tumor cell invasiveness through pathways such as activating G protein-coupled receptors (GPCRs) or inhibiting histone deacetylases (HDACs)25. In melanoma and pancreatic ductal adenocarcinoma (PDAC) models, microbiota-derived formate (the simplest SCFA) was found to exacerbate pulmonary metastasis26.
Similarly, the genus Faecalibacterium, a key SCFA-producing bacterium, was enriched in metastatic groups (A1: 13.60%; B1: 16.15%), contrasting with its lower abundance in N (8.42%). It might also be the reason why the genus Faecalibacterium was enriched in metastatic groups16,21,27,28. The higher Escherichia_Shigella (4.28% vs. 3.07%) in LNM + supports its putative pro-metastatic role via endotoxin production16,18. Conversely, Bacteroides depletion in metastatic groups (A1: 8.87%; B1: 7.54% vs. N: 10.90%) suggests a possible protective role, consistent with studies reporting gut dysbiosis in thyroid cancer7,29. In A1 patients, the marked enrichment of Gammaproteobacteria and Roseburia emphasizes taxa-specific links to central lymph node metastasis, which may be mediated by immune modulation or metabolic pathways13,19. Notably, the incremental rise in Faecalibacterium (12.36%→15.55%) and Bacteroides (7.87%→10.29%) with LNM burden contrasts with their typical anti-inflammatory roles, suggesting context-dependent functions in cancer progression. The depletion of Lachnospiraceae (6.49%→4.22%), a butyrate producer linked to immune modulation in metastatic groups, supports its putative protective role against tumor dissemination30,31. Strikingly, Lachnospiraceae_bacterium_GAM79 showed metastasis-stage-dependent enrichment (highest in T2), mirroring findings in other cancers where specific strains promote epithelial-mesenchymal transition32.
Our fecal metabolomics analysis revealed notable metabolic alterations in thyroid cancer patients with (Y2) and without (W2) lymph node metastasis, central (A1) and lateral cervical (B1) metastasis, and in those categorized by the extent of LNM, possibly indicating tumor-driven metabolic reprogramming. Our findings align with emerging evidence that thyroid cancer metastasis involves extensive metabolic rewiring, particularly in pathways like tyrosine metabolism and lysine biosynthesis33,34. In thyroid cancer patients with (Y2) and without (W2) lymph node metastasis, the five identified diagnostic metabolites (osmanthuside A, niacin, megastigmene derivative, toluene-cis-dihydrodiol, and alpha-tocopherol) represent novel potential biomarkers. Alpha-tocopherol’s discriminative capacity is intriguing, given its known antioxidant properties and prior associations with thyroid cancer redox balance35,36. Niacin’s (vitamin B3) identification echoes recent findings about NAD + metabolism’s role in tumor progression37. Niacin (vitamin B3) and its metabolites play important roles in NAD + metabolism, which is crucial for cancer cell survival and metastasis. The dysregulation of nicotinate/nicotinamide pathways has been associated with metastasis in esophageal squamous cell carcinoma (ESCC), where increased levels of 1-methylnicotinamide (MNA) were observed in patients with lymph node metastasis38. Nicotinamide (NAM), the amide form of niacin, showed synergistic effects when combined with gemcitabine (GEM) in mouse models of pancreatic cancer. This combination improved efficacy, survival, and modulated stromal architecture and tumor-infiltrating immune cells39.
Meanwhile, Niacin (vitamin B3) is converted to NAD + through the Preiss-Handler pathway or salvage synthesis pathway40. Studies have shown that niacin supplementation increases tissue NAD + levels, improves mitochondrial function and metabolism41, and enhances DNA repair capacity by activating Sirt1 (an NAD+-dependent deacetylase)42. In the LN metastasis-positive group, decreased niacin levels may lead to insufficient NAD + synthesis, thereby impairing DNA repair function38.
The plant-derived osmanthuside A and megastigmene compounds may reflect dietary phytochemical interactions with cancer metabolism43. Among the most significantly altered metabolites in the composition analysis of patients with central and lateral cervical lymph node metastasis, Oxyphencyclimine, N(6) − Methyllysine, and leukotriene-F4 may serve as potential biomarkers. Leukotriene-F4, an eicosanoid involved in inflammation, aligns with reported perturbations in arachidonic acid metabolism in TC44,45. The clinical potential of Gingero, Hexazinone, and 5beta − 1,3,7(11) − Eudesmatrien − 8−one for differentiating metastatic subtypes is highlighted by their discriminative power (AUCs: 0.74–0.75). These metabolites, particularly those linked to bile acid and lipid metabolism, may reflect tumor-host interactions or microbial contributions, as gut microbiota-derived secondary bile acids influence cancer progression46,47. In F1vsT2, the marked down-regulation of metabolites like 3-Methoxytyramine (AUC = 0.75) and Ganoderiol I (AUC = 0.77) suggests their potential as diagnostic markers, consistent with research identifying differential metabolites in TC biofluids48. The metabolites validated by ROC, such as Thapsigargin with an AUC of 0.80, are consistent with research highlighting the use of metabolomics for TC classification49,50. However, their mechanistic roles warrant further investigation. Especially, the down-regulation of 214 DEMs in T1 (vs. F1) and 106 DEMs in T2 (vs. F1) suggests a progressive suppression of metabolic pathways as LNM burden increases, aligning with prior findings that metastatic TC exhibits distinct metabolite profiles linked to tumor aggressiveness51,52. The progressive metabolite suppression from F1 to T2 may reflect gut microbiota dysbiosis, as gut microbes modulate host metabolism and TC progression53. Additionally, the observed DEM patterns resemble gut metabolite shifts in other malignancies, suggesting conserved metabolic adaptations during metastasis54. Our findings provide a foundation for developing non-invasive biomarkers and therapeutic strategies targeting the gut-thyroid axis. The identified metabolites could potentially be utilized to anticipate lymph node metastasis preoperatively, refine risk stratification tools, and guide personalized treatment strategies.
Furthermore, our study reveals intricate correlations between gut microbiota composition and metabolite profiles in thyroid cancer patients with varying lymph node metastasis (LNM) patterns. The observed associations, such as the positive correlation between Actinobacillus and butyl (S)-3-hydroxybutyrate glucoside in the A1/B1 groups, align with emerging evidence that gut microbial dysbiosis influences thyroid cancer progression through metabolite-mediated pathways5,10. The strong linkage between 13-cis-Retinol and Catenisphaera/Papillibacter further supports the role of microbial-retinoid interactions in tumor microenvironment regulation, as retinoids have been implicated in thyroid cancer differentiation and metastasis suppression55. In the F1/T1 groups, the positive correlations of Lactonifactor with 3-Oxo-alpha-ionol and Caffeinol suggest that microbial metabolism of polyphenols may influence LNM. Caffeinol, a derivative of dietary polyphenols, exhibits anti-tumor properties, and its association with Atopobiaceae underscores the potential of gut microbiota to convert dietary components into bioactive metabolites affecting cancer dissemination8,16. The significant correlation between Thermoanaerobacterales_bacterium and 3-Oxo-alpha-ionol highlights the involvement of thermophilic bacteria in generating metabolites that may be associated with metastasis, depending on their abundance and host context13,18. Notably, our stratification by LNM number (≤ 5, 5–10, > 10) revealed progressive shifts in microbial-metabolite networks, mirroring clinical observations that metastatic burden correlates with altered gut ecology56,57. These findings resonate with studies linking gut microbiota diversity to thyroid cancer aggressiveness58, where specific taxa (e.g., Synergistes, Tepidimonas) may serve as biomarkers for metastatic risk53,54. The association of Enterodiol (a phytoestrogen metabolite) with Synergistes suggests estrogen-microbiota crosstalk, which could explain the higher thyroid cancer incidence in females59. The microbial-metabolite signatures identified here could refine pre-operative LNM prediction. For instance, Actinobacillus-butyrate interactions might predict central LNM, while Lactonifactor-polyphenol correlations could indicate lateral LNM11,60. These insights align with proposals to integrate microbiota profiling into diagnostic algorithms for thyroid cancer61. Despite the novel insights provided by this study through multi-omics analysis into the potential links between thyroid cancer lymph node metastasis (LNM) and gut microbiota and metabolites, several limitations exist in the study design. First, although we excluded participants with recent antibiotic or probiotic use, smoking, and other factors that could influence gut microbiota, we did not fully control for all potential confounding variables. In particular, dietary intake, dietary habits, exercise habits, lifestyle, and medications, which are known to significantly impact gut microbiota and metabolite profiles, were not specifically recorded or controlled in this study. Furthermore, the relatively small sample size and the fact that the study population was primarily from the Sichuan Basin region may limit the generalizability of our results. Future validation in larger and more diverse cohorts is necessary to ensure the broad applicability of these findings. Meanwhile, longitudinal cohort studies should be conducted by collecting fecal samples from thyroid cancer patients before and after treatment, and researchers could observe trends in gut microbiota and metabolite changes to better understand their roles in disease progression. Future work will involve prospectively testing these biomarkers in a larger, blinded cohort to evaluate their accuracy and reliability for pre-operative LNM prediction. In addition, the sample collection time point for this study was 1 month after surgery and before ¹³¹I treatment; at this time, the primary lesion had been removed, and the microbiota characteristics may mainly reflect the systemic or local inflammation, nutritional status, and drug (such as thyroid hormone replacement) effects after or before the surgery, rather than the direct interaction between the tumor and the microbiota. Therefore, the observed correlation should be cautiously interpreted as ‘microbiota characteristics related to the disease state or the surgical-treatment trajectory’, rather than ‘microbiota characteristics coexisting with the tumor’.
Conclusion
Our comprehensive multi-omics study has shed light on the intricate relationships between gut microbiota, metabolites, and lymph node metastasis (LNM) in thyroid cancer. We have identified significant alterations in gut microbiota composition and metabolite profiles associated with LNM, central and lateral neck metastasis, and metastatic burden. The distinctive microbial and metabolic signatures discovered have the potential to serve as non-invasive biomarkers for pre-operative LNM prediction and could aid in refining risk-stratification tools. The gut-thyroid axis emerged as a crucial modulator of thyroid cancer progression and metastasis. Specific taxa and metabolites, such as Faecalibacterium, Escherichia_Shigella, Proteobacteria, osmanthuside A, niacin, and alpha-tocopherol, were found to be differentially abundant in LNM patients. Notably, certain metabolites like leukotriene-F4 and 3-Methoxytyramine showed significant differences between central and lateral neck metastasis groups, and the number of lymph node metastases also impacted gut metabolite composition. Our findings provide a foundation for developing microbiome or metabolite-based therapeutic strategies targeting the gut-thyroid axis. Overall, this study advances our understanding of the microbiota-metabolite axis in thyroid cancer metastasis and offers novel risk stratification and therapeutic intervention targets.
Data availability
The sequencing data were deposited to the Sequence Read Archive National Center for Biotechnology Information (NCBI) with accession number PRJNA1049645.
References
Xu, B. et al. A UPLC Q-Exactive orbitrap mass Spectrometry-Based metabolomic study of serum and tumor tissue in patients with papillary thyroid cancer. Toxics 11 (1). https://doi.org/10.3390/toxics11010044 (2022).
Du, J. et al. Risk factors for central lymph node metastasis in patients with papillary thyroid carcinoma: a retrospective study. Front. Endocrinol. (Lausanne). 14, 1288527. https://doi.org/10.3389/fendo.2023.1288527 (2023).
Xu, L. et al. Development of a colloidal gold immunochromatographic strip for rapid detection of Cyfra 21 – 1 in lymph node metastasis of thyroid cancer. Front. Bioeng. Biotechnol. 10, 871285. https://doi.org/10.3389/fbioe.2022.871285 (2022).
Zhou, J., Li, D. X., Gao, H. & Su, X. L. Relationship between subgroups of central and lateral lymph node metastasis in clinically node-negative papillary thyroid carcinoma. World J. Clin. Cases. 10 (12), 3709–3719. https://doi.org/10.12998/wjcc.v10.i12.3709 (2022).
Lu, G. et al. Alterations of gut Microbiome and metabolite profiles associated with anabatic lipid dysmetabolism in thyroid cancer. Front. Endocrinol. (Lausanne). 13, 893164. https://doi.org/10.3389/fendo.2022.893164 (2022).
Xie, Z., Zhou, J., Zhang, X. & Li, Z. Clinical potential of microbiota in thyroid cancer therapy. Biochim. Biophys. Acta Mol. Basis Dis. 1870 (2), 166971. https://doi.org/10.1016/j.bbadis.2023.166971 (2024).
Ludgate, M. E., Masetti, G. & Soares, P. The relationship between the gut microbiota and thyroid disorders. Nat. Rev. Endocrinol. 20 (9), 511–525. https://doi.org/10.1038/s41574-024-01003-w (2024).
Gorini, F. & Tonacci, A. Tumor microbial communities and thyroid cancer Development-The protective role of antioxidant nutrients: application strategies and future directions. Antioxid. (Basel). 12 (10). https://doi.org/10.3390/antiox12101898 (2023).
Zhan, S. et al. Asporin interacts with HER2 to promote thyroid cancer metastasis via the MAPK/EMT signaling pathway. Front. Oncol. 12, 762180. https://doi.org/10.3389/fonc.2022.762180 (2022).
Zhang, H., Li, Y. & Li, L. Blood metabolites reflect the effect of gut microbiota on differentiated thyroid cancer: a Mendelian randomization analysis. BMC Cancer. 25 (1), 368. https://doi.org/10.1186/s12885-025-13598-y (2025).
Kang, J. G., Choi, J. E. & Kang, S. H. Risk factors for level V metastasis in patients with N1b papillary thyroid cancer. World J. Surg. Oncol. 20 (1), 327. https://doi.org/10.1186/s12957-022-02782-0 (2022).
Xu, Y. & Zhang, C. Prediction of lateral neck metastasis in patients with papillary thyroid cancer with suspicious lateral lymph ultrasonic imaging based on central lymph node metastasis features. Oncol. Lett. 28 (4), 472. https://doi.org/10.3892/ol.2024.14605 (2024).
Xie, M. et al. The influence of Lactobacillus Johnsonii on tumor growth and lymph node metastasis in papillary thyroid carcinoma. Commun. Biol. 8 (1), 419. https://doi.org/10.1038/s42003-025-07856-9 (2025).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Wu, F., Ji, P., Yang, H., Zhu, X. & Wu, X. Interpretation of the effects of rumen acidosis on the gut microbiota and serum metabolites in calves based on 16S rDNA sequencing and non-target metabolomics. Front. Cell. Infect. Microbiol. 14, 1427763. https://doi.org/10.3389/fcimb.2024.1427763 (2024).
Fang, L. & Ning, J. Recent advances in gut microbiota and thyroid disease: pathogenesis and therapeutics in autoimmune, neoplastic, and nodular conditions. Front. Cell. Infect. Microbiol. 14, 1465928. https://doi.org/10.3389/fcimb.2024.1465928 (2024).
Jiao, J. et al. Saliva Microbiome changes in thyroid cancer and thyroid nodules patients. Front. Cell. Infect. Microbiol. 12, 989188. https://doi.org/10.3389/fcimb.2022.989188 (2022).
Ataollahi, H., Hedayati, M., Zia-Jahromi, N., Daneshpour, M. & Siadat, S. D. Investigating the role of the intratumoral Microbiome in thyroid cancer development and progression. Crit. Rev. Oncol. Hematol. 204, 104545. https://doi.org/10.1016/j.critrevonc.2024.104545 (2024).
Yuan, L. et al. Tumor Microbiome diversity influences papillary thyroid cancer invasion. Commun. Biol. 5 (1), 864. https://doi.org/10.1038/s42003-022-03814-x (2022).
Martín, R. et al. Langella, faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol. Rev. 47 (4). https://doi.org/10.1093/femsre/fuad039 (2023).
He, Z. et al. Influence of postoperative insulin resistance on Short-Term outcomes of radical gastrectomy for gastric cancer: A Microbiome and Metabolome-Based prospective cohort study. Ann. Surg. Oncol. 31 (13), 8638–8650. https://doi.org/10.1245/s10434-024-16125-8 (2024).
Simadibrata, D. M., Lesmana, E., Gunawan, J., Quigley, E. M. & Simadibrata, M. A systematic review of gut microbiota profile in COVID-19 patients and among those who have recovered from COVID-19. J. Dig. Dis. 24 (4), 244–261. https://doi.org/10.1111/1751-2980.13195 (2023).
Karpiński, P. et al. Gut microbiota alterations in schizophrenia might be related to stress exposure: findings from the machine learning analysis. Psychoneuroendocrinology 155, 106335. https://doi.org/10.1016/j.psyneuen.2023.106335 (2023).
Zhu, X. et al. Gut microbiota-dependent modulation of pre-metastatic niches by Jianpi Yangzheng Decoction in the prevention of lung metastasis of gastric cancer. Phytomedicine 128, 155413. https://doi.org/10.1016/j.phymed.2024.155413 (2024).
Li, S. et al. Short-chain fatty acids and cancer. Trends Cancer. 11 (2), 154–168. https://doi.org/10.1016/j.trecan.2024.11.003 (2025).
Prajnamitra, R. P. et al. Gut microbiota-derived formate exacerbates pulmonary metastasis in cancer. Theranostics 15 (15), 7693–7708. https://doi.org/10.7150/thno.108873 (2025).
Gomes, S., Rodrigues, A. C., Pazienza, V. & Preto, A. Modulation of the tumor microenvironment by Microbiota-Derived Short-Chain fatty acids: impact in colorectal cancer therapy. Int. J. Mol. Sci. 24 (6). https://doi.org/10.3390/ijms24065069 (2023).
Zhang, Y., Tang, N., Zhou, H. & Zhu, Y. The role of microbial metabolites in endocrine tumorigenesis: from the mechanistic insights to potential therapeutic biomarkers. Biomed. Pharmacother. 172, 116218. https://doi.org/10.1016/j.biopha.2024.116218 (2024).
Virili, C. et al. The relationship between thyroid and human-associated microbiota: A systematic review of reviews. Rev. Endocr. Metab. Disord. 25 (1), 215–237. https://doi.org/10.1007/s11154-023-09839-9 (2024).
Li, W., Huang, Y., Tong, S., Wan, C. & Wang, Z. The characteristics of the gut microbiota in patients with pulmonary tuberculosis: A systematic review. Diagn. Microbiol. Infect. Dis. 109 (3), 116291. https://doi.org/10.1016/j.diagmicrobio.2024.116291 (2024).
Li, J. et al. Effect and correlation of Rosa Roxburghii Tratt fruit vinegar on Obesity, dyslipidemia and intestinal microbiota disorder in High-Fat diet mice. Foods 11 (24). https://doi.org/10.3390/foods11244108 (2022).
Ma, X. et al. Alternations in the human skin, gut and vaginal microbiomes in perimenopausal or postmenopausal vulvar lichen sclerosus. Sci. Rep. 14 (1), 8429. https://doi.org/10.1038/s41598-024-58983-y (2024).
Narasimhamurthy, R. K. et al. Dattaram Mumbrekar, Low-Dose radiation induces alterations in fatty acid and tyrosine metabolism in the mouse hippocampus: insights from integrated multiomics. ACS Chem. Neurosci. 15 (18), 3311–3320. https://doi.org/10.1021/acschemneuro.4c00231 (2024).
Kuang, A., Kouznetsova, V. L., Kesari, S. & Tsigelny, I. F. Diagnostics of thyroid cancer using machine learning and metabolomics. Metabolites 14 (1). https://doi.org/10.3390/metabo14010011 (2023).
Blatt-Janmaat, K. et al. Impact of in vitro phytohormone treatments on the metabolome of the leafy liverwort radula complanata (L.) Dumort. Metabolomics 19 (3), 17. https://doi.org/10.1007/s11306-023-01979-y (2023).
Wan, Y., Li, G., Cui, G., Duan, S. & Chang, S. Reprogramming of thyroid cancer metabolism: from mechanism to therapeutic strategy. Mol. Cancer. 24 (1), 74. https://doi.org/10.1186/s12943-025-02263-4 (2025).
Imran Khan, M. Exploration of metabolic responses towards hypoxia mimetic DMOG in cancer cells by using untargeted metabolomics. Saudi J. Biol. Sci. 29 (10), 103426. https://doi.org/10.1016/j.sjbs.2022.103426 (2022).
Huang, Q. et al. Multi-omics analysis reveals NNMT as a master metabolic regulator of metastasis in esophageal squamous cell carcinoma. NPJ Precis Oncol. 8 (1), 24. https://doi.org/10.1038/s41698-024-00509-w (2024).
Selvanesan, B. C. et al. Nicotinamide combined with gemcitabine is an Immunomodulatory therapy that restrains pancreatic cancer in mice. J. Immunother Cancer. 8 (2). https://doi.org/10.1136/jitc-2020-001250 (2020).
Curran, C. S. & Kopp, J. B. The complexity of nicotinamide adenine dinucleotide (NAD), hypoxic, and Aryl hydrocarbon receptor cell signaling in chronic kidney disease. J. Transl Med. 21 (1), 706. https://doi.org/10.1186/s12967-023-04584-8 (2023).
Beltrà, M. et al. NAD(+) repletion with niacin counteracts cancer cachexia. Nat. Commun. 14 (1), 1849. https://doi.org/10.1038/s41467-023-37595-6 (2023).
Horimatsu, T. et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: role of NAD+/nicotinamide. Cardiovasc. Res. 116 (14), 2226–2238. https://doi.org/10.1093/cvr/cvz303 (2020).
Yang, H. et al. Integrative metabolomic and transcriptomic analysis reveals the mechanism of specific color formation in Phoebe Zhennan Heartwood. Int. J. Mol. Sci. 23 (21). https://doi.org/10.3390/ijms232113569 (2022).
Sun, Z. et al. Integrated proteomic and metabolomic analysis of plasma reveals regulatory pathways and key elements in thyroid cancer. Mol. Omics. 19 (10), 800–809. https://doi.org/10.1039/d3mo00142c (2023).
Kim, Y. H. et al. Integrative Multi-omics analysis reveals different metabolic phenotypes based on molecular characteristics in thyroid cancer. Clin. Cancer Res. 30 (4), 883–894. https://doi.org/10.1158/1078-0432.Ccr-23-2025 (2024).
Li, M. et al. Therapeutic mechanisms of the medicine and food homology formula Xiao-Ke-Yin on glucolipid metabolic dysfunction revealed by transcriptomics, metabolomics and microbiomics in mice. Chin. Med. 18 (1), 57. https://doi.org/10.1186/s13020-023-00752-6 (2023).
Wise, J. L. & Cummings, B. P. The 7-α-dehydroxylation pathway: an integral component of gut bacterial bile acid metabolism and potential therapeutic target. Front. Microbiol. 13, 1093420. https://doi.org/10.3389/fmicb.2022.1093420 (2022).
Razavi, S. A., Khorsand, B., Salehipour, P. & Hedayati, M. Metabolite signature of human malignant thyroid tissue: A systematic review and meta-analysis. Cancer Med. 13 (8), e7184. https://doi.org/10.1002/cam4.7184 (2024).
Kumari, S., Makarewicz, A. & Klubo-Gwiezdzinska, J. Emerging potential of metabolomics in thyroid Cancer-A comprehensive review. Cancers (Basel). 17 (6). https://doi.org/10.3390/cancers17061017 (2025).
Shi, L. et al. Distinguishing benign and malignant thyroid nodules using plasma trimethylamine N-oxide, carnitine, choline and betaine. J. Cancer Res. Clin. Oncol. 150 (3), 142. https://doi.org/10.1007/s00432-024-05666-w (2024).
Bolf, E. L. et al. Dasatinib and Trametinib promote Anti-Tumor metabolic activity. Cells 12 (10). https://doi.org/10.3390/cells12101374 (2023).
Abooshahab, R. et al. Thyroid cancer cell metabolism: A glance into cell culture system-based metabolomics approaches. Exp. Cell. Res. 435 (2), 113936. https://doi.org/10.1016/j.yexcr.2024.113936 (2024).
Huang, W. et al. Modulation of intestinal flora: a novel immunotherapeutic approach for enhancing thyroid cancer treatment. Probiotics Antimicrob. Proteins. https://doi.org/10.1007/s12602-025-10471-z (2025).
Liu, Q., Sun, W. & Zhang, H. Interaction of gut microbiota with endocrine homeostasis and thyroid cancer. Cancers (Basel). 14 (11). https://doi.org/10.3390/cancers14112656 (2022).
Jiang, W., Lu, G., Gao, D., Lv, Z. & Li, D. The relationships between the gut microbiota and its metabolites with thyroid diseases. Front. Endocrinol. (Lausanne). 13, 943408. https://doi.org/10.3389/fendo.2022.943408 (2022).
Zhu, F. et al. Mendelian randomization suggests a causal relationship between gut dysbiosis and thyroid cancer. Front. Cell. Infect. Microbiol. 13, 1298443. https://doi.org/10.3389/fcimb.2023.1298443 (2023).
Hu, S. et al. Causal relationship between gut microbiota and differentiated thyroid cancer: a two-sample Mendelian randomization study. Front. Oncol. 14, 1375525. https://doi.org/10.3389/fonc.2024.1375525 (2024).
Zhou, J., Zhang, X., Xie, Z. & Li, Z. Exploring reciprocal causation: bidirectional Mendelian randomization study of gut microbiota composition and thyroid cancer. J. Cancer Res. Clin. Oncol. 150 (2), 75. https://doi.org/10.1007/s00432-023-05535-y (2024).
An, J., Kwon, H. & Kim, Y. J. The role of blood Microbiome in the development of thyroid cancer in breast cancer survivors. Cancers (Basel). 15 (18). https://doi.org/10.3390/cancers15184492 (2023).
Kang, S. K. et al. A retrospective study of factors affecting contralateral central-neck lymph node metastasis in unilateral papillary thyroid carcinoma. Asian J. Surg. 46 (9), 3485–3490. https://doi.org/10.1016/j.asjsur.2022.10.081 (2023).
Zhang, W. et al. Integrated gene profiling of fine-needle aspiration sample improves lymph node metastasis risk stratification for thyroid cancer. Cancer Med. 12 (9), 10385–10392. https://doi.org/10.1002/cam4.5770 (2023).
Funding
This research was supported by Technology Research and Development Innovation project of Chengdu Science and Technology Bureau (2024-YF05-01876-SN); Chengdu Medical Research Project (2024113); China Nuclear Medical Science and Technology Innovation Project (ZHYLZD2025014) and Open subject of Sichuan Provincial Clinical Research Center for Radiation and Therapy (2024ZX07).
Author information
Authors and Affiliations
Contributions
Xue Jiang and Yuhong Shi as the co-correspondence authors, designed the study and provided the sources of all the samples, and wrote the manuscript; Dongkun Xu and Jia Chen as the co-first author performed the experiments and helped performed the data analyses, and analyzed the data; Dongkun Xu and Jia Chen contributed equally. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of The Second Affiliated Hospital of Chengdu Medical College. All human samples were obtained with informed consent in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, D., Chen, J., Shi, Y. et al. Gut microbiota and metabolite profiles in thyroid cancer lymph node metastasis: a multi-omics analysis. Sci Rep 15, 40624 (2025). https://doi.org/10.1038/s41598-025-24397-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-24397-7
