
Abstract
The accumulation of lipids by algae makes them attractive for carbon-neutral fuel production; however, the industrial-scale production of algal lipids has yet to be achieved. Currently, researchers are trying to improve the lipid productivity of algal strains using genome editing for molecular breeding with CRISPR-Cas9, which allows the efficient alteration of genomic information. However, CRISPR-based gene modification via double-strand breaks sometimes induces unintended large deletions that are toxic to host cells. Here, we applied the cytidine base editor combined with an episomal vector backbone containing a centromere and autonomous replication sequence to the microalga Nannochloropsis oceanica. The cytosine base editor introduces cytidine-to-thymidine base substitutions using deaminase without double-strand breaks, and an episomal vector enables plasmid removal after base substitution. We succeeded in inducing cytidine-to-thymidine substitution at the six target sites of five endogenous genes. The base substitution activity ranged from 29.2% to 47.6% on cytidine bases at the 16th to 19th positions from the protospacer adjacent motifs. The removal of base editor plasmids was also detected, which is essential for constructing transgene-free strains. Our results provide insights into the applicability of further technologies in the genetic modification of microalgae.
Similar content being viewed by others

Introduction
Algal biodiesels have been proposed as potential candidates for sustainable new sources of energy. Because the production process involves the assimilation of carbon dioxide via photosynthesis, biodiesels can potentially be used as a feedstock for carbon-neutral fuels1,2,3. However, the high cost of producing biodiesel from algae compared with that of traditional fossil fuels hinders its practical use4,5. To address this issue, researchers are searching for high-performing algal strains with enhanced lipid productivity or a desirable lipid composition for biofuel production2,3,6.
Along with the search approach to identify suitable algal strains, the molecular breeding of algae using genome editing techniques is in progress7,8,9,10,11,12. To date, the most widely used genome editing tool is the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-CRISPR-associated (Cas) 9 (CRISPR-Cas9) system13, along with its variants, because of its high mutagenesis activity in various organisms and the relative ease of constructing CRISPR-Cas9 expression vectors. Recent advancements in genome databases for microalgae strains, including Nannochloropsis14,15, have enabled molecular breeding that targets specific genes in these organisms.
While large-scale outdoor cultivation in open systems is essential for the low-cost production of substances using algae8, the transformation process often involves conventional plasmid vectors, which could leave the transgene sequence in the host genome as a footprint. Owing to strict regulations imposed by many countries on the use of such GMOs, there is a critical need for the development of genetic modification techniques that ultimately result in the absence of foreign genes8,16. Recently, it was reported that a gene knockout system utilizing episomally retained and removable plasmid vectors worked efficiently in some algal species. This system with yeast centromere and autonomous replication sequence (CEN/ARS) elements is maintained episomally in the nuclei of diatoms and Nannochloropsis17,18,19,20,21. Episome is the extra-chromosomal elements in the nucleus. Generally, introduced vectors are usually inserted into endogenous chromosomes. However, episomal vectors are maintained in the nucleus, and are not inserted into the chromosomes. The CEN/ARS is the fusion sequence of budding yeast centromere and autonomous replication sequence, and is essential for maintaining episomal nucleus localization of vectors in budding yeast22. These episomal vectors can be removed from cells under non-selective conditions, and this property has been exploited to develop transiently inducible CRISPR-Cas9 and TALEN systems. Presently, the introduction of indels to microalgae, followed by frameshift mutations with the genome editing tools TALEN or CRISPR-Cas9, is considered a highly reliable experimental technique. However, many genes crucial for cellular survival—such as those involved in lipid biosynthesis—are essential, and their deletion would be lethal. Such lethal gene mutations could result in a complete loss of function, making the resulting mutants difficult to analyze.
Furthermore, indels generated from the repair error of DNA double-strand breaks (DSBs), which sometimes induce toxic mutations such as off-target cutting or unexpected large deletions, may pose a significant risk to cell viability23. To prevent such adverse effects, the development of a DSB-free mutagenesis approach with nuclease activity-restricted Cas9 combined with various effector proteins is required24. One such tool is the CRISPR-Cas9-based base editor (BE)25,26,27. In these systems, a deaminase fused with a Cas9 protein with restricted nuclease activity enables base editing at a target sequence. Adenine base editors (ABE) effectively convert adenine to guanine (A-to-G)25, and cytosine base editors (CBE) also effectively convert cytosine to thymine (C-to-T)26,27. Additionally, transversion or various other types of base editing systems have been reported28. These reactions do not introduce DSBs and induce base substitutions within a narrow editing window of a few base pairs, making them a promising genome editing tool with minimal genome damage and reduced off-target effects. Base substitution-based genome editing is expected to have a lower probability of inducing frameshift mutations in target genes, making it a valuable tool for analyzing mutants of essential genes. Base editing systems are applied in various plants29 and prokaryotic algae30,31,32, but have not been reported in eukaryotic microalgae7,33.
In this study, we combined a removable plasmid vector and a CRISPR base editor to establish a genome editing method with lower risks of transgene integration and DSBs in the microalga Nannochloropsis oceanica. Among microalgae, this species has one of the highest capacities for lipid accumulation34. Furthermore, a foundational experimental platform, which includes whole genomic information and protocols for earlier models of genome editing tools, specifically TALEN and CRISPR-Cas9, is established for this species20,21. Transfection with a removable vector successfully introduced mutagenesis in the non-essential genes nitrate reductase (NoNR)20,21,35,36 and lysophosphatidic acid acyltransferase 1 (LPAT1)37 in N. oceanica. Mutagenesis of genes crucial for lipid metabolism—including delta-9 fatty acid desaturase, d9FAD (which introduces a double carbon bond in the delta9 locus of stearic acid and turns it into oleic acid)38, seipin (related to lipid droplet formation and stabilization)39, and LDSP (lipid droplet surface protein)40—has also been performed. As a result, we successfully introduced mutations into six target sites of five target genes. We also established a system for constructing transgene-free base substitution mutants by removing the vectors used for base editing.
Results
Construction of all-in-one ARS base editing (ArBE) vectors for double-strand break (DSB)-free and transgene-free genome editing
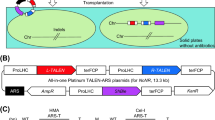
Plasmids containing the CEN/ARS from budding yeast are known to function as episomal vectors in N. oceanica cells20,21,36,41,42. We previously established all-in-one TALEN vectors containing CEN/ARS, which can be used for removable vector systems in N. oceanica20. To establish safer genome editing systems, we aimed to develop a double-strand break-free (DSB-free) genome editing system using removable vectors. This is because mutagenesis via DSB comes with potential risks27, such as inducing unexpected mutations and sometimes causing toxicity in host cells. We selected the base editing system as a DSB-free genome editing system because it is relatively simple to construct, and the removable vectors containing the CRISPR-Cas9 system have already been developed. We adopted the Target-AID system, which exhibits high cytidine (C) to thymidine (T) substitution activity in budding yeast27. This system uses PmCDA1 deaminase from sea lamprey fused with Cas9 (D10A) nickase protein and uracil DNA glycosylase inhibitor (UGI). PmCDA1 deaminase removes the amino group of C and converts it to uridine (U). The resultant U is recognized as T. Furthermore, as reported, the Target-AID system using nCas9(D10A) has the highest base editing activity among other Target-AID systems using nuclease-dead Cas9 or nCas9(H840A)27. Additionally, UGI effectively suppresses unintended C-to-G substitutions27, which induces an endogenous repair system of the abasic lesion because UGI blocks the removal of uracil43 and the downstream repair pathway. The graphical outline of the transgene-free base editing system is shown in Fig. 1A. To construct removable base editing vectors, the Cas9 and sgRNA expression cassettes were cloned into the pMD20-ARS vectors, which act as the backbone for all-in-one ARS-TALEN vectors20. The Cas9 and sgRNA expression cassettes were cloned from pNOC-ARS-CRISPR-v2 (Addgene, #99863). The DNA sequences coding nCas9 (D10A), PmCDA1, and UGI were cloned from pCMV-nCas-PmCDA1-ugi pH1-gRNA(HPRT) (Addgene, #79620), which is the base editing plasmid for human cells. The resultant plasmids, named the all-in-one ARS base editing (ArBE) vectors, are shown in Fig. 1B. In this vector, proteins of nCas9(D10A)-PmCDA1-UGI were driven using ProRibi, bidirectional promoter, and LDSP terminator, which is the same transcription system as for all-in-one CRISPR-Cas9 vectors21. In addition, sgRNAs were transcribed using ProRibi promoter and FCP, terminator. To remove 5′ and 3′ modifications, which obstruct sgRNA function, hammerhead (HH) ribozyme and hepatitis delta virus (HDV) ribozyme, which have self-cleaving activity, were used. The zeocin resistance gene or hygromycin resistance gene was used as an antibiotic marker and expressed using LHC promoter and FCP terminator, which is the same transcription system as in all-in-one TALEN vectors20,44. The construction of all-in-one ArBE vectors for each target is shown in Supplementary Fig. S1. First, the sequences of sgRNA for target sites were inserted into the pMD20-sgRNAm00-KanR plasmids to create pMD20-sgRNA-KanR-sgRNA plasmids. Next, the fragments containing sgRNA and ribozymes were cut out from pMD20-sgRNA-KanR-sgRNA plasmids and inserted into pMD20-ARS base editing vectors. The resultant plasmids were complete all-in-one ArBE vectors for each target site. The information of sequence of nCas9(D10A)-PmCDA1-UGI is shown in Data Availability Statement.

Scheme of transgene-free base editing system and all-in-one ARS base editor (ArBE) vectors. (A) Scheme of transgene-free and double-strand break (DSB)-free genome editing using ArBE vectors. gDNA: Genomic DNA; A: Adenine base; C: Cytidine base; G: Guanine base; T: Thymidine base. (B) Map of ArBE vectors. terLDSP: Terminator of the endogenous gene, LDSP; UGI: uracil DNA glycosylase inhibitor protein; nCas9(D10A): A Cas9 nickase with an aspartic acid-to-alanine substitution at the 10th aspartic acid residue; ProRibi: Bidirectional promoter; HH: Hammerhead ribozyme that has self-cleavage activity; sgRNA: single-guide RNA; HDV: Hepatitis delta virus ribozyme that has self-cleavage activity; terFCP: Terminator of endogenous gene, FCP; ShBle: Zeocin-resistance gene; ProLHC: Promoter of the endogenous gene, LHC; AmpR: Ampicillin-resistance gene cassette; ARS: Centromere and autonomous replication sequence (CEN/ARS).
Base editing using all-in-one ARS base editing (ArBE) vectors in Nannochloropsis oceanica
First, we performed a proof-of-concept experiment to evaluate the activity of the ArBE vectors. The nitrate reductase gene, NoNR, was selected as the target gene for ArBE because its mutant phenotypes are easy to detect, and it has been used as a target gene for the demonstration of genome editing efficiency in numerous reports on N. oceanica20,21,35,44,45,46. The target sites in NoNR were selected at the exon–intron junctions (5′-GT-intron-AG-3′). The sequences GT located at the exon–intron junctions and AG located at the intron–exon junctions in the sense strand are essential for splicing47. Therefore, a gene disruption experiment using ArBE was conducted in an attempt to substitute C bases in the antisense strand at these junctions with T bases to disrupt the splicing junction sites and subsequently to inactivate the NoNR gene. The sgRNA_NRJ1 was designed to target the region between exon 1 and intron 1, while sgRNA_NRJ2 was designed to target the region spanning intron 2 and exon 2 (Fig. 2A). The ArBE vectors were introduced into N. oceanica cells, and the colonies that emerged on selective plates were collected. We extracted mixed genomic DNA from all colonies that emerged on the selective plates and analyzed the PCR products of the target-site sequence using ABI sequencing. The resultant sequence chromatogram showed that the 16th and 17th G bases from the protospacer adjacent motif (PAM) (Fig. 2A) of sgRNA_NRJ1 had main G peaks and small A peaks, which were not detected in the WT chromatogram (Fig. 2B). These extra-small peaks were defined as “subpeaks.” The chromatogram patterns of the ABI sequence data were analyzed using the Web tool EditR (https://moriaritylab.shinyapps.io/editr_v10/)48 (Fig. 2C). EditR analysis showed that approximately 46% of the 16th G base of sgRNA_NRJ1 and approximately 30% of the 17th G base exhibited G-to-A base substitution: the 16th and 17th C bases of the complementary strand were substituted with T. Similarly, for sgRNA_NRJ2, approximately 13%, 10%, and 14% G-to-A substitutions were observed at the 17th, 18th, and 20th G bases, respectively. The target sites in single colonies transformed with ArBE vectors targeting NRJ1 or NRJ2 were analyzed using ABI sequencing (Fig. 2D). The transformant NRJ1-1 and NRJ1-2 strains exhibited G-to-A substitutions at the 16th base from the PAM. These base substitutions induced an amino acid substitution from arginine (CGC) at the 72nd codon to histidine (CAC). The NRJ2-1 and − 2 strains exhibited G-to-A substitutions at the 17th base from the PAM. These mutations disrupted the AG sequence at the intron–exon junctions essential for splicing. Thus, our results successfully demonstrated base substitution in an endogenous gene using the base editing system in N. oceanica.

Analysis of base substitution at the NoNR target site in ArBE-induced strains (A) Target site sequences of the endogenous nitrate reductase gene, NoNR, for ArBE vectors. Underlined “GT” and “AG” represent the junction sites of exon and intron in the NoNR gene. PAMs: Protospacer adjacent motifs. (B) Chromatogram data from ABI sequencing of WT genomic DNAs which indicated “WT” or mixed genomic DNA which indicated “BE” and extracted from colonies that emerged on selective plates containing ArBE vectors targeting NoNR. Red boxes show the positions of the intron–exon junctions. Arrows indicate the position of gRNAs. (C) Data analysis of base editing at sgRNA base positions using chromatogram data of ABI sequencing by EditR. Base positions indicate the distance of sgRNA spacer bases from the PAMs. (D) Chromatogram data of ABI sequencing using single colonies that emerged on selective plates of ArBE vectors for NoNR. Underbars show the position of intron–exon junctions. The white text on the black background indicates the bases substituted by ArBE vectors.
The LPAT1 gene was selected as the next target sequence. LPAT1 is an acyltransferase involved in membrane lipid synthesis. The sgRNA was designed to target the cytidine-rich sequence of the LPAT1 exon to investigate the width of the editing window of this base editing system. We constructed ArBE vectors and introduced them into N. oceanica cells. We also extracted the mixed genomic DNA from all colonies that emerged on selective plates and performed sequencing with PCR products. The resultant sequence chromatogram data showed that the 16th to 20th G bases from the PAM of LPAT1 sgRNA had subpeaks (Fig. 3A). The chromatogram patterns of the ABI sequencing data were analyzed using EditR (Fig. 3B). EditR analysis showed that approximately 24%, 43%, 72%, 61%, and 14% of the 16th, 17th, 18th, 19th, and 20th G bases, respectively, were substituted with an A base. Single colonies of transformants were collected, and the target site sequences were analyzed (Fig. 3C). EditR analysis using ABI sequencing data of single colonies was performed (Supplementary Fig. S2). The L1 strain exhibited 89%, 90%, and 84% G-to-A substitutions at the 17th, 18th, and 19th bases, respectively. The L2 strain exhibited G-to-A substitution at rates of 33%, 35%, 77%, and 62% for the 16th, 17th, 18th, and 19th bases, respectively. The L3 strain also exhibited G-to-A substitution at 22%, 34%, 79%, 61%, and 10% for the 16th, 17th, 18th, 19th, and 20th bases, respectively. The L4 strain showed 23% and 15% G-to-A substitution as subpeaks at the 18th and 19th bases, respectively. Additionally, base substitution was successfully achieved in the other three endogenous genes: LDSP, d9FAD, and seipin (Supplementary Fig. S3). ABI sequencing of mixed genomic DNA extracted from colonies transformed with ArBE vectors targeting LDSP was analyzed using EditR, which showed C-to-T substitution rates of 18%, 32%, 78%, and 58% at the 15th, 16th, 17th, and 18th C bases, respectively. Furthermore, C-to-T substitutions were observed at the 16th, 17th, 18th, and 19th C bases in d9FAD (32%, 28%, 29%, and 5%, respectively) and the 16th, 18th, 19th, and 20th C bases in seipin (11%, 69%, 62%, and 11%, respectively). The position dependency of C-to-T substitution efficiency of mixed genomic DNA using ArBE vectors at endogenous target genes is shown in Table 1.

Analysis of base substitution at the LPAT1 target site in ArBE-induced strains. (A) ABI-sequencing chromatogram data from WT genomic DNAs or mixed genomic DNA extracted from mixed colonies that emerged on selective plates of ArBE vectors for LPAT1. (B) Data analysis of base editing on sgRNA base positions using chromatogram data of ABI sequencing by EditR. (C) Chromatogram data of ABI sequencing from single colonies that emerged on selective plates of ArBE vectors for LPAT1. The white text on a black background indicates the bases substituted by the ArBE vectors.
Clearance of all-in-one ARS base editing (ArBE) vectors from Nannochloropsis oceanica transformants
To construct transgene-free mutants, the clearance of transgenes from host cells is essential. Vectors containing CEN/ARS are known to behave as episomal vectors in diatoms17,18,19 and some N. oceanica strains20,21. We applied the vector clearance procedure reported in our previous study20 to the ArBE-induced transformants to validate the vector removability. The transformants of ArBE vectors were incubated in F2N, a standard medium without antibiotic agents. Under these conditions, these vectors may be lost spontaneously. We cultured the NoNR mutant cells in F2N liquid medium for 14 days, and 3,000 of the resultant cultured cells were spread on F2N solid plates. We collected the colonies that emerged on the F2N plates and verified the clearance of the plasmid vector using PCR with primers for amplification of the deaminase domain of ArBE vectors (Fig. 4A). The full gel images of Fig. 4A are shown in Supplementary Figure S4. The resultant base-edited mutants using ArBE vectors targeting NRJ1 or NRJ2 did not show PCR bands for the deaminase gene, indicating successful vector clearance.

Analysis of ArBE vector clearance from transformants. (A) Vector clearance was confirmed by electrophoresis of PCR products amplified with a primer pair targeting the deaminase sequence to detect residual ArBE plasmid. TUB: Tubulin-beta, an endogenous gene used as a control. B1: A mutant strain of nitrate reductase described in a previous report23. NRJ1 mix: The mixed genomic DNA extracted from mixed colonies emerged on selective plates of ArBE vectors for NoNR. NRJ1-1,2: The candidate strains of vector clearance derived from transformants of ArBE vectors for the NRJ1 site. NRJ2-1,2: The candidate strains of vector clearance derived from transformants of ArBE vectors for the NRJ2 site. (B) Phenotypic analysis of vector clearance strains using three types of solid plates. The strains were spotted onto regular F2N plates (NH4+), F2N without ammonium (NH4–), and F2N + 1/2 seawater containing hygromycin (Hyg). To visualize the spot marks, we used a gray background to take pictures of F2N 50% seawater containing hygromycin (Hyg gray). WT: Wild type; HygR: Strain with hygromycin resistance. (C) The measurement of colony intensity of F2N plate photo (NH4+, Fig. 4B) and F2N without ammonium plate photo (NH4–, Fig. 4B) using the image processing software ImageJ (https://imagej.net/ij/).
To further validate both vector clearance and disruption of the NoNR function, we performed phenotypic analyses of the vector-removed candidates of the NoNR base-edited mutants (Fig. 4B). The intensity of spots of NH4+ or NH4– was measured using the image processing software Image J (https://imagej.net/ij/) (Fig. 4C). NoNR mutants are known to exhibit chlorosis49 in a medium containing nitrate as the sole nitrogen source (NH4Cl–). The N. oceanica strains were cultured in F2N liquid medium and then spotted onto standard F2N solid plates containing both nitrate and ammonium as nitrogen sources (NH4Cl+), F2N 50% seawater plates containing hygromycin (Hyg), and F2N plates lacking ammonium (NH4Cl–). Wild-type N. oceanica cells can grow on either standard F2N or F2N – NH4Cl plates. In contrast, NoNR mutant strains (NRJ1-1, NRJ1-2, NRJ2-1, and NRJ2-2) exhibited chlorosis on the F2N – NH4Cl plates. On hygromycin-containing F2N plates, WT and NoNR mutant strains showed no growth. These results confirm that ArBE vectors successfully disrupted NoNR function, and these vectors can be removed from host cells following mutagenesis.
Discussion
In this study, we demonstrated a transgene-free and DSB-free method for gene disruption and mutagenesis in endogenous genes using ArBE vectors in N. oceanica. While base editing in prokaryotic cells has been reported previously, to the best of our knowledge our report represents the first instance of base editing in eukaryotic algae.
LPAT1 is a non-essential endogenous gene related to membrane lipid synthesis. It has been reported that the LPAT1 disruption mutant exhibited a slightly pale-green color and little exchange of the fatty acid composition of membrane lipids37. The mutant strains of LPAT1 induced by ArBE have an amino acid substitution (G188K), which is within the acyltransferase domain of LPAT1. It is likely to be difficult to detect the phenotype of LPAT1 base-edited strains because they are estimated to exhibit a very small change or none at all.
The total efficiency of C-to-G substitution using ArBE systems is compiled in Table 1. The substitution efficiency depended on the sgRNAs. The statistical significance of total C-to-T substitution efficiency of each position from PAMs was tested using Tukey’s multiple comparison test; however, no significant difference was detected. On average, the highly efficient base substitution activity ranged from 29.2% to 47.6% on C bases at the 16th to 19th positions from PAMs. Base substitution at the 15th to 20th bases from PAMs was detected. The editing window50 of the ArBE system was estimated to be at the 15th − 20th positions from PAMs because C-to-T substitutions were detected in C bases at the 15th − 20th positions from PAMs. This is consistent with the Target-AID system27 because the ArBE system is based on it, the editing window of which is also C bases at the 15th − 20th positions from PAMs. Additionally, no significant undesirable C-to-G or C-to-A substitutions were detected in EditR analysis. These findings demonstrated that an ArBE vector can induce C-to-T base substitution in six target sites of five target genes in N. oceanica. However, subpeaks were detected in colonies with introduction of the ArBE vector targeting LPAT1. Most strains exhibited ABI-sequencing chromatograms showing subpeaks within the editing window, rather than distinct patterns corresponding to specific genotypes. This can likely be explained by genome editing occurring at different time points in individual cells during colony growth, as discussed in a previous report40. Meanwhile, the off-target effects of each ArBE vector were not assayed, which we consider to be an important issue for future study.
Gene disruption in N. oceanica has been performed using homologous recombination (HR) because this species has high HR activity35. Meanwhile, HR-mediated gene disruption requires long homology arms (HAs) and marker gene insertion. However, long HAs are not convenient, and marker gene insertion is inconvenient for creating transgene-free mutant strains. Transgene-free genome editing systems offer important benefits in terms of safety and regulatory compliance. They can reduce concerns regarding biological containment regulations by minimizing the risk of unintended vector insertion, which can potentially lead to cytotoxicity by disrupting essential genes. The generation of transgene-free mutations using HR was reported, but that system requires transformation twice and a FACS system36. Because the formation of a colony in which the plasmid has been introduced takes a long time and transformation sometimes induces unexpected mutation, transgene-free mutation using HR is inefficient. In contrast, the CRISPR-Cas9 system does not require long HAs and has high mutagenesis efficiency in N. oceanica21,45,46,51,52. The endogenous DNA repair mechanisms mainly consist of HR and non-homologous end joining (NHEJ). Moreover, NHEJ imperfectly repairs the DSBs and induces insertions or deletions (indels), resulting in genetic modifications53. Cas12a also has high mutagenesis activity, as high as that of Cas936,45. In addition, the TALEN system has TALE DNA binding domains and a FokI nuclease domain. The mutagenesis activity of TALEN is also very high in N. oceanica44. There are effective gene disruption tools via DSB in N. oceanica, which can be used to create transgene-free strains using an episomal vector system20,21; however, such DSBs sometimes induce unexpected large deletions and cytotoxicity. Meanwhile, a DSB-free system in N. oceanica was reported, specifically, the CRISPRi system. This system consists of nuclease-dead Cas9 connected to Krüppel associated box (KRAB), a transcriptional repressor domain54, and exhibited 85% reduction of endogenous gene expression36. The CRISPRi system is useful enough for the phenotypic analysis of gene-repressed strains and is safe because it does not induce DSBs, but it is not suitable for creating transgene-free strains. In contrast, our ArBE system can realize gene disruption in a transgene-free and DSB-free manner simultaneously, which is a distinguishing feature.
Moreover, marker recycling, which can be achieved via a removable, transgene-free system, is indispensable for inducing mutations multiple times. Marker removal has been demonstrated (Fig. 3), but marker recycling has not. The plasmid-removed strains at least exhibited sensitivity for hygromycin in a plate assay. It is thus possible to use hygromycin resistance gene as an antibiotic selection marker for renewed plasmid transformation. In contrast, hygromycin sensitivity does not definitively indicate a transgene-free status, because it is possible that plasmid fragments that do not confer hygromycin resistance are inserted into the genome of host cells. To definitively confirm a transgene-free status in host cells, whole-genome sequencing may be required. Additionally, this base editing system offers a significant advantage for multiplex mutagenesis because the transcription of sgRNA utilizes self-cleaving ribozymes55, which facilitate the concurrent transcription of multiple sgRNA36,41. In contrast, multiplex mutagenesis that relies on DSBs is not ideal; it sometimes results in large deletions between target sites when using the multiplex CRISPR-Cas9 system7,41. These large deletions can be toxic to cells and are generally undesirable. Therefore, base editing systems that do not induce DSBs represent safer and more suitable alternatives for a multiplex mutagenesis approach. Our relatively safe, vector-removable base editing system provides a robust and broadly applicable platform for the construction of “high-performance” algal strains.
Taken together, these results highlight the utility of an efficient and removable cytosine base editing system in microalgae, potentially facilitating further applications and advances in the molecular breeding of these organisms.
Experimental procedures
Materials and culture conditions
The experimental organism used in this study was N. oceanica NIES-2145, which was provided by the National Institute for Environmental Studies (NIES) in Japan. The culture conditions for N. oceanica were set to continuous light at an intensity of 50 µmol photons·m− 2·s− 1, maintained at a temperature of 25 °C, and grown in F2N medium or on F2N plates containing 0.8% agar35. For antibiotic screening, F2N plates containing 50% seawater were used, with an antibiotic concentration of 2 µg·mL− 1 of zeocin (Invitrogen, Waltham, US) or 100 µg·mL− 1 of hygromycin (FUJIFILM Wako, Tokyo, Japan)37,56.
Construction of all-in-one ARS base editing (ArBE) plasmids
The procedure for the construction of all-in-one ARS base editing (ArBE) plasmids is shown in the Supplementary Information and Supplementary Fig. 1.
Transformation
The ArBE plasmids were introduced into algal cells by electroporation using an ELEPO21 super electroporator (NEPAGENE, Ichikawa, Japan) as described in Kurita et al.20. Transformants were selected using F2N 50% seawater medium containing 2 µg⋅mL− 1 zeocin or 100 µg⋅mL− 1 hygromycin.
Treatment of plasmid clearance
Plasmid clearance was performed as in a previous report20. Plasmid clearance was assessed by PCR amplification of the deaminase-Cas9 sequence (1,828 bp) using the primers Target-AID-cloning-R and Cas9-seq-1-R to detect ArBE plasmids. As an internal control, a 288-bp fragment of the endogenous tubulin gene was amplified using the TUB-F and TUB-R primers.
Genomic DNA extraction
Mixed genomic DNA was extracted from the colonies that emerged on antibiotic-selective plates. The extraction of mixed genomic DNA was performed as described in a previous report20. We also performed the DNA extraction from isolated colonies using the Kaneka Easy DNA Extraction Kit version 2 (KANEKA, Osaka, Japan), following the product instructions.
ABI sequencing
The method of ABI sequencing is shown in the Supplementary Information.
Spot test
Spot tests were conducted as previously described20,21,35,36. Candidate strains were cultivated for 7 days in 2 mL of F2N medium in 12-well plates. After incubation, 5 µL (3.6 × 104 cells· µL–¹) of the cultured cells were spotted onto standard F2N plates containing either regular F2N, F2N without ammonium, or F2N prepared with 50% strength seawater and supplemented with 100 µg· L–¹ of hygromycin. The intensity of spots was quantified using Image J (https://imagej.net/ij/).
Data availability
The sequence of nCas9(D10A)-PmCDA1-UGI was deposited into the DNA Databank of Japan, DDBJ (http://www.ddbj.nig.ac.jp) under accession number LC890537. The direct link of the sequence of nCas9(D10A)-PmCDA1-UGI in DDBJ is shown here: https://getentry.ddbj.nig.ac.jp/getentry/na/LC890537/?format=flatfile&filetype=html&trace=true&show_suppressed=false&limit=10.
References
Abo, B. O., Odey, E. A., Bakayoko, M. & Kalakodio, L. Microalgae to biofuels production: a review on cultivation, application and renewable energy. Rev. Environ. Health. 34, 91–99. https://doi.org/10.1515/reveh-2018-0052 (2019).
Hu, Q. et al. Microalgal triacylglycerols as feedstocks for biofuel production: perspectives and advances. Plant. J. 54, 621–639. https://doi.org/10.1111/j.1365-313X.2008.03492.x (2008).
Wood, D. A. Microalgae to biodiesel: Review of recent progress. Bioresource Technol. Rep. https://doi.org/10.1016/j.biteb.2021.100665 (2021).
Chen, H., Li, T. & Wang, Q. Ten years of algal biofuel and bioproducts: gains and pains. Planta 249, 195–219. https://doi.org/10.1007/s00425-018-3066-8 (2019).
Thanigaivel, S., Priya, A. K., Dutta, K., Rajendran, S. & Vasseghian, Y. Engineering strategies and opportunities of next generation biofuel from microalgae: A perspective review on the potential bioenergy feedstock. Fuel https://doi.org/10.1016/j.fuel.2021.122827 (2022).
Venkata Subhash, G. et al. Challenges in microalgal biofuel production: A perspective on techno economic feasibility under biorefinery stratagem. Bioresour Technol. 343, 126155. https://doi.org/10.1016/j.biortech.2021.126155 (2022).
Jeong, B. R., Jang, J. & Jin, E. Genome engineering via gene editing technologies in microalgae. Bioresourc Technol. 373, 128701. https://doi.org/10.1016/j.biortech.2023.128701 (2023).
Kurita, T., Iwai, M., Ohta, H., Sakuma, T. & Yamamoto, T. Genome editing for biodiesel production in oleaginous microalga, Nannochloropsis species. Gene Genome Edit. https://doi.org/10.1016/j.ggedit.2023.100027 (2023).
Patel, V. K. et al. CRISPR-Cas9 system for genome engineering of photosynthetic microalgae. Mol. Biotechnol. 61, 541–561. https://doi.org/10.1007/s12033-019-00185-3 (2019).
Poliner, E., Farre, E. M. & Benning, C. Advanced genetic tools enable synthetic biology in the oleaginous microalgae Nannochloropsis Sp. Plant. cell. Rep. 37, 1383–1399. https://doi.org/10.1007/s00299-018-2270-0 (2018).
Sharma, P. K., Saharia, M., Srivstava, R., Kumar, S. & Sahoo, L. Tailoring microalgae for efficient biofuel production. Front. Mar. Sci. 5, ARTN. https://doi.org/10.3389/fmars.2018.00382 (2018).
Tanwar, A., Sharma, S. & Kumar, S. Targeted genome editing in algae using CRISPR/Cas9. Indian J. Plant. Physiol. 23, 653–669. https://doi.org/10.1007/s40502-018-0423-3 (2018).
Jinek, M. et al. A programmable Dual-RNA-Guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. https://doi.org/10.1126/science.1225829 (2012).
Estrada-Graf, A. et al. Genome assembly and annotation of microalga Nannochloropsis oceanica C018. Microbiol. Resour. Announc. 14, e0088324. https://doi.org/10.1128/mra.00883-24 (2025).
Jinkerson, R. E., Radakovits, R. & Posewitz, M. C. Genomic insights from the oleaginous model Alga Nannochloropsis Gaditana. Bioengineered 4, 37–43. https://doi.org/10.4161/bioe.21880 (2013).
Tang, D. Y. Y. et al. Green technology for the industrial production of biofuels and bioproducts from microalgae: a review. Environ. Chem. Lett. 18, 1967–1985. https://doi.org/10.1007/s10311-020-01052-3 (2020).
Diner, R. E., Bielinski, V. A., Dupont, C. L., Allen, A. E. & Weyman, P. D. Refinement of the diatom episome maintenance sequence and improvement of conjugation-based DNA delivery methods. Front. Bioeng. Biotechnol. https://doi.org/10.3389/fbioe.2016.00065 (2016).
Diner, R. E. et al. Diatom centromeres suggest a mechanism for nuclear DNA acquisition. Proc. Natl. Acad. Sci. USA. 114, E6015–E6024. https://doi.org/10.1073/pnas.1700764114 (2017).
Karas, B. J. et al. Designer diatom episomes delivered by bacterial conjugation. Nat. Com. 6, 6925. https://doi.org/10.1038/ncomms7925 (2015).
Kurita, T. et al. Genome editing with removable TALEN vectors harboring a yeast centromere and autonomous replication sequence in oleaginous microalga. Sci. Rep. 12, 2480. https://doi.org/10.1038/s41598-022-06495-y (2022).
Poliner, E., Takeuchi, T., Du, Z. Y., Benning, C. & Farre, E. M. Nontransgenic Marker-Free gene disruption by an episomal CRISPR system in the oleaginous Microalga, Nannochloropsis oceanica CCMP1779. ACS Synth. Biol. 7, 962–968. https://doi.org/10.1021/acssynbio.7b00362 (2018).
Sikorski, R. S. & Hieter, P. A. System of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces ceratisiae. Genetics 122, 19–27. https://doi.org/10.1093/genetics/122.1.19 (1989).
Davies, B. The technical risks of human gene editing. Hum. Reprod. 34, 2104–2111. https://doi.org/10.1093/humrep/dez162 (2019).
Doudna, J. A. The promise and challenge of therapeutic genome editing. Nature 578, 229–236. https://doi.org/10.1038/s41586-020-1978-5 (2020).
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. https://doi.org/10.1038/nature24644 (2017).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. https://doi.org/10.1038/nature17946 (2016).
Nishida, K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science https://doi.org/10.1126/science.aaf8729 (2016).
Xu, F. et al. Breaking genetic shackles: the advance of base editing in genetic disorder treatment. Front. Pharmacol. 15, 1364135. https://doi.org/10.3389/fphar.2024.1364135 (2024).
Azameti, M. K. & Dauda, W. P. Base editing in plants: Applications, Challenges, and future prospects. Front. Plant. Sci. 12, 664997. https://doi.org/10.3389/fpls.2021.664997 (2021).
Wang, S. Y., Li, X., Wang, S. G. & Xia, P. F. Base editing for reprogramming Cyanobacterium Synechococcus elongatus. Metab. Eng. 75, 91–99. https://doi.org/10.1016/j.ymben.2022.11.005 (2023).
Li, X. D. et al. Development of a base editor for convenient and multiplex genome editing in cyanobacteria. Commun. Biol. 7, 994. https://doi.org/10.1038/s42003-024-06696-3 (2024).
Lee, M., Heo, Y. B. & Woo, H. M. Cytosine base editing in cyanobacteria by repressing archaic type IV uracil-DNA glycosylase. Plant. J. 113, 610–625. https://doi.org/10.1111/tpj.16074 (2023).
Nievergelt, A. P. Genome editing in the green Alga chlamydomonas: past, present practice and future prospects. Plant. J. 122, e70140. https://doi.org/10.1111/tpj.70140 (2025).
Chisti, Y. Biodiesel from microalgae. Biotechnol. Adv. 25, 294–306. https://doi.org/10.1016/j.biotechadv.2007.02.001 (2007).
Kilian, O., Benemann, C. S., Niyogi, K. K. & Vick, B. High-efficiency homologous recombination in the oil-producing Alga Nannochloropsis Sp. Proc. Natl. Acad. Sci. USA. 108, 21265–21269. https://doi.org/10.1073/pnas.1105861108 (2011).
Naduthodi, M. I. S. et al. Comprehensive genome engineering toolbox for microalgae Nannochloropsis oceanica based on CRISPR-Cas systems. ACS Synth. Biol. 10, 3369–3378. https://doi.org/10.1021/acssynbio.1c00329 (2021).
Nobusawa, T., Hori, K., Mori, H., Kurokawa, K. & Ohta, H. Differently localized lysophosphatidic acid acyltransferases crucial for triacylglycerol biosynthesis in the oleaginous Alga Nannochloropsis. Plant. J. 90, 547–559. https://doi.org/10.1111/tpj.13512 (2017).
Poliner, E. et al. A toolkit for Nannochloropsis oceanica CCMP1779 enables gene stacking and genetic engineering of the eicosapentaenoic acid pathway for enhanced long-chain polyunsaturated fatty acid production. Plant. biotechnol. J. 16, 298–309. https://doi.org/10.1111/pbi.12772 (2018).
Liu, L. et al. Down-regulation of SEIPIN transcription attenuated the triacylglycerol accumulation in Nannochloropsis oceanica. J. Oceanol. Limnol. 43, 187–195. https://doi.org/10.1007/s00343-024-3282-y (2024).
Vieler, A., Brubaker, S. B., Vick, B. & Benning, C. A. Lipid droplet protein of Nannochloropsis with functions partially analogous to plant oleosins. Plant. Physiol. 158, 1562–1569. https://doi.org/10.1104/pp.111.193029 (2012).
Wang, Q. et al. Genome engineering of Nannochloropsis with hundred-kilobase fragment deletions by Cas9 cleavages. Plant. J. https://doi.org/10.1111/tpj.15227 (2021).
Zhang, P. et al. Exploring a blue-light-sensing transcription factor to double the peak productivity of oil in Nannochloropsis oceanica. Nat. Com. 13, 1664. https://doi.org/10.1038/s41467-022-29337-x (2022).
Wang, Z. G., Smith, D. G. & Mosbaugh, D. W. Overproduction and characterization of the uracil-DNA glycosylase inhibitor of bacteriophage PBS2. Gene 99, 31–37. https://doi.org/10.1016/0378-1119(91)90030-f (1991).
Kurita, T. et al. Efficient and multiplexable genome editing using platinum TALENs in oleaginous microalga, Nannochloropsis oceanica NIES-2145. Genes Cells. 25, 695–702. https://doi.org/10.1111/gtc.12805 (2020).
Naduthodi, M. I. S. et al. CRISPR-Cas ribonucleoprotein mediated homology-directed repair for efficient targeted genome editing in microalgae Nannochloropsis oceanica IMET1. Biotechnol. Biofuels. 12, 66. https://doi.org/10.1186/s13068-019-1401-3 (2019).
Wang, Q. et al. Genome editing of model oleaginous microalgae Nannochloropsis spp. By CRISPR/Cas9. Plant. J. 88, 1071–1081. https://doi.org/10.1111/tpj.13307 (2016).
Breathnach, R., Benoist, C., O’Hare, K., Gannon, F. & Chambon, P. Ovalbumin gene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc. Natl. Acad. Sci. USA. 75, 4853–4857. https://doi.org/10.1073/pnas.75.10.4853 (1978).
Kluesner, M. G. et al. EditR: A method to quantify base editing from Sanger sequencing. CRISPR J. 1, 239–250. https://doi.org/10.1089/crispr.2018.0014 (2018).
Chen, X. et al. One-stage anammox and thiocyanate-driven autotrophic denitrification for simultaneous removal of thiocyanate and nitrogen: pathway and mechanism. Water Res. 265, 122268. https://doi.org/10.1016/j.watres.2024.122268 (2024).
Ravindran, S. Fixing genome errors one base at a time. Nature 575, 553–555. https://doi.org/10.1038/d41586-019-03536-x (2019).
Verruto, J. et al. Unrestrained markerless trait stacking in Nannochloropsis Gaditana through combined genome editing and marker recycling technologies. Proc. Natl. Acad. Sci. USA. 115, E7015–E7022. https://doi.org/10.1073/pnas.1718193115 (2018).
Ajjawi, I. et al. Lipid production in Nannochloropsis Gaditana is doubled by decreasing expression of a single transcriptional regulator. Nat. Biotechnol. 35, 647–652. https://doi.org/10.1038/nbt.3865 (2017).
Yang, H. et al. Methods favoring homology-directed repair choice in response to CRISPR/Cas9 induced-double strand breaks. Int. J. Mol. Sci. https://doi.org/10.3390/ijms21186461 (2020).
Zhang, G. et al. TRAPT: a multi-stage fused deep learning framework for predicting transcriptional regulators based on large-scale epigenomic data. Nat. Commun. 16, 3611. https://doi.org/10.1038/s41467-025-58921-0 (2025).
Wang, Y. et al. Enhanced RNA secondary structure prediction through integrative deep learning and structural context analysis. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaf533 (2025).
Iwai, M., Hori, K., Sasaki-Sekimoto, Y., Shimojima, M. & Ohta, H. Manipulation of oil synthesis in Nannochloropsis strain NIES-2145 with a phosphorus starvation-inducible promoter from chlamydomonas reinhardtii. Front. Microbiol. 6, 912. https://doi.org/10.3389/fmicb.2015.00912 (2015).
Acknowledgements
We thank Dr. Tetsushi Sakuma for the helpful discussion.
Funding
There was no funding for this study.
Author information
Authors and Affiliations
Contributions
K.M. performed the experiments and wrote the manuscript. T.Y. provided technical instructions on genome editing and project guidance. T.K. supervised this work, designed the study, and aided in the construction of plasmids. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moroi, K., Yamamoto, T. & Kurita, T. Double-strand break-free and transgene-free genome editing in the microalga Nannochloropsis oceanica using removable vectors containing the CRISPR base editing system. Sci Rep 15, 42431 (2025). https://doi.org/10.1038/s41598-025-26657-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26657-y
