
Abstract
Lipopolysaccharide (LPS) can trigger an inflammatory response in lung epithelial cells, which may contribute to the development of Acute Respiratory Distress Syndrome (ARDS). Epigallocatechin-3- gallate (EGCG), a bioactive constituent in green tea, is widely acknowledged for its anti-inflammatory characteristics. This study explored the anti-inflammatory effects of EGCG and its potential molecular mechanisms in mouse lung epithelial-12 (MLE-12) cells. MLE-12 cells were pre-exposed to increasing EGCG concentrations (5, 7.5, and 10 µM) for 24 h, followed by exposure to LPS (20 ng/mL) to induce inflammation After induction, cell and supernatant samples were collected. A range of detection methods were applied to systematically assess the anti-inflammatory activities and the latent mechanisms of EGCG, comprising real-time polymerase chain reaction assay (RT-PCR), enzyme-linked immunosorbent assays (ELISA), western blotting, the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP), transcriptome sequencing, immunofluorescence, co-immunoprecipitation, and confocal microscopy imaging. EGCG exhibited anti-inflammatory effects in LPS-stimulated MLE-12 cells by inhibiting the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α. Its primary mechanism involves robust attachment to 67-kDa laminin receptor (67LR), which modulates the interaction between 67LR and the JAK2 protein within the JAK2/STAT3 signaling pathway. This results in a considerable diminishment in LPS-induced production and expression of inflammatory-promoting cytokines in MLE-12 cells. EGCG attenuates the inflammatory reaction in MLE-12 cells by binding to 67LR and inhibiting the JAK2/STAT3 signaling pathway. The current study explored the effects of EGCG on LPS-induced inflammation in MLE-12 cells, which may offer some new perspectives in inflammation research and broadening our comprehension of the fundamental mechanisms inflammation-related diseases. Considering EGCG’s role in suppressing inflammation in MLE-12 cells, it is reasonable to advocate, from a preventive perspective, the inclusion of EGCG-rich foods as a dietary component for helping alleviate the probability of inflammation-associated disorders.
Similar content being viewed by others

Introduction
ARDS is a life-endangering respiratory disorder that affects approximately 10–20% of patients in intensive care units, and its incidence continues to rise1,2. Despite certain improvements in treatment, mortality remains high at 30 to 40%3,4. The etiology of ARDS is multifactorial, involving bacterial, viral, and other insults. Among these, LPS is often utilized in experimental models to study ARDS pathogenesis, as it effectively induces inflammation and lung injury5. Upon binding to receptors on alveolar epithelial cells, LPS activates multiple signaling pathways, leading to the secretion of pro-inflammatory mediators including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), which contribute to alveolar epithelial cell damage6,7,8. These mediators damage alveolar epithelial cells and reduce surfactant production, leading to alveolar collapse, intrapulmonary shunting, and ultimately respiratory failure9,10. Current therapeutic strategies for ARDS primarily focus on two aspects. The first is respiratory support, which relies on lung-protective ventilation strategies using mechanical ventilators11,12. The second aspect is anti-inflammatory therapy, mainly glucocorticoids. However, glucocorticoid use can may be associated with several adverse effects, including increased infection risk, hyperglycemia, and osteoporosis13,14. Consequently, it becomes critical to unearth safer and more effective anti-inflammatory agents, alongside identifying key inflammatory targets to advance ARDS treatment.
EGCG is the paramount plentiful and biologically efficacious catechin within green tea15. It exerts diverse biological effects, including anticancer, antioxidant, anti-inflammatory, and antibacterial activities16,17. By modulating multiple intracellular signaling pathways, EGCG inhibits the secretion of inflammatory mediators and scavenges free radicals, highlighting its potential for the control and prophylaxis of multiple disorders18,19. Against the backdrop of ARDS, EGCG has gained increasing research attention20. Evidence suggests that EGCG may improve the inflammatory microenvironment in the lungs and reduce tissue damage by suppressing inflammatory signaling pathways and alleviating oxidative stress, thus contributing positively to ARDS management and prognosis21,22. Nevertheless, the precise role and underlying mechanisms of EGCG in regulating inflammation during ARDS remain insufficiently understood.
The 67-kDa laminin receptor (67LR), a high-avidity laminin receptor expressed in a wide range of cell types23, contributes to essential biological phenomena such as cell attachment, growth, and viability24,25. More recently, increasing attention has been drawn to the activity of 67LR in modulating inflammation across various diseases26,27. Interactions between 67LR and its ligands activate complex intracellular signaling pathways that regulate inflammatory responses28. Multiple research have demonstrated that 67LR considerably contributes to the progression of airway inflammatory diseases like chronic obstructive pulmonary disease (COPD), asthma, pneumonia, and carcinoma of the lung29,30. Notably, 67LR regulates the adhesion and binding between inflammatory cells and airway or alveolar epithelial cells, as well as the crosstalk between cells and the extracellular matrix31,32. In addition, 67LR directly influences the inflammatory state of airway and alveolar epithelial cells by modulating the secretion of pro-inflammatory factors like TNF-α and IL-633. Earlier investigations have indicated that EGCG precisely binds to 67LR, exerting biological impacts via the activation or inhibition of downstream signaling cascades34,35. Nonetheless, the specific role played by the EGCG/67LR interaction and its regulation of downstream signaling in ARDS-related inflammatory responses remain unclear.
The Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway is a critical intracellular transduction cascade that maintains normal cellular functions and homeostasis36. Upon detection of extracellular stimuli such as cytokines, growth factors, or pathogen-associated molecular patterns, JAK2, a key non-receptor tyrosine kinase, is rapidly activated37. Activated JAK2 phosphorylates STAT3, which then dimerizes and migrates into the nucleus, where it regulates the expression of genes involved in cell growth, differentiation, apoptosis, and inflammation38. Given its central role, the JAK2/STAT3 pathway is a focus of biomedical research on modulating inflammation39. In ARDS, pathogenic factors activate the JAK2/STAT3 pathway within alveolar epithelial cells, leading to inflammation, impairment of barrier function, as well as immune dysregulation40,41. Studies have shown that EGCG can inhibit JAK2/STAT3 activation, thus reducing the production of inflammatory mediators and exerting anti-inflammatory effects42,43. Nevertheless, the precise regulatory role of the EGCG/67LR axis in modulating the JAK2/STAT3 pathway and its potential clinical applications require further exploration.
During this study, we show that EGCG safeguards MLE-12 cells against LPS-trigger inflammation. Our results indicate that these protective effects could be mediated by targeting 67LR. Mechanistic analyses revealed that EGCG significantly reduces LPS stimulates the production of pro-inflammatory cytokines, e.g., IL-6, TNF-α, and IL-1β, via specific interaction with 67LR. Notably, engagement of 67LR by EGCG suppresses the JAK2/STAT3 signaling pathway, representing a novel anti-inflammatory mechanism. These discoveries shed new light on the molecular mechanisms of EGCG’s protective effects in acute lung injury and identify 67LR as a promising target for developing strategies to mitigate inflammation in MLE-12 cells.
Materials and methods
Reagents and cell culture
EGCG (Cat. No. M4208) was purchased from Abmole Bioscience, Inc. LPS (Cat. No. L2630-100 mg) was obtained from Sigma-Aldrich Co., LLC. Coumermycin A1 (Cat. No. 4434-05-3) and JAK/IN-21 (Cat. No. 2445499-20-5) were purchased from MedChemExpress Co., Ltd. Colivelin TFA (Cat. No. 2803948-60-7) and Stattic (Cat. No. 19983-44-9) were obtained from Shanghai Macklin Biochemical Co., Ltd.
MLE-12 cells (passages 8–18) were acquired from the Meisen Chinese Tissue Culture Collections (CTCC-003-0020, Zhejiang Province, China). Cells were maintained in DMEM/F-12 medium (Cat. No. C11330500BT) supplemented with 2% Trans-Serum® EQ fetal bovine serum, insulin, and transferrin to prepare a complete medium. Cultures were maintained at 37 °C with 5% CO2 until they reached 80% confluence.
CCK-8 assay
Cell viability was evaluated utilizing the Cell Counting Kit-8 (CCK-8, Cat. No. K101842133EF5E). In brief, 10,000 cells suspended in 100 µL of culture medium were inoculated into each well of a 96-well microplate and cultured for 8 to 10 h. Following treatment or transfection, 10 µL of CCK-8 reagent was introduced into each well, and the plate was nurtured at 37 °C for 2 h. Absorbance was then ascertained at a wavelength of 450 nm using a Bio-Tek Synergy H1 Multi-Mode Microplate Reader (Alfa Aesar, a Gibco Company, USA).
Enzyme-linked immunosorbent assays
Supernatants from MLE-12 cells treated with EGCG and LPS were harvested and preserved at − 80 °C. The concentrations of IL-6 (Cat. No. EM0121), IL-1β (Cat. No. EM0109), and TNF-α (Cat. No. EM0183) were quantified using ELISA kits (Fine Biotech Co., Ltd., Wuhan, China) in line with the manufacturer’s guidelines. Absorbance was assayed at a wavelength of 450 nm using a Bio-Tek Synergy H1 Multi-Mode Microplate Reader.
Western blotting assay
MLE-12 cells that had undergone treatment with EGCG and LPS were rinsed twice with ice-cold phosphate-buffered saline (PBS) and harvested. Cells were lysed in high-strength RIPA buffer supplemented with protease and phosphatase inhibitors. After centrifugation, total protein concentrations were ascertained using a BCA kit (Cat. No. P0010). Equalized whole-cell lysates were separated by SDS–PAGE and translocated onto the surface of PVDF membranes. Membranes were sealed with 5% BSA and kept in contact with primary antibodies (see Supplementary Table S1) at 4 °C overnight (8–10 h). Subsequent to washing using PBST, membranes were exposed to the corresponding secondary antibodies at 22–26 °C (specificities listed in Supplementary Table S1). Protein bands were visualized employing an ECL kit (Oriscience Biotechnology Co., Ltd., Chengdu, China, Cat. No. PD203), and images were captured and analyzed using a Tanon 5200CE system (Shanghai, China) and ImageJ software (National Institutes of Health, USA).
Real-time polymerase chain reaction assay
MLE-12 cells incubated with EGCG and LPS were rinsed twice with ice-cold PBS to remove residual impurities. Total RNA was isolated utilizing Trizol reagent (Cat. No. 15596018CN) as per the manufacturer’s instructions. RNA was transformed into cDNA via a reverse transcription kit (Bio-Rad Laboratories, California, USA, Cat. No. 1708891). Quantitative RT–PCR was executed by means of 2× M5 Hiper Real-time PCR Supermix (Cat. No. MF797-01). Primer sequences are available in Supplementary Table S2. PCR conditions Commenced with an initial denaturation at 95 °C for 1 min, succeeded by 40 cycles of 95 °C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 45 s, with a final extension at 72°C for 1 min. Relative mRNA levels of 67LR, IL-1β, IL-6, and TNF-α were calculated with the 2−ΔΔCt method, with GAPDH acted as the internal control.
A comprehensive network pharmacology approach to elucidate the mechanisms of active components in traditional Chinese medicine for regulating inflammatory responses.
Following the approach of Madikyzy et al.44,45,46,47, the Simplified Molecular-Input Line-Entry System (SMILES) structure of EGCG was retrieved from the TCMSP and PubChem databases (Figure S2A). Potential targets of EGCG were predicted, and a gene database was constructed. An inflammation-related target library was compiled by searching DisGeNET, GeneCards, and the Online Mendelian Inheritance in Man (OMIM) databases, and overlapping targets were identified. A protein–protein interaction (PPI) network was formed with the assistance of the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING). Finally, gene ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted using the Database for Annotation, Visualization, and Integrated Discovery (DAVID), and the data outputs were illustrated as a bubble chart.
Transcriptome sequencing
Following the protocol of Kim et al.48, total RNA was retrieved and purified from MLE-12 cells treated with EGCG and LPS. Cells were flushed twice with ice-cold PBS, and RNA was extracted by utilizing the phenol–chloroform method. For every 1 mL of TRIzol-treated lysate, 0.2 mL of chloroform was added, followed by vortexing, allowing it to rest, and then centrifuging to separate and precipitate RNA. mRNA was captured using Epi™ beads through sequential incubations and magnetic separations. A cDNA library was then constructed, amplified, and purified. Library quality was assessed prior to sequencing, which was performed on a NovaSeq PE150 platform. Raw sequencing data were trimmed and processed to generate clean data for subsequent gene identification and analysis.
Immunofluorescence and co-immunoprecipitation
IF was performed adopting the approach of Yu et al.49. MLE-12 cells treated with EGCG and LPS were cultured in 6-well plates with glass coverslips until reaching 30–40% confluence. Cells underwent washing and fixation with 4% paraformaldehyde (Cat. No. G1101-500ML), followed by permeabilization and occupying non-specific sites with 5% BSA and 0.1% Triton X-100. Cells were then kept in contact with (8–10 h) at 4 °C with primary antibodies targeting 67LR, p-STAT3, JAK2, and p-JAK2. Subsequent to washing, cells were incubated with a fluorophore-conjugated secondary antibody (Cat. No. SA00003-2) for 1 h at 22–26 °C in the dark. Nuclei were counterstained by adding 5 µL of DAPI (Cat. No. C1002) per well and incubating for 5 min without light exposure. Cells underwent a triple-wash procedure with PBS for 5 min each. The coverslips were carefully removed and allowed to dry, after which approximately 5 µL of anti-fade mounting medium (Cat. No. P0126-25 mL) was applied. Fluorescence visualization was carried out via a Zeiss AX10 microscope or a confocal microscope (Olympus, Tokyo, Japan) to visualize the results.
Co-IP was performed following the method of Liu et al.50. MLE-12 cells were seeded in 10-cm culture dishes and cultured until reaching 80–85% confluence. The medium was then substituted by DMEM-12 containing chloroquine, followed by transfection with 67LR-FLAG and JAK-HA plasmids (Supplementary Materials 1.7.1 and 1.7.2). After 48 h, fluorescence expression was confirmed, and cells were lysed for collection of the supernatant. Primary antibodies for immunoprecipitation, including HA, FLAG, and isotype control IgG, were added and incubated. Whole-cell lysates were then separated by SDS–PAGE and transferred onto PVDF membranes. Membranes were sequentially blocked and incubated with primary and secondary antibodies. Protein interactions were visualized and quantified using a Tanon imaging system and analyzed with ImageJ software.
Statistical analysis
All experiments were executed in at least three independent replicates to ensure reproducibility. Statistical analyses were conducted using Tukey’s multiple comparison test in GraphPad Prism software (version 9.5.1; GraphPad Software, San Diego, CA) to assess differences between groups. Data are rendered as the means ± SEMs, and P < 0.05 was judged statistically significant.
Results
EGCG alleviates inflammation induced by LPS in MLE-12 cells
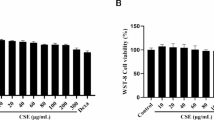
CCK-8 assay results indicated that EGCG concentrations between 0 and 10 µM did not significantly affect the viability of MLE-12 cells (Figure S1A). Consequently, 5, 7.5, and 10 µM concentrations of EGCG were used for the subsequent experiments.
RT–PCR and ELISA analyses showed that LPS treatment led to an increase IL-1β, IL-6, and TNF-α expressions compared to the control group, whereas EGCG pretreatment effectively these elevations (Fig. 1A–F). These findings indicate that EGCG protects MLE-12 cells from LPS-induced inflammation, with the most pronounced protective effect observed at 7.5 µM. However, at a higher concentration of 10 µM, EGCG may exert cytotoxic effects, compromising cell membrane integrity. Because the cell membrane serves as a critical barrier between the intracellular and extracellular environments, its damage can trigger inflammatory responses, resulting in elevated levels of inflammatory markers. At an EGCG concentration of 10 µM, cellular feedback mechanisms that maintain homeostasis might be activated, disrupting the original balance and triggering compensatory responses that can result in the upregulation of inflammatory markers. In addition, EGCG metabolism may vary with dose; at 10 µM, cells can generate specific metabolites with certain characteristics, which cause EGCG to lose its anti - inflammatory effect.

Epigallocatechin-3-gallate (EGCG) alleviates the inflammation induced by Lipopolysaccharid (LPS) in mouse lung epithelial-12 cells. (A–C) Levels of IL-1β, IL-6, and TNF-α in cell supernatants were quantified by enzyme-linked immunosorbent assays (ELISA). (D–F) Cellular levels of IL-1β, IL-6, and TNF-α were assayed by real-time polymerase chain reaction assay (RT-PCR). Data are presented as means ± SEMs (N = 3, # P < 0.05, ## P < 0.01, ### P < 0.001, and #### P < 0.0001 versus the control group; * P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 versus the LPS-treated group).
EGCG reduces inflammatory responses induced by targeting 67LR
Previous studies have proposed 67LR as a membrane binding site for EGCG, may mediating its physiological effects upon binding51,52. In this study, MLE-12 cells were pre-incubated with EGCG prior to stimulation by LPS investigate its effect on 67LR. The RT–PCR results showed revealed no statistically significant disparities in 67LR abundance between the LPS or EGCG pretreatment groups versus the control group (Fig. 2A). In contrast, western blot analysis revealed that EGCG treatment downregulated 67LR protein levels (Fig. 2B,C). To assess whether EGCG’s inhibitory effect on LPS-induced inflammation is dependent on 67LR, cells were stably transfected to either overexpress 67LR (OE-67LR) or knock down 67LR (KD-67LR). Transfection success ratio was validated by RT–PCR and western blot analyses (Fig. 2D–F). To assess inflammatory cytokine expression, MLE-12 cells were pre-treated with EGCG before stimulation with LPS. RT–PCR results showed that the inhibitory effect of EGCG was hardly detectable in KD-67LR cells but seemed to be enhanced in OE-67LR cells (Fig. 3A–C). These findings were further supported by ELISA measurements (Fig. 3D–F). Together, these outcomes imply that EGCG significantly suppresses LPS-evoked inflammatory cytokine expression via 67LR.

Intervention with Epigallocatechin-3-gallate leads to differential changes in the expression of 67-kDa laminin receptor protein and mRNA. (A) 67-kDa laminin (67LR) levels were evaluated by RT-PCR. (B) 67LR protein levels were assessed by western blotting. (C) Relative 67LR protein band intensities were quantified, normalized to the Tubulin band, and analyzed using ImageJ software. (D) 67LR mRNA levels following lentiviral transfection were assessed by RT–PCR. (E) 67LR protein levels following lentiviral transfection were evaluated by western blotting. (F) Relative protein band intensities were quantified, normalized to the Tubulin band, and analyzed with ImageJ. OE-67LR indicates cells transfected with the 67LR-overexpressing lentiviral vector, with OE-NC as the control. KD-67LR represents the 67LR-knockdown cells, with KD-NC as the knockdown control. Data are presented as the means ± SEMs (N = 3, ns P > 0.05, ## P < 0.01, ### P < 0.001, and #### P < 0.0001 versus the control group; * P < 0.05 and *** P < 0.001 versus the LPS-treated group).

EGCG attenuates inflammatory responses by targeting 67LR. (A–C) The levels of IL-1β, IL-6, and TNF-α in cell supernatants were measured by ELISA. (D–F) Cellular levels of IL-1β, IL-6, and TNF-α were evaluated by RT–PCR. Data are presented as the means ± SEMs (N = 3, # P < 0.05, ## P < 0.01, ### P < 0.001, and #### P < 0.0001 versus the control group; * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001 versus the LPS-treated group; & P < 0.05, && P < 0.01, and &&& P < 0.001 versus the LPS + EGCG 7.5 µM group).
EGCG mitigates inflammatory responses through the JAK2/STAT3 signaling pathway
The mechanisms underlying EGCG’s anti-inflammatory effects were investigated using a systematic bioinformatics approach. Initially, 104 EGCG-associated genes were identified from the TCMSP and SwissTargetPrediction databases. A comprehensive dataset of 16,290 inflammation-related genes was then compiled from DisGeNET, OMIM, and GeneCards. Venn diagram analysis revealed 96 overlapping genes potentially involved in EGCG’s anti-inflammatory activity (Figure S2B). PPI network analysis using STRING and Cytoscape identified key hub genes, including STAT3, Pik3ca, Map2k1, and STAT1, which exhibit high connectivity within anti-inflammatory signaling pathways (Figure S2C). This analysis revealed that these genes were associated with 437 biological processes, 70 cellular components, and 107 molecular functions (Figures S3.A–C). KEGG pathway enrichment analysis further highlighted the relevance of these genes (Fig. 4A), prominently identifying the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway as highly enriched. This evidence provides support for the hypothesis that the JAK/STAT pathway is likely to serve as a key mechanism by which EGCG mediates its anti-inflammatory actions.
Differential gene expression analysis was performed the control versus LPS groups, alongside the comparison of the LPS with LPS + EGCG groups. Venn diagram analysis identified 20 overlapping genes (Figure S4A). A PPI network was constructed via the STRING database to explore potential functional connections among these genes. After the removal of non-relevant nodes and topological analysis in Cytoscape, several key genes, including JAK2, JAK1, STAT1, and STAT3, were highlighted for their high connectivity and central roles in anti-inflammatory signaling pathways (Figure S4B). MetaScape enrichment analysis uncovered that these genes may participate in 175 biological activities, 20 cellular components, and 37 molecular functions (Figures S5A–C). KEGG pathway analysis further indicated substantial enrichment of the JAK/STAT pathway, providing support for the hypothesis that it may play a crucial role in EGCG-mediated anti-inflammatory effects, consistent with the network pharmacology results (Fig. 4B).

KEGG enrichment analysis of EGCG based on network pharmacology and transcriptomics. (A) KEGG enrichment analysis of the EGCG through network pharmacology. (B) KEGG enrichment analysis was conducted for the differentially expressed genes among the control group, the LPS group, and the LPS + EGCG group through transcriptomic analysis.
To further explore the possible molecular mechanisms underlying this regulatory interplay, we examined protein expression profiles of key JAK/STAT pathway effectors. Quantitative analysis via western blotting and immunofluorescence showed that LPS stimulation markedly increased the phosphorylation of JAK2 and STAT3, indicating their activation in response to LPS-induced inflammation (Fig. S5A–F). Importantly, pretreatment with 7.5 µM EGCG effectively reversed these effects, significantly reducing LPS-induced phosphorylation of both JAK2 and STAT3 (Fig. 5A–F). These results suggest that EGCG mediates its anti-inflammatory effects, to some extent, via suppressing the phosphorylation and activation of JAK2 and STAT3. Further analysis revealed that the expression levels of p-JAK1, p-STAT1, p-STAT2, p-STAT4, and p-STAT5 were relatively low and showed minimal correlation with the inflammatory regulatory network (Figures S6A–G), indicating that these molecules are unlikely to serve as primary targets of EGCG in this context. Collectively, these findings highlight the specific roles of JAK2 and STAT3 within the JAK/STAT pathway in mediating LPS-induced inflammation and in the inhibitory effects of EGCG.

Modulations of EGCG on p-JAK2 and p-STAT3 in the JAK2/STAT3 signaling pathway in LPS-induced inflammation. MLE-12 cells were plated onto 6-well plates and grown to 30–40% confluence, followed by a 24-h pretreatment with EGCG and subsequent 10-h exposure to LPS. (A) The protein levels of JAK2, p-JAK2, STAT3, and p-STAT3 were measured by western blotting. (B,C). The relative intensities of p-JAK2/JAK2 and p-STAT3/STAT3 were analyzed using ImageJ software. (D) The protein levels of JAK2, p-JAK2, STAT3, and p-STAT3 were assessed by immunofluorescence (scale bar = 20 μm). (E,F) The MFI of p-JAK2 and p-STAT3 proteins was analyzed via Image J software. Data are displayed as the means ± SEMs (N = 3, ### P < 0.001 an #### P < 0.0001 versus the control group; * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001 versus the LPS-treated group).
Contemporary investigations have accented the roles of specific compounds in modulating the JAK2/STAT3 signaling pathway. Coumermycin A1 acts as a JAK-2 agonist36,37, while JAK-IN-21 functions as a JAK-2 inhibitor38. Colivelin TFA and Stattic have been identified as a STAT3 agonist and inhibitor39,40,41. The modulatory effects of these compounds on the JAK2/STAT3 signaling pathway have been well established. CCK-8 assays were used to determine their safe concentrations (Figures S7A–D), and western blot analysis provided evidence suggesting their effective regulation of p-JAK2 and p-STAT3 protein levels (Figs. 6A–D). Additionally, LPS-induced MLE-12 inflammation models were treated with these compounds for 48 h to assess their functional impact. RT–PCR and ELISA showed that the agonist augmented the expression of IL-1β, IL-6, and TNF-α, while the inhibitors respectively decreased these markers (Fig. 7A–F). These discoveries indicate that EGCG possibly have an anti-inflammatory effect on LPS-initiated MLE-12 cell inflammation models through suppressing the JAK2/STAT3 signaling pathway.

EGCG regulates and improves the phosphorylation levels of proteins in the JAK2/STAT3 signaling pathway via its agonists and inhibitors. MLE-12 cells were inoculated in 6-well plates and grown to 60% confluence. The cells were incubated with Coumermycin A1, JAK-IN-21, Colivelin TFA, and Stattic in culture medium for 48 h. The medium was then refreshed, followed by a 24-h pretreatment with EGCG, and finally exposure to LPS for 10 h. (A–B) Protein levels of JAK2, p-JAK2, STAT3, and p-STAT3 were measured by western blotting. (C–D) Relative intensities of p-JAK2/JAK2 and p-STAT3/STAT3 were analyzed via ImageJ software. The data are presented as the means ± SEMs (N = 3, #### P < 0.0001 versus the control group; ** P < 0.01 and **** P < 0.001 versus the LPS-treated group; & P < 0.05, &&& P < 0.001, &&&& P < 0.0001 versus the LPS + EGCG 7.5 µM group).

EGCG inhibited inflammation through the JAK2/STAT3 signaling pathway. MLE-12 cells were subjected to the experimental procedure shown in Fig. 6. After 48 h, the medium was refreshed, and the cells were subsequently administered with EGCG and LPS. (A–C) Levels of IL-1β, IL-6, and TNF-α in cell supernatants were quantified by ELISA. (D–F) Cellular levels of IL-1β, IL-6, and TNF-α were measured by RT–PCR. The data are presented as means ± SEMs (N = 3, #### P < 0.0001 versus the control group; *** P < 0.001 and **** P < 0.0001 versus the LPS-treated group; & P < 0.05, && P < 0.01, &&& P < 0.001, and &&&& P < 0.0001 versus the LPS + EGCG 7.5 µM group).
JAK2 is a direct target of 67-LR
To explore the possible molecular mechanisms by which 67LR regulates the JAK2/STAT3 signaling pathway in MLE-12 cells, we first used the Nucleotide BLAST tool and identified complementary sequences in the 3′-UTR of the JAK2 gene that matched 67LR (Figure S8). These results indicate a potential direct interaction between 67LR and JAK2 in MLE-12 cells. Co-IP experiments further provided evidence suggesting the presence of a stable complex between 67LR and JAK2 (Fig. 8A), indicating a functional interplay between these two proteins in cellular physiological processes. To delve deeper into the intracellular spatial relationship 67LR and JAK2 in relation to each other, we carried out immunofluorescence confocal microscopy. The results showed pronounced spatial co-localization of 67LR and JAK2 (Fig. 8B). Importantly, following EGCG treatment, quantitative image analysis demonstrated that the interaction between 67LR and JAK2 was potentiated (Figs. 8B–E). This interaction likely facilitates the repressive influence of 67LR on the JAK2/STAT3 signaling pathway.

JAK2 is a direct target of 67-LR. MLE-12 cells were implanted in 6-well plates and grown to 80–85% confluence, followed by transfection with 67LR-FLAG and JAK-HA plasmids. (A) The interaction between 67LR and JAK2 was assessed by a co-immunoprecipitation (co-IP) assay. The MLE-12 cells were sown in 6-well plates at a density of 200 × 104 cells per well and grown to 30–40% confluence, pretreated with EGCG for 24 h, and subsequently exposed to LPS for 10 h. (B) Spatial co-localization of 67LR and JAK2 proteins was visualized by immunofluorescence (scale bar = 10 μm). (C–E) Fluorescence intensity of 67LR–JAK2 colocalization was quantified using ImageJ software.
EGCG ameliorated LPS-induced inflammation via the 67LR-mediated inhibition of the JAK2/STAT3 signaling pathway
RT–PCR and ELISA analyses were used to assess inflammatory markers. Knockdown of 67LR via siRNA-mediated gene silencing, combined with JAK2 agonist administration, appeared to upregulate the expression of inflammation-related molecules. In contrast, overexpression of 67LR through lentiviral transduction, together with JAK2 inhibitor treatment, seemed to reduce inflammatory marker expression (Fig. 9A–F). These results evince that EGCG modulates the interaction between 67LR and the JAK2/STAT3 signaling pathway, thus effectively suppressing inflammatory responses.

EGCG ameliorated LPS-induced inflammation via the 67LR-mediated inhibition of the JAK2/STAT3 signaling pathway. MLE-12 cells were transfected with lentiviruses (OE-67LR or KD-67LR). The cells were then cultured with Coumermycin A1 or JAK-IN-21, followed by co-treatment with EGCG and LPS. (A–C) Levels of IL-1β, IL-6, and TNF-α in cell supernatants were evaluated by ELISA. (D–F) The levels of IL-1β, IL-6, and TNF-α were assessed by RT–PCR. The data are rendered as the means ± SEMs (N = 3, #### P < 0.0001 versus the control group; * P < 0.05, *** P < 0.001, and **** P < 0.0001 versus the LPS-treated group; & P < 0.05, && P < 0.01, and &&&& P < 0.0001 versus the LPS + EGCG 7.5 µM group).
Discussion
ARDS is a common and life-threatening condition that often arises secondary to critical illnesses such as viral infections, severe burns, and septic shock53. Many ARDS patients are susceptible to concurrent Gram-negative bacterial infections54. When LPS, a key constituent of the Gram-negative bacterial cell wall, enters the bloodstream, alveolar epithelial cells are among the first to be damaged55. These damaged cells activate complex intracellular signaling pathways, upregulating inflammation-related genes and promoting the secretion of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6. This can trigger a cytokine storm, exacerbating ARDS56. Currently, clinical care for ARDS relies primarily on supportive therapies, including but not limited to mechanical ventilation and fluid management, which do not address the underlying inflammatory damage57. Therefore, investigating the inflammatory mechanisms in alveolar epithelial cells and developing drugs to block the inflammatory cascade are essential.
The exploration of bioactive compounds from natural sources has garnered significant academic interest58. EGCG, a principal bioactive constituent of green tea, is well recognized for its anti-inflammatory properties59. The MLE-12 cell line, derived from mouse alveolar epithelial cells, is widely used to model ARDS-related epithelial cell damage60. Exposure of MLE-12 cells to 20 ng/mL LPS effectively simulates the inflammatory damage characteristics of ARDS61,62. In this study, we focused on the key pathological features of ARDS and investigated the cytoprotective actions of EGCG on lung epithelial cells. Our experiments demonstrated that 7.5 µM EGCG exerted a pronounced anti-inflammatory effect against LPS-induced inflammation, consistent with previous studies showing EGCG’s potent ability to inhibit inflammatory responses in various models63. These findings confirm EGCG’s anti-inflammatory activity and provide valuable experimental evidence to support further investigation of its mechanisms and potential applications in treating inflammation-related diseases.
The research on anti-inflammatory mechanisms aims to develop highly effective treatments for inflammation-related diseases while minimizing side effects. EGCG has shown considerable promise on account of its potent anti-inflammatory properties, which are associated with 67LR64. This receptor, expressed at the surface of various cell types, binds specifically to EGCG65,66. Upon entering the body, EGCG interacts with cells by attaching to 67LR with high specificity owing to its unique chemical structure67. This binding reduces the release of pro-inflammatory factors, thus alleviating the inflammatory response66. In our study, we examined the interaction between EGCG and 67LR and found that treatment of MLE-12 cells with EGCG led to a significant decrease in 67LR levels. Results from rescue experiments demonstrate that EGCG exerts its anti-inflammatory effects primarily through binding to 67LR. Notably, the anti-inflammatory effect of EGCG was was absent from cells with knockdown of 67LR, confirming its essential contribution to the anti-inflammatory response initiated by EGCG. The mechanisms by which EGCG exerts its anti-inflammatory effects notably differ from traditional approaches, such as regulating immune system function and addressing oxidative stress. For instance, some traditional anti-inflammatory drugs reduce the synthesis and release of pro-inflammatory mediators by directly inhibiting the corresponding biosynthetic enzymes67. However, these synthetic enzymes participate in numerous essential physiological processes, such as maintaining gastrointestinal mucosal integrity and regulating renal blood flow68. Consequently, the use of drugs targeting these enzymes to control inflammation often results in side effects, including gastrointestinal ulcers, bleeding, and impaired renal function69. Glucocorticoids, another class of anti-inflammatory agents, reduce inflammation by suppressing immune cell activity and inhibiting the release of inflammatory factors70. Nevertheless, long-term glucocorticoid use can weaken the immune system, increase susceptibility to infections, and lead to metabolic disorders, osteoporosis, and other adverse effects71. Antioxidants exert anti-inflammatory effects primarily by reducing oxidative stress and scavenging reactive oxygen species (ROS) and reactive nitrogen species (RNS), thus exerting an anti-inflammatory effect72. However, the anti-inflammatory effects of antioxidants are relatively limited, and their mechanisms are generally singular, making it challenging to comprehensively control complex inflammatory processes73. In contrast, EGCG specifically targets inflammatory signal transduction by binding to 67LR. This minimizes the disruption of normal cellular functions and reduces the risk of side effects. Clinically, the high specificity and low toxicity of EGCG present promising opportunities for treating inflammation-related diseases, offering patients safe and effective therapeutic options and providing new targets for the development of innovative anti-inflammatory drugs, thereby thus the field of anti-inflammatory therapy.
The JAK2/STAT3 signaling pathway is a well-characterized cytokine signal transduction mechanism that plays important roles in numerous physiological and pathological cellular processes74, including inflammatory responses, apoptosis, proliferation, and differentiation75. Several studies have shown that this pathway is considerably activated during cytokine storms in ARDS, and its inhibition can effectively mitigate the cytokine storm caused by different underlying conditions76. Clinically, drugs targeting the JAK2/STAT3 pathway, such as baricitinib, ruxolitinib, and tofacitinib, have been employed as novel therapeutic strategies for ARDS77. In MLE-12 cells, we observed significant activation of the intracellular JAK2/STAT3 signaling pathway. Additionally, basic research indicates that EGCG can may inhibit this pathway. In inflammatory cell models, EGCG treatment appears to reduce the phosphorylation levels of JAK2 and STAT3, thus limiting the formation of STAT3 dimers and their ability to enter the nucleus and bind to DNA. STAT3’s reduced DNA-binding capacity subsequently inhibits the transcription and expression of downstream inflammation-related genes. Consequently, the synthesis and release of pro-inflammatory cytokines may be decreased, effectively controlling the cellular inflammatory response78,79. Our study confirmed these findings, confirming the anti-inflammatory effect of EGCG of inhibiting the JAK2/STAT3 signaling pathway. Additionally, prior research showed that EGCG exerts anti-inflammatory effects via 67LR. In our study, we further revealed that EGCG suppresses inflammation by targeting the JAK2/STAT3 signaling pathway. Through comprehensive experiments, we provided evidence suggesting that EGCG binds to 67LR, thus inhibiting the JAK2/STAT3 signaling pathway, and exerting its anti-inflammatory effects. Supplementary experiments further confirmed this critical finding. Collectively, these results provide robust experimental evidence supporting the development of novel EGCG-based anti-inflammatory therapeutics and establish a solid theoretical foundation for future drug design.
This study has several limitations. First, we primarily focused on cellular-level experiments, which may not fully reflect in vivo conditions. Consequently, discrepancies could exist between the observed cell-based effects and actual physiological responses. Future studies involving well-designed animal experiments and clinical trials are necessary to validate the translational potential of these findings. Second, the investigation of signaling pathways was not comprehensive. While this study focused on the 67LR/JAK2/STAT3 pathway, in vivo inflammatory responses are governed by a complex network of multiple signaling pathways and molecular interactions. Therefore, other unexamined pathways may also contribute to EGCG’s anti-inflammatory effects. Future research will further investigate the interactions between the 67LR/JAK2/STAT3 pathway and other signaling cascades to comprehensively elucidate the molecular mechanisms underlying EGCG’s anti-inflammatory effects and provide a stronger theoretical foundation for its clinical application. Additionally, this study did not explore the potential of combination therapies. In clinical practice, combination treatment is often employed to enhance therapeutic efficacy. Future experiments could evaluate EGCG in conjunction with other anti-inflammatory agents to assess potential synergistic effects, thus offering a basis for optimizing clinical treatment strategies. These investigations are essential for confirming the in vivo anti-inflammatory efficacy and safety of EGCG, as well as for clarifying the specific role of the 67LR/JAK2/STAT3 pathway in the inflammatory cascade. In summary, this study provides a novel perspective on the anti-inflammatory mechanisms of EGCG, identifying the 67LR/JAK2/STAT3 pathway as a critical target for its action. Future research will further elucidate EGCG’s anti-inflammatory properties, offer new strategies for the treatment of inflammation-related diseases.
Conclusion
This study suggests that EGCG may attenuates the inflammatory response in MLE-12 cells by specifically interacting with the 67LR receptor and inhibiting the activation of the JAK2/STAT3 signaling pathway. In the context of available research, the data obtained from our research provide evidence suggesting a potential cellular-level anti-inflammatory mechanism of EGCG in the context of ARDS, which may offer a theoretical basis for the development of treatments for related inflammatory diseases.
Data availability
All quantitative data (western blot gray values, raw Ct values of qPCR, cell viability assays, immunofluorescence, confocal microscopy, network pharmacology, transcriptomics, and lentiviral transfection data) have been publicly uploaded to Figshare with the DOIs: https://doi.org/10.6084/m9.figshare.29650847 and https://doi.org/10.6084/m9.figshare.29650925.
References
Bellani, G. et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. Jama 315, 788–800. https://doi.org/10.1001/jama.2016.0291 (2016).
Gorman, E. A., O’Kane, C. M. & McAuley, D. F. Acute respiratory distress syndrome in adults: diagnosis, outcomes, long-term sequelae, and management. Lancet 400, 1157–1170. https://doi.org/10.1016/s0140-6736(22)01439-8 (2022).
Yu, Y. & Qiu, L. Nanotherapy therapy for acute respiratory distress syndrome: a review. Front. Med. (Lausanne). 11, 1492007. https://doi.org/10.3389/fmed.2024.1492007 (2024).
Horie, S. et al. Emerging pharmacological therapies for ARDS: COVID-19 and beyond. Intensive Care Med. 46, 2265–2283. https://doi.org/10.1007/s00134-020-06141-z (2020).
You, Q. et al. MiR-802 alleviates lipopolysaccharide-induced acute lung injury by targeting Peli2. Inflamm. Res. 69, 75–85. https://doi.org/10.1007/s00011-019-01295-z (2020).
Wang, J. et al. Interplay between pulmonary epithelial stem cells and innate immune cells contribute to the repair and regeneration of ALI/ARDS. Transl Res. 272, 111–125. https://doi.org/10.1016/j.trsl.2024.05.012 (2024).
Ding, N., Li, P., Li, H., Lei, Y. & Zhang, Z. The ROCK-ezrin signaling pathway mediates LPS-induced cytokine production in pulmonary alveolar epithelial cells. Cell. Commun. Signal. 20, 65. https://doi.org/10.1186/s12964-022-00879-3 (2022).
Tao, H., Xu, Y. & Zhang, S. The role of macrophages and alveolar epithelial cells in the development of ARDS. Inflammation 46, 47–55. https://doi.org/10.1007/s10753-022-01726-w (2023).
Cattel, F. et al. Use of exogenous pulmonary surfactant in acute respiratory distress syndrome (ARDS): role in SARS-CoV-2-related lung injury. Respir Physiol. Neurobiol. 288, 103645. https://doi.org/10.1016/j.resp.2021.103645 (2021).
Mikolka, P. et al. The synthetic surfactant CHF5633 restores lung function and lung architecture in severe acute respiratory distress syndrome in adult rabbits. Lung 202, 299–315. https://doi.org/10.1007/s00408-024-00689-z (2024).
Haitsma, J. J. & Lachmann, B. Lung protective ventilation in ARDS: the open lung maneuver. Minerva Anestesiol. 72, 117–132 (2006).
Fan, E., Brodie, D. & Slutsky, A. S. Acute respiratory distress syndrome: advances in diagnosis and treatment. Jama 319, 698–710. https://doi.org/10.1001/jama.2017.21907 (2018).
Vettorazzi, S. et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat. Commun. 6, 7796. https://doi.org/10.1038/ncomms8796 (2015).
Guo, H. et al. Peptide-guided delivery improves the therapeutic efficacy and safety of glucocorticoid drugs for treating acute lung injury. Mol. Ther. 31, 875–889. https://doi.org/10.1016/j.ymthe.2023.01.003 (2023).
Kim, H. S., Quon, M. J. & Kim, J. A. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2, 187–195. https://doi.org/10.1016/j.redox.2013.12.022 (2014).
Safwat, M. A., Kandil, B. A., Elblbesy, M. A., Soliman, G. M. & Eleraky, N. E. Epigallocatechin-3-gallate-loaded gold nanoparticles: Preparation and evaluation of anticancer efficacy in Ehrlich tumor-bearing mice. Pharmaceuticals (Basel). 13. https://doi.org/10.3390/ph13090254 (2020).
Alam, M. et al. Epigallocatechin-3-gallate therapeutic potential in human diseases: molecular mechanisms and clinical studies. Mol. Biomed. 5, 73. https://doi.org/10.1186/s43556-024-00240-9 (2024).
Xiao, J. et al. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through TGF/SMAD, PI3 K/Akt/FoxO1, and NF-kappa B pathways. Eur. J. Nutr. 53, 187–199. https://doi.org/10.1007/s00394-013-0516-8 (2014).
Lakshmi, S. P., Reddy, A. T., Kodidhela, L. D. & Varadacharyulu, N. C. Epigallocatechin gallate diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells. Life Sci. 259, 118260. https://doi.org/10.1016/j.lfs.2020.118260 (2020).
Liu, R. et al. Mitochondrial DNA-Induced inflammatory responses and lung injury in thermal injury rat model: protective effect of epigallocatechin gallate. J. Burn Care Res. 38, 304–311. https://doi.org/10.1097/bcr.0000000000000501 (2017).
Almatroodi, S. A. et al. Epigallocatechin-3-Gallate (EGCG), an active compound of green tea attenuates acute lung injury regulating macrophage polarization and Krüpple-Like-Factor 4 (KLF4) expression. Molecules 25 https://doi.org/10.3390/molecules25122853 (2020).
Mehmood, S. et al. Epigallocatechin gallate: Phytochemistry, bioavailability, utilization challenges, and strategies. J. Food Biochem. 46, e14189. https://doi.org/10.1111/jfbc.14189 (2022).
Fatehullah, A. et al. Interactions of the 67 kDa laminin receptor and its precursor with laminin. Biosci. Rep. 30, 73–79. https://doi.org/10.1042/bsr20090023 (2009).
Pesapane, A., Ragno, P., Selleri, C. & Montuori, N. Recent advances in the function of the 67 kDa laminin receptor and its targeting for personalized therapy in cancer. Curr. Pharm. Des. 23, 4745–4757. https://doi.org/10.2174/1381612823666170710125332 (2017).
Yamada, S. et al. Epigallocatechin-3-O-gallate up-regulates microRNA-let-7b expression by activating 67-kDa laminin receptor signaling in melanoma cells. Sci. Rep. 6, 19225. https://doi.org/10.1038/srep19225 (2016).
Hong Byun, E., Fujimura, Y., Yamada, K. & Tachibana, H. TLR4 signaling inhibitory pathway induced by green tea polyphenol epigallocatechin-3-gallate through 67-kDa laminin receptor. J. Immunol. 185, 33–45. https://doi.org/10.4049/jimmunol.0903742 (2010).
Byun, E. B., Choi, H. G., Sung, N. Y. & Byun, E. H. Green tea polyphenol epigallocatechin-3-gallate inhibits TLR4 signaling through the 67-kDa laminin receptor on lipopolysaccharide-stimulated dendritic cells. Biochem. Biophys. Res. Commun. 426, 480–485. https://doi.org/10.1016/j.bbrc.2012.08.096 (2012).
Chen, J. et al. The laminin receptor modulates granulocyte-macrophage colony-stimulating factor receptor complex formation and modulates its signaling. Proc. Natl. Acad. Sci. U S A. 100, 14000–14005. https://doi.org/10.1073/pnas.2334584100 (2003).
Ménard, S., Tagliabue, E. & Colnaghi, M. I. The 67 kDa laminin receptor as a prognostic factor in human cancer. Breast Cancer Res. Treat. 52, 137–145. https://doi.org/10.1023/a:1006171403765 (1998).
Mokra, D., Adamcakova, J. & Mokry, J. Green tea polyphenol (-)-Epigallocatechin-3-Gallate (EGCG): A time for a new player in the treatment of respiratory diseases? Antioxid. (Basel). 11. https://doi.org/10.3390/antiox11081566 (2022).
Formisano, P. et al. PED/PEA-15 interacts with the 67 kD laminin receptor and regulates cell adhesion, migration, proliferation and apoptosis. J. Cell. Mol. Med. 16, 1435–1446. https://doi.org/10.1111/j.1582-4934.2011.01411.x (2012).
Nelson, J. et al. The 67 kDa laminin receptor: structure, function and role in disease. Biosci. Rep. 28, 33–48. https://doi.org/10.1042/bsr20070004 (2008).
Cloutier, G., Sallenbach-Morrissette, A. & Beaulieu, J. F. Non-integrin laminin receptors in epithelia. Tissue Cell. 56, 71–78. https://doi.org/10.1016/j.tice.2018.12.005 (2019).
Hsu, S. C., Wu, N. P., Lu, Y. C. & Ma, Y. H. Laminin receptor-mediated nanoparticle uptake by tumor cells: interplay of epigallocatechin gallate and magnetic force at nano-bio interface. Pharmaceutics. 14 https://doi.org/10.3390/pharmaceutics14081523 (2022).
Fujimura, Y. et al. The involvement of the 67 kDa laminin receptor-mediated modulation of cytoskeleton in the degranulation Inhibition induced by epigallocatechin-3-O-gallate. Biochem. Biophys. Res. Commun. 348, 524–531. https://doi.org/10.1016/j.bbrc.2006.07.086 (2006).
Zhong, Y. et al. The bidirectional role of the JAK2/STAT3 signaling pathway and related mechanisms in cerebral ischemia-reperfusion injury. Exp. Neurol. 341, 113690. https://doi.org/10.1016/j.expneurol.2021.113690 (2021).
Sopjani, M., Morina, R., Uka, V., Xuan, N. T. & Dërmaku-Sopjani, M. JAK2-mediated intracellular signaling. Curr. Mol. Med. 21, 417–425. https://doi.org/10.2174/1566524020666201015144702 (2021).
Huang, B., Lang, X. & Li, X. The role of IL-6/JAK2/STAT3 signaling pathway in cancers. Front. Oncol. 12, 1023177. https://doi.org/10.3389/fonc.2022.1023177 (2022).
Jaśkiewicz, A., Domoradzki, T. & Pająk, B. Targeting the JAK2/STAT3 pathway-can we compare it to the two faces of the god janus? Int. J. Mol. Sci. 21 https://doi.org/10.3390/ijms21218261 (2020).
Xu, G. et al. Berberine administrated with different routes attenuates inhaled LPS-induced acute respiratory distress syndrome through TLR4/NF-κB and JAK2/STAT3 Inhibition. Eur. J. Pharmacol. 908, 174349. https://doi.org/10.1016/j.ejphar.2021.174349 (2021).
Qi, L. et al. Mechanism of Qingdai in alleviating acute lung injury by inhibiting the JAK2/STAT3 signaling pathway. J. Inflamm. Res. 17, 11403–11417. https://doi.org/10.2147/jir.S498299 (2024).
Mao, L. et al. Green tea polyphenol (-)-epigallocatechin gallate (EGCG) attenuates neuroinflammation in palmitic acid-stimulated BV-2 microglia and high-fat diet-induced obese mice. Int. J. Mol. Sci. 20 https://doi.org/10.3390/ijms20205081 (2019).
Zhu, L. et al. Astaxanthin ameliorates lipopolysaccharide-induced acute lung injury via inhibition of inflammatory reactions and modulation of the SOCS3/JAK2/STAT3 signaling pathways in mice. Food Funct. 13, 11638–11651. https://doi.org/10.1039/d2fo02182j (2022).
Madikyzy, M. et al. Honghua extract mediated potent inhibition of COVID-19 host cell pathways. Sci. Rep. 12, 14296. https://doi.org/10.1038/s41598-022-15338-9 (2022).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kim, Y. et al. Seq-scope protocol: repurposing illumina sequencing flow cells for high-resolution spatial transcriptomics. bioRxiv. https://doi.org/10.1101/2024.03.29.587285 (2024).
Yu, J. et al. Liang-Ge-San protects against viral infection-induced acute lung injury through inhibiting α7nAChR-mediated mitophagy. Phytomedicine 135, 156231. https://doi.org/10.1016/j.phymed.2024.156231 (2024).
Liu, G. Z., Xu, X. W., Tao, S. H., Gao, M. J. & Hou, Z. H. HBx facilitates ferroptosis in acute liver failure via EZH2 mediated SLC7A11 suppression. J. Biomed. Sci. 28, 67. https://doi.org/10.1186/s12929-021-00762-2 (2021).
Bao, S. et al. Epigallocatechin gallate (EGCG) suppresses lipopolysaccharide-induced Toll-like receptor 4 (TLR4) activity via 67 kDa laminin receptor (67LR) in 3T3-L1 adipocytes. J. Agric. Food Chem. 63, 2811–2819. https://doi.org/10.1021/jf505531w (2015).
Li, Y. F. et al. Epigallocatechin-3-gallate inhibits matrix metalloproteinase-9 and monocyte chemotactic protein-1 expression through the 67-κDa laminin receptor and the TLR4/MAPK/NF-κB signalling pathway in lipopolysaccharide-induced macrophages. Cell. Physiol. Biochem. 43, 926–936. https://doi.org/10.1159/000481643 (2017).
Bos, L. D. J. & Ware, L. B. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet 400, 1145–1156. https://doi.org/10.1016/s0140-6736(22)01485-4 (2022).
Müller, A. M., Cronen, C., Müller, K. M. & Kirkpatrick, C. J. Heterogeneous expression of cell adhesion molecules by endothelial cells in ARDS. J. Pathol. 198, 270–275. https://doi.org/10.1002/path.1186 (2002).
Zhang, H. et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int. J. Biol. Sci. 18, 3337–3357. https://doi.org/10.7150/ijbs.69141 (2022).
Cui, Y. et al. MiR-29a-3p improves acute lung injury by reducing alveolar epithelial cell PANoptosis. Aging Dis. 13, 899–909. https://doi.org/10.14336/ad.2021.1023 (2022).
Kaku, S. et al. Acute respiratory distress syndrome: Etiology, pathogenesis, and summary on management. J. Intensive Care Med. 35, 723–737. https://doi.org/10.1177/0885066619855021 (2020).
Danciu, C. Natural bioactive compounds, vegetal extracts and modern pharmaceutical formulations: new insights into the anti-cancer mechanism of action. Anticancer Agents Med. Chem. 20, 1754–1755. https://doi.org/10.2174/187152062015200911152012 (2020).
Ohishi, T., Goto, S., Monira, P., Isemura, M. & Nakamura, Y. Anti-inflammatory action of green tea. Antiinflamm Antiallergy Agents Med. Chem. 15, 74–90. https://doi.org/10.2174/1871523015666160915154443 (2016).
Huang, X., Zhu, W., Zhang, H., Qiu, S. & Shao, H. SARS-CoV-2 N protein induces alveolar epithelial apoptosis via NLRP3 pathway in ARDS. Int. Immunopharmacol. 144, 113503. https://doi.org/10.1016/j.intimp.2024.113503 (2025).
Cui, Y. R. et al. Beneficial effects of aloperine on inflammation and oxidative stress by suppressing necroptosis in lipopolysaccharide-induced acute lung injury mouse model. Phytomedicine 100, 154074. https://doi.org/10.1016/j.phymed.2022.154074 (2022).
Xu, B. et al. Luteoloside ameliorates sepsis-induced acute lung injury via AMPK-ULK1 pathway-mediated autophagy. Histol. Histopathol. 18922 https://doi.org/10.14670/hh-18-922 (2025).
Kawai, K. et al. Epigallocatechin gallate induces apoptosis of monocytes. J. Allergy Clin. Immunol. 115, 186–191. https://doi.org/10.1016/j.jaci.2004.10.005 (2005).
Xu, M. J. et al. Epigallocatechin-3-gallate inhibits TLR4 signaling through the 67-kDa laminin receptor and effectively alleviates acute lung injury induced by H9N2 swine influenza virus. Int. Immunopharmacol. 52, 24–33. https://doi.org/10.1016/j.intimp.2017.08.023 (2017).
Wang, H., Liu, W., Yu, F. & Lu, L. Identification of (-)-epigallocatechin-3-gallate as a potential agent for blocking infection by grass carp reovirus. Arch. Virol. 161, 1053–1059. https://doi.org/10.1007/s00705-016-2751-9 (2016).
Wang, T. et al. (-)-Epigallocatechin gallate targets Notch to attenuate the inflammatory response in the immediate early stage in human macrophages. Front. Immunol. 8, 433. https://doi.org/10.3389/fimmu.2017.00433 (2017).
Moore, B. C. & Simmons, D. L. COX-2 inhibition, apoptosis, and chemoprevention by nonsteroidal anti-inflammatory drugs. Curr. Med. Chem. 7, 1131–1144. https://doi.org/10.2174/0929867003374273 (2000).
Vonkeman, H. E. & van de Laar, M. A. Nonsteroidal anti-inflammatory drugs: adverse effects and their prevention. Semin Arthritis Rheum. 39, 294–312. https://doi.org/10.1016/j.semarthrit.2008.08.001 (2010).
Bacchi, S., Palumbo, P., Sponta, A. & Coppolino, M. F. Clinical pharmacology of non-steroidal anti-inflammatory drugs: a review. Antiinflamm Antiallergy Agents Med. Chem. 11, 52–64. https://doi.org/10.2174/187152312803476255 (2012).
Xu, J., Wang, B. & Ao, H. Corticosterone effects induced by stress and immunity and inflammation: mechanisms of communication. Front. Endocrinol. (Lausanne). 16, 1448750. https://doi.org/10.3389/fendo.2025.1448750 (2025).
Oray, M., Abu Samra, K., Ebrahimiadib, N., Meese, H. & Foster, C. S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 15, 457–465. https://doi.org/10.1517/14740338.2016.1140743 (2016).
Shaik-Dasthagirisaheb, Y. B. et al. Role of vitamins D, E and C in immunity and inflammation. J. Biol. Regul. Homeost. Agents. 27, 291–295 (2013).
Shakoor, H. et al. Immune-boosting role of vitamins D, C, E, zinc, selenium and omega-3 fatty acids: could they help against COVID-19? Maturitas 143, 1–9. https://doi.org/10.1016/j.maturitas.2020.08.003 (2021).
Huang, S. P. et al. Broussonin E suppresses LPS-induced inflammatory response in macrophages via inhibiting MAPK pathway and enhancing JAK2-STAT3 pathway. Chin. J. Nat. Med. 17, 372–380. https://doi.org/10.1016/s1875-5364(19)30043-3 (2019).
Chen, X. M., Yu, Y. H., Wang, L., Zhao, X. Y. & Li, J. R. Effect of the JAK2/STAT3 signaling pathway on nerve cell apoptosis in rats with white matter injury. Eur. Rev. Med. Pharmacol. Sci. 23, 321–327. https://doi.org/10.26355/eurrev_201901_16779 (2019).
Gajjela, B. K. & Zhou, M. M. Calming the cytokine storm of COVID-19 through Inhibition of JAK2/STAT3 signaling. Drug Discov Today. 27, 390–400. https://doi.org/10.1016/j.drudis.2021.10.016 (2022).
Li, W. et al. Classic signaling pathways in alveolar injury and repair involved in Sepsis-Induced ALI/ARDS: new research progress and prospect. Dis. Mark. 2022, 6362344. https://doi.org/10.1155/2022/6362344 (2022).
Qin, Y. et al. Lipopolysaccharide induces epithelial-mesenchymal transition of alveolar epithelial cells cocultured with macrophages possibly via the JAK2/STAT3 signaling pathway. Hum. Exp. Toxicol. 39, 224–234. https://doi.org/10.1177/0960327119881678 (2020).
Zhang, X. S. et al. Caprylic acid improves lipid Metabolism, suppresses the inflammatory response and activates the ABCA1/p-JAK2/p-STAT3 signaling pathway in C57BL/6J mice and RAW264.7 cells. Biomed. Environ. Sci. 35, 95–106. https://doi.org/10.3967/bes2022.014 (2022).
Acknowledgements
This study acknowledges the significant technical support and resource provision from the Southwest Medical University’s Advanced Precision Instrument platform, Cellular and Molecular platform, and Animal Center. These platforms provided comprehensive assistance in the use of equipment, experimental operations, and data analysis, thereby laying a solid foundation for the smooth implementation and high-quality completion of the research.
Funding
This investigation was financially supported by the Yibin Science and Technology program (Grant No. 2023SF002) and the Jiang’an County Bureau of Economic Commerce, Informatization, and Science and Technology (Grant No. 2023SF01), the Natural Science Foundation of Sichuan Province (No. 2022NSFSC0046), the Sichuan Science and Technology program (No. 2022YFS0631), the Luzhou Science and Technology program (No. 2023JYJ049), the School-level Scientific Research project of Southwestern Medical University (No. 2023ZD007), the Research Start-up Fund of Affiliated Hospital of Southwest Medical University and the Fund of the “Tianfu Talent” project.
Author information
Authors and Affiliations
Contributions
The conception and design of the study were undertaken by Shiming Fan, Xianming Fan, Xiaoqing Fan, and Dan Luo. The experimental procedures were executed by Shiming Fan and Xiaoqing Fan, while the statistical analysis and data interpretation were performed by Shiming Fan and Dan Luo. The manuscript was initially drafted by Shiming Fan. All authors critically reviewed the manuscript and ultimately approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, S., Fan, X., Ma, N. et al. Epigallocatechin-3-gallate ameliorates lipopolysaccharide-induced inflammation via the 67LR/JAK2/STAT3 signaling pathway. Sci Rep 15, 42738 (2025). https://doi.org/10.1038/s41598-025-26875-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-26875-4
